

Abstract
Genetic code reprogramming is a powerful approach to controlled protein modification. A remaining challenge, however, is the generation of vacant codons. We targeted the initiation machinery of E. coli, showing that restriction of the formyl donor or inhibition of the formyl transferase during in vitro translation is sufficient to prevent formylation of the acylated initiating tRNA and thereby create a vacant initiation codon that can be reprogrammed by exogenously charged tRNA. Our approach conveniently generates peptides and proteins tagged N‐terminally with non‐canonical functional groups at up to 99 % reprogramming efficiency, in combination with decoding the AUG elongation codons either with native methionine or with further reprogramming with azide‐ and alkyne‐containing cognates. We further show macrocyclization and intermolecular modifications with these click handles, thus emphasizing the applicability of our method to current challenges in peptide and protein chemistry.
Keywords: Bioconjugation, cyclic peptides, genetic code reprogramming, protein modification, protein engineering
An approach is presented for the facile generation of a vacant initiation codon, based on preventing formylation of the endogenous initiator. This can be achieved by either removing its substrate or adding an inhibitor, and it enables the generation of peptides and proteins reprogrammed with multiple and diverse functional groups and reactive handles.
Genetic code reprogramming provides many opportunities in the generation of biomolecules with novel and useful properties. Examples include antibody‐drug conjugates, conjugated vaccines, mechanistic studies, and generating new catalysts.[ 1 , 2 , 3 ] Reprogramming of the genetic code requires a tRNA charged with an unnatural amino acid and a vacant codon. Multiple approaches have been developed for the first requirement and these are generally robust. For tRNA aminoacylation in cells or in vivo the most common approach is by engineered tRNA and amino‐acyl tRNA synthetase pairs, [4] while in vitro more chemoenzymatic approaches are possible such as by ligation of a pdCpA‐amino acid with a truncated tRNA, [5] or by acylation of an activated amino acid onto in vitro generated tRNA by a catalytic RNA called flexizyme. [6] Mis‐recognition of near cognates by the endogenous amino‐acyl tRNA synthetases also provides a convenient approach, [7] where possible. With flexizyme‐mediated acylation a broad range of moieties have proven amenable to ribosomal translation, with the N‐terminus proving particularly permissive and allowing boron clusters, [8] exotic peptide fragments, [9] and even foldamers. [10] In addition, the N‐terminus of proteins is a common target for chemical modification. [11] Approaches for generating a vacant initiating codon are thus particularly valuable.
One crucial challenge that remains only partially solved is an efficient way to generate a vacant codon to reprogram. The most common current approach is reprogramming of a stop codon, [12] and is also compatible with initiation. [13] However, this is in competition with release factors, which in many cases results in low efficiency and truncated products. Sense codon reprogramming during in vitro translation can efficiently provide a vacant codon, but requires the use of a reconstituted recombinant translation system to allow for omission of canonical amino acids and prevents generation of sequences with all 20 canonical amino acids.[ 6 , 14 ] There clearly remains a demand for alternative approaches.
Recently, during efforts to generate multiply reprogrammed peptides using an in vitro translation system [15] we observed that methionine near‐cognates such as homopropargylglycine (Hpg) and azidohomoalanine (Aha) competed with flexizyme‐charged initiator aminoacyl‐tRNA (Ac‐Phe‐tRNAfMet), giving a mixture of reprogrammed and formyl‐Hpg‐initiated products (Figure 2). While not unexpected, as initiation with these near‐cognates has been reported before, [16] it lead us to investigate ways of improving the ratio of desired (reprogrammed) product and to enable access to a broader range of N‐terminal non‐canonical amino acids.
Figure 2.
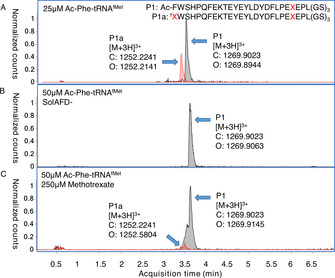
UPLC‐MS extracted‐ion chromatograms (EIC) from analyses of translation tests with reprogrammed initiation. A) Competition between 25 μM Ac‐Phe‐tRNAfMet and endogenous initiators (69:31 P1/P1 a). B) As for (A) but using SolAFD‐ and with 50 μM Ac‐Phe‐tRNAfMet (100 % P1, byproduct undetectable). C) As for (A), but with 250 μM methotrexate and 50 μM Ac‐Phe‐tRNAfMet (93:7 P1/P1 a). X=homopropargylglycine, f=N‐formylation.
Given that omitting the methionine cognate is not an option in this case, we turned our attention to the bacterial formylation apparatus. In bacterial initiation, the enzyme methionyl‐tRNA formyltransferase (MTF) is responsible for transferring a formyl group from 10‐formyltetrahydrofolate (10‐CHO‐THF) to methionyl‐tRNAfMet generated by methionyl‐tRNA synthetase (MetRS). [17] This formylated methionyl‐tRNA (fMet‐tRNAfMet) is then recognized by initiation factors (IF1, IF2‐GTP, and IF3) and the ribosome to form the pre‐30S initiation complex.[ 18 , 19 ] Typically the N‐terminal methionine and its formyl group are removed following translation in a series of enyzmatic steps by peptide deformylase (PDF) and methionine amino peptidase (MAP). In some cases, however, the N‐terminal processing is attenuated and this can be exploited by changing the identity of the second amino acid in the protein sequence to provide proteins that retain a methionine analogue. [20] It should also be noted that there also exist eubacteria that do not require formylation for initiation of protein synthesis. [21] Nonetheless, for Escherichia coli the formyl moiety is an important recognition factor for the initiation process, [22] and so we reasoned that if we could disrupt the activity of MTF we could prevent generation of fMet‐tRNAfMet or its ana‐logues and thus bias initiation in favor of an exogenously charged aminoacyl‐tRNAfMet. To that end, in the current work we explore the possibility of preventing the formylation process in initiation of bacterial in vitro translation, either by substrate restriction or by addition of an inhibitor, to create vacant start codons without removal of any canonical amino acids (Figure 1).
Figure 1.
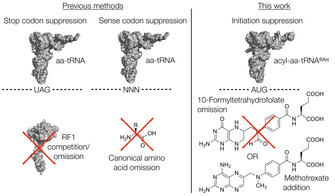
Previous and current approaches to vacant codon creation. aa=amino acid, RF1=release factor 1.
Translation of a model peptide was carried out in the “PURExpress” in vitro translation sytem (a commercialised version of the PURE system [15] ) with homopropargylglycine substituted for methionine and supplemented with 25 μM initiator aminoacyl‐tRNA charged with N‐acetyl phenylalanine by means of flexizyme [6] (an aminoacylating ribozyme), Ac‐Phe‐tRNAfMet. This gave a mixture of reprogrammed and formyl‐Hpg‐initiated products in a ratio of roughly 2:1 (Figure 2 A and S1). To prevent MTF activity, and thus hopefully suppress this undesired formylated product, we removed its source of the formyl group by using a previously reported custom energy solution (SolAFD‐) [6] that omits 10‐CHO‐THF. Using this in an otherwise identical translation to above, still substituting homopropargylglycine for methionine and adding 25 μM of Ac‐Phe‐tRNAfMet, changed the ratio of desired product to side product to 92:8, while also changing the nature of the side product to the non‐formylated form (Figure S2). Increasing the concentration of Ac‐Phe‐tRNAfMet to 50 μM was found to give only the desired product P1, with all initiation side‐products being undetectable by UPLC‐MS (Figure 2 B and S3). Simple removal of 10‐CHO‐THF from the translation is thus sufficient for reprogramming of the initiation codon even in the presence of a substrate for MetRS, provided that sufficient amounts of exogenous aminoacyl‐tRNAfMet are added.
As an alternative to 10‐CHO‐THF omission, we also investigated whether MTF activity could be suppressed by a competitive inhibitor to achieve the same result. This removes the need for a custom energy solution, allowing reprogramming with only simple commercial reagents. One candidate inhibitor was found in methotrexate, a chemotherapy drug that targets dihydrofolate reductase and which is a close analogue of 10‐CHO‐THF. This has previously been shown to be a modest inhibitor of algal MTF. [23] Adding 25 μM Ac‐Phe‐tRNAfMet and 100 μM methotrexate to a translation reaction using the commercial translation kit components gave an improved fraction of reprogrammed initiation product (83 %, Figure S4), with the only detectable side product still being initiation with formylated homopropargylglycine (17 %). This reaction contains the substrates 10‐CHO‐THF and homopropargylglycine as well as the enzymes MetRS and MTF, and so should be formylation‐competent. Increasing the methotrexate concentration to 250 μM and the Ac‐Phe‐tRNAfMet concentration to 50 μM gave a further improvement to 93 % fractional yield of the fully reprogrammed product (Figure 2 C and S5). This indicates that initiation reprogramming by MTF suppression by addition of an inexpensive commercially available inhibitor is compatible with the presence of all natural translation components.
An additional trace side product that could be detected in some cases was a peptide starting from the second amino acid (“initiator‐truncated”), which is also observed for vacant codon creation by methionine omission in cases of less efficient reprogramming.[ 10 , 24 ] A control reaction using the custom SolAFD‐ but with no flexizyme‐mediated reprogramming (still containing the Met analogue Hpg) afforded peptide product with the initiating amino acid present in the formylated form (P2 a; 25 %), to our surprise, as well as the free amine (P2 b; 63 %) and the initiator‐truncated peptide (P2 c; 12 %; Figure 3 A and S6). To investigate whether this initiator‐truncated product can be exploited as a further alternative to initiation with formyl‐methionine, we tested whether addition of excess un‐acylated in vitro‐transcribed tRNAfMet to the translation mix would improve the yield and purity of initiator‐truncated product. An additional 50 μM tRNAfMet indeed gave clean production of an abundant amount of this initiator‐truncated product (Figure 3 B and S7). Although accurate absolute quantification of the product is not possible with the UPLC‐MS method used, this reaction gave no detectable peptide side products and peak area was comparable to the initiation‐reprogrammed translations above. Free tRNAfMet is clearly initiation‐competent in the PURE system, and addition of an excess is a practical solution to its low efficiency. This is consistent with reported upregulation of expression of tRNAfMet as an adaptation to deletion or inhibition of MTF in E. coli, allowing initiation of protein synthesis to resume. [25] Uncharged tRNA addition thus provides an alternative approach for production of peptides with a free amine at the N‐terminus, as compared to post‐translational removal of the initiating formyl methionine by the enzymes peptide deformylase and methionine aminopeptidase (PDF/MAP). [26] Our approach does not require any additional proteins to be purified and also removes restrictions imposed by MAP's substrate scope for the amino acid in the second position. [27]
Figure 3.
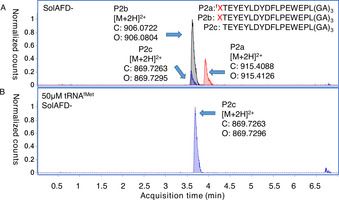
EIC from UPLC‐MS analyses of translation without reprogramming initiation. A) Control translation in the absence of both formyl donor and reprogrammed initiator tRNA (25:63:12 P2 a/P2 b/P2 c). B) As for (A) but with additional uncharged tRNAfMet. X=homopropargylglycine, f=N‐formylation.
Encouraged by these results, we next investigated the scope of the 10‐CHO‐THF omission approach for initiation with amino acids bearing different functional groups as N‐acyl modifications. In addition to simple capping by an acetyl group, we investigated a chloroacetyl group for thioether head‐to‐sidechain cyclisation with cysteine, [28] an aminomethylbenzyl group for fluorogenic oxidative cyclization/derivatization reactions with hydroxytryptophan, [29] a fluorescein derivative, and biotin. For decoding of the elongation AUG codon we included two “clickable” near cognates of methionine, homopropargylglycine and azidohomoalanine. In all cases a good ratio of desired product to side‐products was obtained, and in comparable yields to a non‐reprogrammed translation by peak area in UPLC‐MS (Table 1 and S1, Figures S1–S18). Methionine is also able to be included in this approach (Table 1 entry 9 and Figure S17), allowing translation of otherwise completely native sequences with only initiation reprogrammed and showing that generation of the vacant codon is not dependent on the use of a near‐cognate. These reactions were not further optimized with additional exogenous initiating tRNA and observed variations in flexizyme acylation yields were not accounted for (Figure S19), so there remains scope to optimize in cases where complete suppression of the side products is needed. Unlike recently reported approaches to N‐terminal reprogramming that are based on engineered bacteria,[ 30 , 31 ] our approach allows convenient access to a much broader scope of functional groups as it does not need orthogonal tRNA/aminoacyl tRNA synthetase pairs to fill the vacated initiation codon.
Table 1.
Products of translation reprogrammed with various combinations of initiating amino acid and methionine analogues for the peptide sequence (M)WSHPQFEKTEYEYLDYDFLPE(M)EPLGSGSGS.
|
Entry |
Initiator[a] (R) |
Elongator[a] (R′) |
Product ratio (N‐terminus as shown)[b] |
|||
|---|---|---|---|---|---|---|
|
|
|
|
|
|
|
|
|
|
|
|
(reprogrammed) |
(formyl‐Met or analogue) |
(Met or analogue) |
(free amine) |
|
1 |
ClAc‐Tyr |
Hpg |
>99 |
0 |
0 |
0 |
|
2 |
ClAc‐Tyr |
Aha |
>99 |
0 |
0 |
0 |
|
3 |
AMB‐Phe |
Hpg |
77 |
4 |
14 |
5 |
|
4 |
AMB‐Phe |
Aha |
65 |
5 |
14 |
16 |
|
5 |
(5/6)FAM‐Phe |
Hpg |
78 |
4 |
9 |
9 |
|
6 |
(5/6)FAM‐Phe |
Aha |
72 |
7 |
11 |
10 |
|
7 |
Biotin‐Phe |
Hpg |
>99 |
0 |
0 |
0 |
|
8 |
Biotin‐Phe |
Aha |
>99 |
0 |
0 |
0 |
|
9 |
Ac‐Phe |
Met |
>99 |
0 |
0 |
0 |
|
10 |
Pyn‐Phe |
Aha |
>99 |
0 |
0 |
0 |
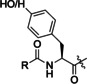
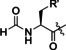
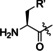
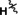
[a] Aha: azidohomoalanine, Hpg: homopropargylglycine, AMB: 4‐aminomethylbenzoic acid, ClAc: chloroacetyl, FAM: carboxyfluorescein, Pyn: 4‐pentynoic acid. [b] Determined by relative peak areas in UPLC‐MS EIC traces. R and R′ reflect the initiator and AUG elongator, respectively.
Given that we were able to show translation of clickable near‐cognates of methionine along with reprogramming of the initiating position, we investigated the application of copper‐catalyzed azide‐alkyne cycloaddition reactions to macrocyclization of the products. [32] A peptide P3 initiated with N‐(4‐pentynoyl) phenylalanine (“Pyn‐Phe”) and containing a downstream azidohomoalanine was treated with CuI directly in the translation mixture. Clean conversion to the macrocyclized product P4 was shown by retention shift of the isobaric product (Figure 4) and further confirmed by treatment with tris(2‐carboxyethyl)phosphine (TCEP), which gave no detectable azide reduction. This click macrocyclization reaction is particularly convenient as it was carried out without any intermediate purification or removal of oxygen, and is anticipated to be compatible with mRNA display as well as further modifications such as chemical S‐glycosylation at cysteine residues. [33]
Figure 4.
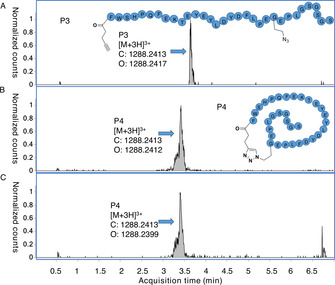
EIC from UPLC‐MS analyses of peptide macrocyclisation following reprogrammed translation. A) Peptide translation with an alkynyl initiator and downstream azidohomoalanine (100 % P3, no detectable side products). B) The same peptide after overnight treatment with 1.2 mM CuI.THPTA and aminoguanidine hydrochloride scavenger. C) Macrocyclized product from (B) treated with 35 mM TCEP for one hour (mass peak for reduction not detectable).
Finally, we tested translation of a small panel of proteins with (+)‐biotinyl phenylalanine as the initiating amino acid and azidohomoalanine reprogramming the elongation AUG codons, with formylation suppressed by methotrexate inhibition of MTF. Chosen as model proteins were a nanobody, a protein used in folding studies (E. coli dihydrofolate reductase), and a carrier protein used in conjugate vaccine development (tetanospasmin toxoid). Protein products were reacted with DBCO‐Sulfo‐Cy3 in a copper‐free click reaction under mild denaturing conditions, then pulled down by magnetic streptavidin beads and analyzed by SDS‐PAGE gel detected by both fluorescence and silver staining (Figure 5 and S20). Pull‐down by biotin confirms the initiation reprogramming while attachment of fluorophore confirms orthogonal reprogramming of the elongator AUG codons, together demonstrating facile generation of doubly reprogrammed proteins amenable to further covalent modification. Quantification of yield was estimated by densitometry of the silver stain band for a “non‐clicked” product referenced to a standard curve of bovine serum albumin to give 40 ng μL−1 (Figure S21), consistent with the manufacturer's estimated yield for non‐reprogrammed translation. We envisage our method to be suited for applications such as generating immobilized proteins for protein interaction experiments, conjugate vaccines carrying multiple epitopes, and generating proteins with multiple fluorophores for conformational studies.
Figure 5.
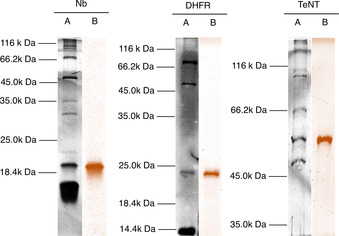
SDS‐PAGE showing translation of proteins under a multiply reprogrammed genetic code (Biotin‐Phe initation and Aha decoding of AUG elongator codons). A: silver staining, B: Cy3 fluorescence (color applied in processing), Nb: nanobody, DHFR: dihydrofolate reductase, TeNT: tetanospasmin toxoid.
We have shown here that suppression of formylation is sufficient to liberate the initiation codon in E. coli‐derived in vitro translation, achievable by removing the formyl donor or by inhibiting the enzyme MTF. We have demonstrated reprogramming of the genetic code on this vacated codon using amino acids acylated with non‐canonical functional groups such as reactive handles, a fluorophore, and biotin, and we have shown this to be applicable to protein translation as well as peptides. This initiation reprogramming is compatible with translation of downstream AUG codons by methionine, or exploiting mis‐acylation of near cognates by MetRS to incorporate clickable handles to provide convenient access to doubly modified proteins. The method we report here provides a novel and practical approach to creating a vacant initiation codon, and allows rapid access to N‐terminal reprogramming with a broad range of chemical functionality using simple molecular biology techniques and easily accessible reagents.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We are grateful for funding support provided by the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska‐Curie grant agreement No. 746631 to SAKJ, and for a CSC scholarship to ML. We thank Prof. P van Bergen en Henegouwen and Prof. GJPH Boons (both of Utrecht University) for providing Nb and TeNT plasmids, respectively.
M. Liu, V. Thijssen, S. A. K. Jongkees, Angew. Chem. Int. Ed. 2020, 59, 21870.
References
- 1. Richardson S. L., Dods K. K., Abrigo N. A., Iqbal E. S., Hartman M. C., Curr. Opin. Chem. Biol. 2018, 46, 172–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Huang Y., Liu T., Synth. Syst. Biotechnol. 2018, 3, 150–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wals K., Ovaa H., Front. Chem. 2014, 2, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.”Directed Evolution of Orthogonal Pyrrolysyl-tRNA Synthetases in Escherichia coli for the Genetic Encoding of Noncanonical Amino Acids”: Schmidt M. J., Summerer D., in Noncanonical Amino Acids, Springer, Berlin, 2018, pp- 91–111. [DOI] [PubMed] [Google Scholar]
- 5. Duca M., Chen S., Hecht S. M., Methods 2008, 44, 87–99. [DOI] [PubMed] [Google Scholar]
- 6. Goto Y., Katoh T., Suga H., Nat. Protoc. 2011, 6, 779–790. [DOI] [PubMed] [Google Scholar]
- 7. Hartman M. C. T., Josephson K., Szostak J. W., Proc. Natl. Acad. Sci. USA 2006, 103, 4356–4361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yin Y., Ochi N., Craven T. W., Baker D., Takigawa N., Suga H., J. Am. Chem. Soc. 2019, 141, 19193–19197. [DOI] [PubMed] [Google Scholar]
- 9. Goto Y., Suga H., J. Am. Chem. Soc. 2009, 131, 5040–5041. [DOI] [PubMed] [Google Scholar]
- 10. Rogers J. M., Kwon S., Dawson S. J., Mandal P. K., Suga H., Huc I., Nat. Chem. 2018, 10, 405–412. [DOI] [PubMed] [Google Scholar]
- 11. Rosen C. B., Francis M. B., Nat. Chem. Biol. 2017, 13, 697–705. [DOI] [PubMed] [Google Scholar]
- 12. Noren C. J., Anthony-Cahill S. J., Griffith M. C., Schultz P. G., Science 1989, 244, 182–188. [DOI] [PubMed] [Google Scholar]
- 13. Mamaev S., Olejnik J., Olejnik E. K., Rothschild K. J., Anal. Biochem. 2004, 326, 25–32. [DOI] [PubMed] [Google Scholar]
- 14. Goto Y., Ohta A., Sako Y., Yamagishi Y., Murakami H., Suga H., ACS Chem. Biol. 2008, 3, 120–129. [DOI] [PubMed] [Google Scholar]
- 15. Shimizu Y., Inoue A., Tomari Y., Suzuki T., Yokogawa T., Nishikawa K., Ueda T., Nat. Biotechnol. 2001, 19, 751–755. [DOI] [PubMed] [Google Scholar]
- 16. Horiya S., Bailey J. K., Temme J. S., Guillen Schlippe Y. V., Krauss I. J., J. Am. Chem. Soc. 2014, 136, 5407–5415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kahn D., Fromant M., Fayat G., Dessen P., Blanquet S., Eur. J. Biochem. 1980, 105, 489–497. [DOI] [PubMed] [Google Scholar]
- 18. Antoun A., Pavlov M. Y., Lovmar M., Ehrenberg M., EMBO J. 2006, 25, 2539–2550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zorzet A., Pavlov M. Y., Nilsson A. I., Ehrenberg M., Andersson D. I., Mol. Microbiol. 2010, 75, 1299–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wiltschi B., Merkel L., Budisa N., ChemBioChem 2009, 10, 217–220. [DOI] [PubMed] [Google Scholar]
- 21. Newton D. T., Creuzenet C., Mangroo D., J. Biol. Chem. 1999, 274, 22143–22146. [DOI] [PubMed] [Google Scholar]
- 22. Varshney U., RajBhandary U. L., J. Bacteriol. 1992, 174, 7819–7826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gambini A., Crosti P., Bianchetti R., Biochim. Biophys. Acta Enzymol. 1980, 613, 73–78. [DOI] [PubMed] [Google Scholar]
- 24. Lee J., Schwieter K. E., Watkins A. M., Kim D. S., Yu H., Schwarz K. J., Lim J., Coronado J., Byrom M., Anslyn E. V., et al., Nat. Commun. 2019, 10, 5097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nilsson A. I., Zorzet A., Kanth A., Dahlström S., Berg O. G., Andersson D. I., Proc. Natl. Acad. Sci. USA 2006, 103, 6976–6981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kawakami T., Ohta A., Ohuchi M., Ashigai H., Murakami H., Suga H., Nat. Chem. Biol. 2009, 5, 888–890. [DOI] [PubMed] [Google Scholar]
- 27. Xiao Q., Zhang F., Nacev B. A., Liu J. O., Pei D., Biochemistry 2010, 49, 5588–5599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Iwasaki K., Goto Y., Katoh T., Suga H., Org. Biomol. Chem. 2012, 10, 5783–5786. [DOI] [PubMed] [Google Scholar]
- 29. Yamagishi Y., Ashigai H., Goto Y., Murakami H., Suga H., ChemBioChem 2009, 10, 1469–1472. [DOI] [PubMed] [Google Scholar]
- 30. Vincent R. M., Wright B. W., Jaschke P. R., ACS Synth. Biol. 2019, 8, 675–685. [DOI] [PubMed] [Google Scholar]
- 31. Tharp J. M., Ad O., Amikura K., Ward F. R., Garcia E. M., Cate J. H. D., Schepartz A., Söll D., Angew. Chem. Int. Ed. 2020, 59, 3122–3126; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 3146–3150. [Google Scholar]
- 32. Sako Y., Morimoto J., Murakami H., Suga H., J. Am. Chem. Soc. 2008, 130, 7232–7234. [DOI] [PubMed] [Google Scholar]
- 33. Jongkees S. A. K., Umemoto S., Suga H., Chem. Sci. 2017, 8, 1474–1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary