

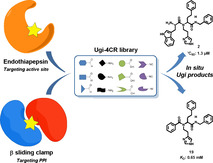
Abstract
Kinetic target‐guided synthesis represents an efficient hit‐identification strategy, in which the protein assembles its own inhibitors from a pool of complementary building blocks via an irreversible reaction. Herein, we pioneered an in situ Ugi reaction for the identification of novel inhibitors of a model enzyme and binders for an important drug target, namely, the aspartic protease endothiapepsin and the bacterial β‐sliding clamp DnaN, respectively. Highly sensitive mass‐spectrometry methods enabled monitoring of the protein‐templated reaction of four complementary reaction partners, which occurred in a background‐free manner for endothiapepsin or with a clear amplification of two binders in the presence of DnaN. The Ugi products we identified show low micromolar activity on endothiapepsin or moderate affinity for the β‐sliding clamp. We succeeded in expanding the portfolio of chemical reactions and biological targets and demonstrated the efficiency and sensitivity of this approach, which can find application on any drug target.
Keywords: drug discovery, kinetic target-guided synthesis, protein–protein interaction inhibitors, protein-templated reactions, Ugi reaction
Screening fast: The Ugi four‐component reaction (Ugi‐4CR) is introduced as a novel reaction in kinetic‐target guided synthesis (KTGS). Novel inhibitors of a model enzyme (aspartic protease endothiapepsin) and new binders of the emerging antibacterial target (β‐sliding clamp DnaN) were successfully identified. This new technique enables a rapid screening of a large number of compounds in early drug discovery.
Introduction
The discovery of new bioactive compounds is a long and expensive process, which calls for the development of new techniques that can speed up hit identification and render it more efficient. In this context, target‐guided synthesis (TGS) is a powerful approach to discover hit compounds by using the biological target itself in ligand selection. Two main methods, dynamic combinatorial chemistry (DCC) and kinetic target‐guided synthesis (KTGS) have emerged. [1] In KTGS, the target is actively involved in ligand selection by assembling its own inhibitors via an irreversible reaction from a library of complementary building blocks, whereas DCC assembles ligands via reversible processes.[ 2 , 3 , 4 ] Only a few protein‐templated reactions for KTGS have been reported so far.[ 5 , 6 , 7 , 8 , 9 , 10 , 11 , 12 , 13 , 14 , 15 , 16 , 17 , 18 ] The azide‐alkyne cyclo‐addition was the first reaction used for KTGS[ 5 , 7 , 8 , 9 ] and is still by far the most widely used. Subsequently, the sulfo‐click reaction between sulfonylazides and thioacids, [10] the amidation between amines and carboxylic acids,[ 11 , 12 ] the alkylation of thiols by halides,[ 13 , 14 ] the thio‐Michael addition [15] and the SN2 opening of epoxides by thiols [16] were added to the portfolio of protein‐templated reactions.[ 6 , 17 , 18 , 19 ] Stringent requirements need to be met by the irreversible reaction: it needs to be compatible with physiological conditions, the building blocks need to be inert towards biomolecules and a substantial difference in reaction rate between the blank and biomacromolecule‐templated reaction is required.
To increase the power and scope of KTGS, it is necessary to expand the number of biocompatible reactions and therefore scaffold diversity. Reactions leading to structural motifs present in numerous protein ligands, irrespective of their target, are particularly desirable. Multicomponent reactions (MCRs) are one‐pot reactions, in which more than two starting materials are incorporated to form a new adduct comprising most of the structural motifs of the starting building blocks. [20] This motivated us and other groups to exploit MCRs for KTGS. The use of Ugi MCRs was first mentioned in a review by Weber but so far no experimental data have been reported. [18] Only recently, MCRs in protein‐templated fragment ligation have re‐emerged by the application of a Mannich‐3CR for the discovery of STAT5 inhibitors, [21] in addition to the on‐target attempts using the Groebke–Blackburn–Bienaymé 3CR for the discovery of urokinase (uPA) inhibitors. [22] Herein, we describe the first use of the Ugi‐4CR in KTGS for the identification of inhibitors. The Ugi four‐component reaction (Ugi‐4CR), [23] is one of the most important MCRs, which affords dipeptide‐like structures from isocyanides, carboxylic acids, aldehydes and amines. [24] Amides are omnipresent in bioactive ligands, which explains why this elegant reaction has found numerous applications in drug discovery, including hit‐ and lead‐identification as well as the generation of large libraries of analogues.[ 20 , 24 ] Owing to the high exploratory power with regard to chemical space and biocompatibility, it represents an attractive reaction for KTGS.
In the Ugi‐4CR‐based KTGS, the enzyme templates the synthesis of its own binders from a pool of four types of building blocks. It follows the same concept as other protein‐templated reactions in terms of simultaneous binding of building blocks to adjacent pockets of the protein target, enabling the assembly of the corresponding binders. The advantages of the new method over other reported protein‐templated reactions are the simultaneous screening of four subpockets and relative ease of accessing structurally complex heterotetrameric binders and their derivatives for further optimization by a one‐pot reaction, starting from diverse and synthetically accessible building blocks (Figure 1).
Figure 1.
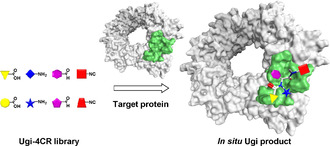
Schematic representation of target‐guided Ugi four‐component reaction, leading to a dipeptide‐like Ugi product from a library of carboxylic acids, amines, aldehydes and isocyanides.
Results and Discussion
In situ Ugi 4‐CR using endothiapepsin
To establish a proof‐of‐concept, we used endothiapepsin as a model target, a member of the class of pepsin‐like aspartic proteases, which play a causative role in several diseases such as malaria, HIV, Alzheimer's disease and hypertension. The high degree of similarity to the corresponding drug targets makes endothiapepsin a convenient model enzyme for the elucidation of the mechanism,[ 25 , 26 , 27 ] and the identification of inhibitors of the related drug targets such as renin [28] and β‐secretase. [29] Moreover, endothiapepsin is a particularly robust enzyme, maintaining its activity for three weeks at room temperature. [30] Eukaryotic aspartic proteases have two catalytic residues (D35 and D219 in endothiapepsin), which form the catalytic dyad at the core of the active site and cleave the substrate's peptide bond using a bound water molecule.[ 25 , 26 ]
We designed a novel potential dipeptide‐like inhibitor 2 based on compound 1, which we had reported previously (IC50=12.8±0.4 μm). [30] Starting from the X‐ray crystal structure of 1 in complex with endothiapepsin (PDB: 4KUP), we used the molecular‐modeling software Moloc [31] and the FlexX docking module in the LeadIT [32] suite for the structure‐based design. Compound 2 features the indolyl moiety as an anchor preserving the most important interactions of 1 in S1 and S2 pockets and in addition benefiting from hydrophobic interactions in S1’ and S2’ pockets (Figure 2).
Figure 2.
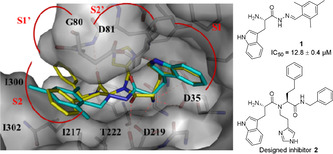
X‐ray crystal structure of endothiapepsin in complex with inhibitor 1 (PDB: 4KUP) [30] superimposed with designed inhibitor 2. Color code: inhibitor 1 skeleton: C: cyan, N: blue, O: red; inhibitor 2 skeleton: C: yellow, N: blue, O: red; protein backbone: gray; dashed lines: H‐bonding interactions below 3.3 Å.
To generate the library, we selected two different building blocks for each of the four components (compounds 3–10, Table 1) with comparable reactivity and solubility. We used benzoic acid as one of the acid components, a negative control for our docking studies as it cannot engage in the same H‐bonding interactions with the catalytic dyad as inhibitor 1. The Ugi‐4CR is favored at high concentrations (0.5–2 m) to afford the corresponding products. [33] The Passerini reaction is the main side reaction of the Ugi‐4CR. In this 3‐CR, an aldehyde, carboxylic acid and isocyanide react to form α‐acyloxyamides. [34] The mechanism and the products formed are similar for both reactions, which enables the simultaneous screening of a higher number of products than for a single reaction.
Table 1.
Selected building blocks 3–10 and 21–24 for the in situ Ugi 4‐CR using endothiapepsin and β‐sliding clamp DnaN.
|
|
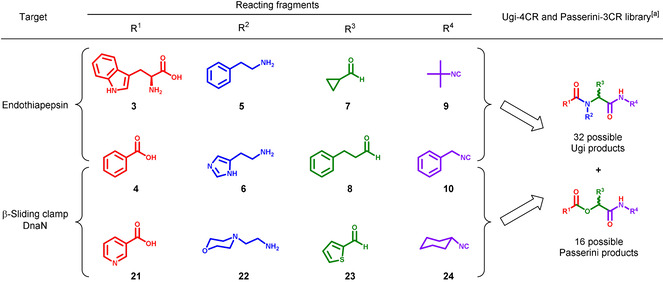
[a] Each library affords 32 possible Ugi and 16 possible Passerini reaction products for each target including the diastereomeric and enantiomeric pairs.
Having selected the building blocks, which are commercially available, we set up two reactions in parallel, a protein‐templated reaction and a blank reaction, in phosphate buffer (pH 6.8) by mixing the carboxylic acids 3 and 4, amines 5 and 6, aldehydes 7 and 8, and the isocyanides 9 and 10 to reach a final concentration of 100 μm of each component in a total volume of 1 mL phosphate buffer containing DMSO 10 % (v/v) (Scheme S1). The optimized reaction conditions have a building‐block concentration of 100 μm to minimize product formation in the background reaction and to reduce the amount of protein used. High stability of endothiapepsin allowed us to use up to 10 % DMSO to assure complete solubility of the building blocks and the prospective Ugi‐4CR products. To the protein‐templated reaction, we added a catalytic amount of endothiapepsin (25 μm final concentration) and analyzed both reaction mixtures after 18 h by using UPLC‐TQD‐SIR (SIR=selective‐ion recording) for each of the possible 48 Ugi and 16 Passerini products considering the bifunctionality of l‐Trp and diastereomers formation. The SIR technique enables fast and sensitive screening of specific molecular weights (MWs) even at very low concentrations. By screening eight MWs per injection using the same concentrations for protein‐templated and reference reactions, we analyzed each reaction in three SIR measurements and detected the formation of two Ugi products 2 and 11 only in the presence of endothiapepsin after 18 h at retention time (tR) 11.66 and 10.78 min, respectively (Figures S1–S4). To demonstrate that the active site of endothiapepsin is required for product formation, we repeated the reaction in the presence of 25 μm bovine serum albumin (BSA) as an orthogonal protein, and in the presence of a strong competitive inhibitor of endothiapepsin, saquinavir (100 μm, K i=48 nm). No product formation was observed in both control experiments, indicating that binding of the components to the active site of endothiapepsin is essential for their formation (Figures S1 and S3). Worth mentioning, other peaks at different retention times also appeared in the SIR chromatograms, which do not correspond to the Ugi‐4CR products. These peaks could arise from fragmentation of bigger molecules or constitute adducts of smaller fragments.
To check for possible protein modifications in the active site of endothiapepsin, we carried out the following experiments in parallel under identical conditions: the whole library mixture 3–10 incubated with endothiapepsin, the individual building blocks 3, 6–8, and 10 incubated with endothiapepsin, and as a reference only endothiapepsin in the reaction buffer. After 18 h, we evaluated the activity of the enzyme in each reaction using an adaptation of the fluorescence‐based assay for HIV protease. [35] The activity was not affected, demonstrating that no modification had occurred in the active site (Figures S25 and S26).
To investigate whether the individual building blocks bind to endothiapepsin, we used saturation‐transfer difference (STD) NMR spectroscopy. STD‐NMR enables the characterization of target‐ligand interactions in solution. STD‐NMR experiments of building blocks 3, 6–8 and 10, which are comprised in the hit compounds’ skeleton formed in the protein‐templated reaction, showed that all building blocks interact with the target except for aldehyde 7. Lack of interactions between endothiapepsin and 7 may be ascribed to its small size (Figures S6–S10). In addition, we performed competition STD‐NMR experiments between each of the above fragments and a known inhibitor of endothiapepsin (bisacylhydrazone 20, IC50=54 nm). [36] The results revealed that compound 20 displaces fragments 3 and 6 from the binding site of the enzyme (Figures S12 and S13). Given that 20 is a strong inhibitor and binds in the active site of the enzyme (PDB: 5HCT), [36] this experiment demonstrates that both 3 and 6 bind to the same pocket of the enzyme as the bisacylhydrazone inhibitor. On the other hand, the signals of fragments 8 and 10 did not disappear in the competition STD‐NMR experiment with 20, indicating that they occupy different subpockets of the active site (Figures S14 and S15). As a result, we conclude that the two fragments 3 and 6 bind in the core of the active site of endothiapepsin just like the bisacylhydrazone 20, whereas fragments 8 and 10 occupy alternative, adjacent subpockets, enabling protein‐templated formation of the products.
As the in situ Ugi‐4CR represents the first example in the field, we synthesized a library of Ugi products to validate its selectivity and to confirm that the formed compounds 2 and 11 are indeed inhibitors of endothiapepsin. The validation library includes all possible eight Ugi‐4CR products 2, 11, 13–18 using l‐Trp as the acid component and one Ugi‐4CR product 19 using benzoic acid (Table 2). Synthesis was accomplished by the reaction of benzoic acid (4) or N‐Boc‐protected l‐Trp (12) with the appropriate amine, aldehyde and isocyanide in methanol at ambient temperature, followed by HCl‐mediated Boc deprotection of the corresponding Ugi products (Scheme S2). The use of N‐Boc Trp was necessary to avoid occurrence of the Ugi 5‐center‐4‐component reaction. [37] We isolated the final compounds as diastereomeric mixtures and used them without further separation.
Table 2.
Inhibitory activities of synthesized compounds selected from the KTGS library on endothiapepsin.
|
|
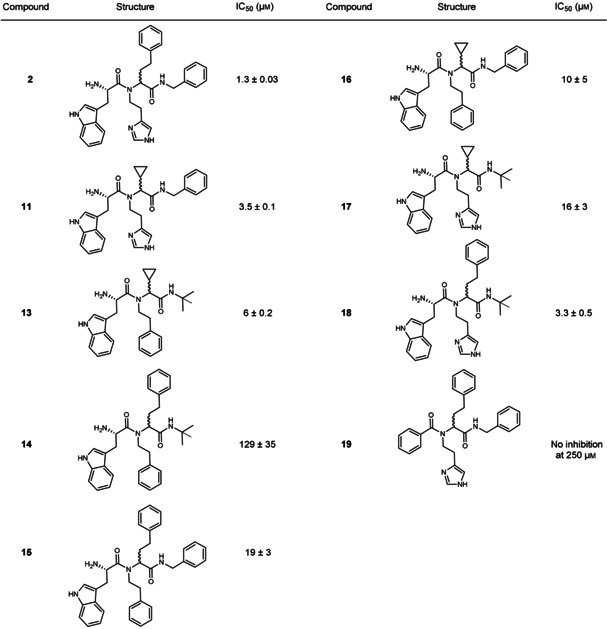
Biochemical evaluation of the library demonstrated that all combinations with l‐Trp as the acid component show activity against endothiapepsin in the IC50 range of 1.3–129 μm. Compounds 2 and 11 are potent inhibitors with IC50 values of 1.3±0.1 μm and 3.5±0.1 μm, respectively (Figures S16 and S18). We could isolate only one diastereomer of inhibitor 2, which showed a slight improvement in activity (IC50=0.89±0.9 μm) (Figure S17). Further attempts to separate diastereomers of hit compounds 2 and 11 by using various separation techniques failed. Interestingly, compounds 18 and 13 show comparable activity to the hit compounds 2 and 11 (Table 2). This could be due to the content ratio of the synthesized diastereomeric mixtures. The non‐observed formation of compounds 18 and 13 by KTGS could be due to a less favorable binding mode and lower affinity of the tert‐butyl isocyanide compared to the benzyl moiety in 2 and 11, which are reflected in the inhibitory activities (IC50 values: 3.3 and 6 μm vs. 1.3 and 3.5 μm, respectively). According to our expectation, the analogue 19 with the benzoic acid component did not show any inhibition at the starting concentration of 250 μm (Table 2). These results demonstrate that the in situ Ugi‐4CR technique is a powerful method, facilitating the selection of the best binders from this library.
In situ Ugi 4‐CR using β sliding clamp DnaN
These results motivated us to investigate the scope of the protein‐templated Ugi‐4CR further. Given that protein–protein interactions (PPIs) are notoriously challenging to inhibit with small molecules, KTGS should represent an invaluable tool to identify a suitable ligand especially when using the Ugi‐4CR. Hence, we chose the underexploited antibacterial target DnaN also known as β sliding clamp. This ring‐shaped homodimer of two β subunits of DNA polymerase III plays a crucial role during DNA synthesis and repair. Upon formation of a stable clamp around the DNA strand, DnaN interacts with linear motifs of protein binding partners including the DNA polymerases I–V, giving high processivity to DNA synthesis.[ 38 , 39 , 40 ] Due to its highly conserved structure in bacteria and its structural divergence from its eukaryotic counterpart, proliferating cell nuclear antigen (PCNA), it represents a promising target to develop new antimicrobial agents acting via a novel mechanism. Interactions of DNA polymerases to DnaN occur at a shallow hydrophobic binding site composed of two subsites I and II (Figure 3). Known synthetic DnaN inhibitors hitherto occupy only subsite I and suffer from limited antibacterial activity.[ 41 , 42 , 43 , 44 ] On the other hand, the natural antibiotics griselimycins (GMs) are tight binders of the mycobacterial sliding clamp, occupying both subsites I and II. [45] We expected the powerful combination of KTGS with the Ugi‐4CR to address this challenge. Inspection of the X‐ray crystal structures of synthetic inhibitors and GMs in complex with DnaN led to the identification of molecular fragments involved in key interactions with DnaN.[ 41 , 42 , 43 , 44 , 45 ] We utilized these building blocks to generate a virtual library of 320 possible Ugi‐4CR and Passerini products including their enantiomers, which we docked into DnaN using MOE software. [46] Top‐ranked ligands such as compound 19 inspired the selection of building blocks for KTGS experiments (Table 1). According to the docking study, compound 19 establishes two hydrophobic interactions with M396 in subsite I and an H‐bond between the benzoyl carbonyl and R183. Importantly, the isocyanide component can be accommodated in subsite II (Figure 3).
Figure 3.
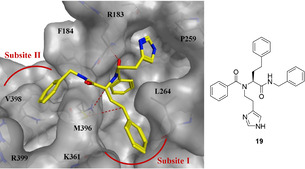
Top‐ranked pose of Ugi‐4CR derivative 19 (yellow) generated by docking into DnaN (PDB code: 5AGV) [45] using the scoring function in MOE. [46] Color code: skeleton: C: yellow, N: blue, O: red; protein backbone and surface: gray; dashed lines: hydrophobic and H‐bonding interactions.
We ran two reactions in parallel, a protein‐templated reaction containing a catalytic amount of Mycobacterium smegmatis DnaN (25 μm) and a blank reaction by mixing the building blocks 4, 6, 8, 10 and 21–24 to reach a final concentration of 50 μm of each component in 1 mL phosphate buffer (pH 7, 5 % DMSO) and analyzed them by LC‐HRMS after 6 h. Notably, due to the high reactivity of the building blocks, lower concentration (50 μm) and shorter reaction time (6 h) were sufficient to detect the change between the protein‐templated and blank reactions. Screening for the exact masses of all possible 16 Ugi‐4CR (19, 25–39) and eight Passerini products (40–47) revealed the amplification of two Ugi products 19 and 25 in the presence of DnaN (Figure 4 and S29–S44). In contrast to the first library used for endothiapepsin, the Ugi derivatives 19 and 25 were also formed in the blank reaction. We observed, however, a marked acceleration of the reaction in the presence of DnaN, leading to an increased amount of the Ugi products compared to the blank reaction (17 to 20‐fold for 19 and 25, respectively) (Figure 4 A). We confirmed the identity of the peaks by comparison of the exact masses, fragmentation patterns, and retention times of the compounds formed in situ with those of synthetic references. Furthermore, we quantified the amount of 19 and 25 formed in KTGS and the blank reactions using the synthetic compounds for external calibration (Figure S27 and S28). The peak area of 19 formed by KTGS corresponds to 129 pm (0.004 % yield) vs. 5 pm in the blank reaction. The concentration of 25 is equal to 362 pm in the presence of DnaN, in a slightly better yield (0.012 %) compared to 19, vs. 8 pm in the background reaction. These results underline the high sensitivity of HRMS for analyzing KTGS reactions allowing the detection of picomolar quantities of the formed products. To verify that compounds 19 and 25 were selectively templated by DnaN, we repeated the KTGS experiment using endothiapepsin as an unrelated protein. Interestingly, no product formation for 19 and 25 was observed indicating a specific DnaN‐templated reaction (Figure 4 D). The absence of a background reaction could be due to a non‐specific binding of the fragments to endothiapepsin. In agreement with this finding, compound 19 did not emerge in the previous KTGS experiment using endothiapepsin and it showed no inhibitory effect (Table 2). To investigate whether the DnaN‐templated reaction occurred in the DNA polymerase binding site or elsewhere, we performed a control experiment in the presence of the tight DnaN binder GM (25 μm) showing no amplification of Ugi products. Thus, binding of the Ugi building blocks indeed takes place in the DNA polymerase interaction site (Figure 4 C). To study the reproducibility of hit formation, we repeated the experiments using a new batch of DnaN. The same results were obtained showing significant amplification of compounds 19 and 25 in KTGS (Figure S45), however with lower yields. In KTGS, the concentration of 19 is 22 pm (0.001 % yield) vs. 1.5 pm in the blank, while the concentration of 25 is 95 pm (0.003 % yield) in KTGS vs. 0.7 pm in the blank. The variation in yields could be attributed to batch‐to‐batch differences in activity, conformation, and purity of the protein or a change in concentration of building blocks in the DMSO stock solutions.
Figure 4.
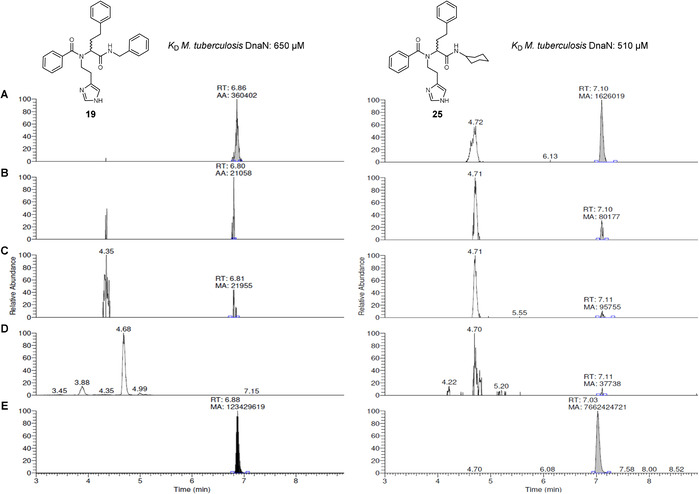
LC‐HRMS Hit identification of Ugi‐4CR derivatives 19 ([M+H]+ = 467.2436, tR 6.88 min) and 25 ([M+H]+ = 459.2749, tR 7.10 min): (A) formation of the hits in the presence of M. smegmatis DnaN after 6 h of incubation (B) traces in buffer without protein after 6 h of incubation; (C) suppression of DnaN‐templated reaction by GM; (D) templated reaction in the presence of endothiapepsin; (E) reference compounds 19 and 25 obtained by synthetic pathway.
As observed for endothiapepsin and also by others, [47] additional peaks were detected in the KTGS experiments even when screening for the exact masses of the Ugi and Passerini derivatives (e.g., Figure 4, Figures S32, S35, and S43). The identity of these peaks was not investigated as they do not match the retention of the synthetic references and do not display any significant amplification.
To evaluate the probability of chemical modification of M. smegmatis DnaN by the building blocks, we incubated the protein in the presence and absence of fragments 4, 6, 8, and 10 for 6 h under KTGS conditions. Determination of the molecular mass of DnaN in both samples using ESI‐MS revealed no change in the mass demonstrating that no modification occurred (Figure S55).
Since the in situ Ugi‐4CR was run at low reactant concentration, we investigated the probability of Ugi‐4CR product formation with water competing as the acid component. [48] Monitoring of the eight possible compounds (48–55) revealed no or minor formation with no difference between DnaN‐templated and blank experiments (Figure S46 and S47).
As some of our fragments bear an activated β‐arylethylamine moiety, for example, compounds 3 and 6, they could undergo Pictet–Spengler cyclization reaction with aldehydes. [49] Therefore, we monitored the formation of the Pictet–Spengler cyclization products by scanning for their exact masses in DnaN‐based KTGS reaction. Interestingly, we detected the formation of a Pictet–Spengler product 56 from histamine 6 and the aldehyde 8 in the DnaN‐templated reaction as well as all control experiments, however with only slight amplification in the presence of protein (Figure S48). Although we did not consider compound 56 as a hit, this finding could be very interesting for further investigations of this reaction in KTGS technique.
In order to demonstrate the selectivity of DnaN during the KTGS experiment, we synthesized all possible 16 Ugi products 19, 25–39 and a representative Passerini compound 40 that contains the same acid, aldehyde and isocyanide components as hit compound 19. We confirmed the structures of Ugi compounds 26, 28 and 35 by X‐ray crystallography (Figure S50).
Screening of compounds 19, 25–40 as racemates for M. smegmatis DnaN binding using surface plasmon resonance (SPR) revealed that compounds 19 and 25 display the highest binding responses (Table S9). The equilibrium dissociation constant (K D) was determined for the hit compounds as well as compound 38, as a negative control. Compounds 19 and 25 showed low affinity (K D values: 650 and 510 μm, respectively) for M. tuberculosis β‐sliding clamp, whereas 38 exhibited much weaker affinity (K D value: 3200 μm) in line with the KTGS results (Table S10). As a result, we did not proceed to the functional assay, which might be hampered by the solubility limit of the compounds. Nevertheless, we confirmed binding of 19 to M. tuberculosis DnaN by STD‐NMR revealing that all aromatic protons are in close proximity to DnaN and contribute to binding (Figure S51). This result is in line with the predicted binding mode for compound 19 (Figure 3). Furthermore, we performed a competitive STD‐NMR experiment for 19 to verify its binding site using the known inhibitor RU7. [43] We observed 30–100 % reduction of signal intensities for 19 in the presence of RU7 (Figure S53), demonstrating that both compounds bind to the same binding pocket, in agreement with our KTGS control experiment using GM.
Subsequent evaluation of the antibacterial activity revealed that hits 19 and 25 inhibit the growth of Escherichia coli TolC and Micrococcus luteus with minimum inhibitory concentration (MIC) values of 128 μm, while none of the compounds shows growth inhibitory effects on M. smegmatis (Table S10). Although showing antibacterial effects on Gram‐positive and the efflux mutant Gram‐negative bacteria, the lack of activity on mycobacteria could hint to a permeability or an efflux issue.
Conclusions
In summary, we described the first in situ ligand assembly using Ugi‐4CR, which enables fast screening of a series of compounds that could be expanded to a large number, circumventing the need for synthesis, purification and evaluation of each individual compound. Starting from commercially available building blocks, we screened 64 compounds (48 Ugi and 16 Passerini) by using a catalytic amount of the target protein, demonstrating the efficiency of the in situ Ugi‐4CR in KTGS for the discovery of two low‐micromolar inhibitors of endothiapepsin. We subsequently applied this powerful strategy to the novel antibacterial target DnaN, resulting in the identification of two binders with decent antibacterial activity. Furthermore, we found that another side reaction, that is, Pictet–Spengler cyclization occurred under the KTGS conditions, which would open a door for studying the utility of this reaction in KTGS. The use of the Ugi‐4CR in hit identification holds great potential, in particular for challenging PPI targets such as DnaN and since it enhances the structural diversity of KTGS products. The results obtained through our novel approach will serve as a starting point to develop new Ugi‐4CR derivatives with improved affinity and antibacterial activity.
Experimental Section
Crystallographic data
Deposition Numbers 1917479, 1917480, and 1917481 contain the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service.
General protocol for Ugi‐4CR‐based KTGS using endothiapepsin
Endothiapepsin (25 μL, 1.0 mm in phosphate buffer 0.1 m, pH 6.8) and the 7 building blocks 4–10 (1 μL each, 100 mm stock solution in DMSO), l‐tryptophan (3) (4 μL, 25 mm stock solution in DMSO) were added to a mixture of DMSO (89 μL) and phosphate buffer (900 μL, 0.1 m, pH 6.8) to give a final concentration of 100 μm of each component in the presence of 25 μm protein. The reaction mixture was allowed to rotate using a rotating mixer at room temperature with 10 rpm. After 18 h, acetonitrile (100 μL) was added to a sample (50 μL) and was centrifuged at 9,727 g for 2 min and the supernatant was collected for analysis. The library was analyzed by UPLC‐TQD‐SIR [electro‐spray ionization (ESI+)] measurement because of its higher sensitivity and greater reliability for product identification. UPLC‐TQD was performed using a Waters Acquity UPLC H‐class system coupled to a Waters TQD. All analyses were performed using a reversed‐phase UPLC column (ACQUITY HSS T3 Column, 130 Å, 1.8 μm, 2.1 mm×150 mm). Positive‐ion mass spectra were acquired using ES ionization, injecting 10 μL of sample; column temperature 35 °C; flow rate 0.3 mL min−1. The eluents, water (A) and acetonitrile (B) contained 0.1 % of formic acid. The library components were eluted with a gradient from 95 %→30 % (A) over 20 min, then at 5 % (A) over 1 min, followed by 5 % (A) for 2 min. The UPLC‐TQD‐SIR method was used to analyze the formation of Ugi products in in situ and blank reactions. [M+H]+ were monitored using the full mass range to ensure correct isotope patterns for all possible potential Ugi products both for in situ Ugi and blank reactions. The Ugi products in the protein‐templated reaction were identified by comparison of their retention time with that of the compound synthesized using conventional methods.
General protocol for Ugi‐4CR‐based KTGS using DnaN
To investigate the DnaN‐templated synthesis, the eight building blocks 4, 6, 8, 10, and 21–24 (1 μL each, 50 mm stock solution in DMSO) and DnaN (25 μL, 1.0 mm in phosphate buffer 0.05 m, pH 7) were added to a mixture of DMSO (42 μL) and phosphate buffer (925 μL, 0.05 m, pH 7) to reach a total reaction volume of 1 mL containing a final concentration of 50 μm of each component and 25 μm protein. The reaction mixture was allowed to rotate using a rotating mixer at room temperature with 10 rpm. After 6 h, acetonitrile (100 μL) was added to a sample (50 μL), which was centrifuged at 9,727 g for 2 min, and the supernatant was collected for analysis. All analyses were performed using a Thermo Scientific Dionex UltiMate 3000 UHPLC+ focused system with a reversed‐phase UPLC column (ACQUITY UPLC BEH C8 Column, 130 Å, 1.7 μm, 2.1 mm×150 mm) coupled to a Thermo Scientific Q Exactive Focus system. Positive‐ion mass spectra were acquired using ES ionization, injecting 4 μL of sample; column temperature 35 °C; flow rate 0.25 mL min−1. Separations were performed using water (A)/acetonitrile (B) with 0.1 % of formic acid. The library components were eluted with a gradient of 90 % (A) for 1 min, then from 90 %→5 % (A) over 9 min, then at 5 % (A) over 2 min, followed by 90 % (A) for 1.8 min for column equilibration. The identities of the hit compounds 19 (tR 6.88 of 14.8 min) and 25 (tR 7.10 of 14.8 min) were confirmed by exact mass and comparison of tR with the authentic synthesized compounds. For the blank reaction, identical building blocks mixture (1 μL each, 50 mm in DMSO) was incubated in buffer (950 μL, 0.05 m, pH 7) without DnaN and subjected to LC‐HRMS analysis. Control experiments using endothiapepsin (25 μL, 1 mm in phosphate buffer 0.05 m, pH 7) instead of DnaN as well as in the presence of the strong inhibitor (25 μL of griselimycin, 1.0 mm in DMSO) were run consecutively as described above. Comparison of the LC/MS chromatograms of these experiments allowed us to determine whether the protein is templating the Ugi‐4CR of the building blocks.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We would like to acknowledge T. D. Tiemersma‐Wegman for help with the UPLC‐TOF‐SIR measurements, Dr. M. Nuzzolo for help with HPLC measurements, P. van der Meulen for useful suggestions and discussions about STD‐NMR spectroscopy, C. Bader for the ESI‐MS measurements, Dr. V. Huch for X‐ray determinations and Prof. G. Klebe, Prof. A. Dömling, Prof. A. J. Minnaard, Prof. R. W. Hartmann and Dr. M. Mondal for fruitful discussions. Funding was granted by the German Federal Ministry for Education and Research (grant 16 GW0207K to A.K.H.H., R.M. and W.B.), by the Helmholtz‐Association's Initiative and Networking Fund, by the Netherlands Organisation for Scientific Research (NWO‐CW, ECHO‐STIP grant 717014004), by the Dutch Ministry of Education, Culture, Science (gravitation program 024.001.035) and by the European Research Council (ERC starting grant 757913; all to A.K.H.H.). Open access funding enabled and organized by Projekt DEAL.
F. Mancini, M. Y. Unver, W. A. M. Elgaher, V. R. Jumde, A. Alhayek, P. Lukat, J. Herrmann, M. D. Witte, M. Köck, W. Blankenfeldt, R. Müller, A. K. H. Hirsch, Chem. Eur. J. 2020, 26, 14585.
A previous version of this manuscript has been deposited on a preprint server (https://doi.org/10.26434/chemrxiv.11769993.v1).
References
- 1. Jaegle M., Wong E. L., Tauber C., Nawrotzky E., Arkona C., Rademann J., Angew. Chem. Int. Ed. 2017, 56, 7358–7378; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 7464–7485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Oueis E., Sabot C., Renard P.-Y., Chem. Commun. 2015, 51, 12158–12169. [DOI] [PubMed] [Google Scholar]
- 3. Unver M. Y., Gierse R. M., Ritchie H., Hirsch A. K. H., J. Med. Chem. 2018, 61, 9395–9409. [DOI] [PubMed] [Google Scholar]
- 4. Bosc D., Camberlein V., Gealageas R., Castillo-Aguilera O., Deprez B., Deprez-Poulain R., J. Med. Chem. 2020, 63, 3817–3833. [DOI] [PubMed] [Google Scholar]
- 5. Mocharla V. P., Colasson B., Lee L. V., Röper S., Sharpless K. B., Wong C.-H., Kolb H. C., Angew. Chem. Int. Ed. 2005, 44, 116–120; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2005, 117, 118–122. [Google Scholar]
- 6. Greasley S. E., Marsilje T. H., Cai H., Baker S., Benkovic S. J., Boger D. L., Wilson I. A., Biochemistry 2001, 40, 13538–13547. [DOI] [PubMed] [Google Scholar]
- 7. Lewis W. G., Green L. G., Grynszpan F., Radić Z., Carlier P. R., Taylor P., Finn M. G., Sharpless K. B., Angew. Chem. Int. Ed. 2002, 41, 1053–1057; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2002, 114, 1095–1099. [Google Scholar]
- 8. Manetsch R., Krasiński A., Radić Z., Raushel J., Taylor P., Sharpless K. B., Kolb H. C., J. Am. Chem. Soc. 2004, 126, 12809–12818. [DOI] [PubMed] [Google Scholar]
- 9. Krasiński A., Radić Z., Manetsch R., Raushel J., Taylor P., Sharpless K. B., Kolb H. C., J. Am. Chem. Soc. 2005, 127, 6686–6692. [DOI] [PubMed] [Google Scholar]
- 10. Hu X., Sun J., Wang H.-G., Manetsch R., J. Am. Chem. Soc. 2008, 130, 13820–13821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gelin M., Poncet-Montange G., Assairi L., Morellato L., Huteau V., Dugue L., Dussurget O., Pochet S., Labesse G., Structure 2012, 20, 1107–1117. [DOI] [PubMed] [Google Scholar]
- 12. Jaegle M., Steinmetzer T., Rademann J., Angew. Chem. Int. Ed. 2017, 56, 3718–3722; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 3772–3776. [Google Scholar]
- 13. Chase J. F., Tubbs P. K., Biochem. J. 1969, 111, 225–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nguyen R., Huc I., Angew. Chem. Int. Ed. 2001, 40, 1774–1776; [PubMed] [Google Scholar]; Angew. Chem. 2001, 113, 1824–1826. [Google Scholar]
- 15. Oueis E., Nachon F., Sabot C., Renard P.-Y., Chem. Commun. 2014, 50, 2043–2045. [DOI] [PubMed] [Google Scholar]
- 16. Maki T., Kawamura A., Kato N., Ohkanda J., Mol. Biosyst. 2013, 9, 940–943. [DOI] [PubMed] [Google Scholar]
- 17. Asaba T., Suzuki T., Ueda R., Tsumoto H., Nakagawa H., Miyata N., J. Am. Chem. Soc. 2009, 131, 6989–6996. [DOI] [PubMed] [Google Scholar]
- 18. Weber L., Drug Discovery Today Technol. 2004, 1, 261–267. [DOI] [PubMed] [Google Scholar]
- 19. Bosc D., Jakhlal J., Deprez B., Deprez-Poulain R., Future Med. Chem. 2016, 8, 381–404. [DOI] [PubMed] [Google Scholar]
- 20. Dömling A., Wang W., Wang K., Chem. Rev. 2012, 112, 3083–3135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wong E. L., Nawrotzky E., Arkona C., Kim B. G., Beligny S., Wang X., Wagner S., Lisurek M., Carstanjen D., Rademann J., Nat. Commun. 2019, 10, 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gladysz R., Vrijdag J., van Rompaey D., Lambeir A.-M., Augustyns K., de Winter H., van der Veken P., Chem. Eur. J. 2019, 25, 12380–12393. [DOI] [PubMed] [Google Scholar]
- 23. Ugi I., Steinbrückner C., Angew. Chem. 1960, 72, 267–268. [Google Scholar]
- 24. Slobbe P., Ruijter E., Orru R. V. A., Med. Chem. Commun. 2012, 3, 1189–1218. [Google Scholar]
- 25. Coates L., Erskine P. T., Mall S., Gill R., Wood S. P., Myles D. A. A., Cooper J. B., Eur. Biophys. J. 2006, 35, 559–566. [DOI] [PubMed] [Google Scholar]
- 26. Coates L., Tuan H. H., Tomanicek S., Kovalevsky A., Mustyakimov M., Erskine P., Cooper J., J. Am. Chem. Soc. 2008, 130, 7235–7237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Coates L., Erskine P. T., Wood S. P., Myles D. A. A., Cooper J. B., Biochemistry 2001, 40, 13149–13157. [DOI] [PubMed] [Google Scholar]
- 28. Cooper J., Quail W., Frazao C., Foundling S. I., Blundell T. L., Humblet C., Lunney E. A., Lowther W. T., Dunn B. M., Biochemistry 1992, 31, 8142–8150. [DOI] [PubMed] [Google Scholar]
- 29. Geschwindner S., Olsson L. L., Albert J. S., Deinum J., Edwards P. D., De Beer T., Folmer R. H. A., J. Med. Chem. 2007, 50, 5903–5911. [DOI] [PubMed] [Google Scholar]
- 30. Mondal M., Radeva N., Köster H., Park A., Potamitis C., Zervou M., Klebe G., Hirsch A. K. H., Angew. Chem. Int. Ed. 2014, 53, 3259–3263; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 3324–3328. [Google Scholar]
- 31. Gerber P. R., Müller K., J. Comp. Aid. Mol. Des. 1995, 9, 251–268. [DOI] [PubMed] [Google Scholar]
- 32.BioSolveIT GmbH, Sankt Augustin. http://www.biosolveit.de/LeadIT/.
- 33. Dömling A., Ugi I., Angew. Chem. Int. Ed. 2000, 39, 3168–3210; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2000, 112, 3300–3344. [Google Scholar]
- 34. Passerini M., Gazz. Chim. Ital. 1921, 51, 126–129. [Google Scholar]
- 35. Toth M. V., Marshall G. R., Int. J. Pept. Protein Res. 2009, 36, 544–550. [DOI] [PubMed] [Google Scholar]
- 36. Mondal M., Radeva N., Fanlo-Virgý H., Otto S., Klebe G., Hirsch A. K. H., Angew. Chem. Int. Ed. 2016, 55, 9422–9426; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 9569–9574. [Google Scholar]
- 37. Demharter A., Hörl W., Herdtweck E., Ugi I., Angew. Chem. Int. Ed. Engl. 1996, 35, 173–175; [Google Scholar]; Angew. Chem. 1996, 108, 185–187. [Google Scholar]
- 38. Yin Z., Kelso M. J., Beck J. L., Oakley A. J., J. Med. Chem. 2013, 56, 8665–8673. [DOI] [PubMed] [Google Scholar]
- 39. Yin Z., Whittell L. R., Wang Y., Jergic S., Liu M., Harry E. J., Dixon N. E., Beck J. L., Kelso M. J., Oakley A. J., J. Med. Chem. 2014, 57, 2799–2806. [DOI] [PubMed] [Google Scholar]
- 40. Yin Z., Whittell L. R., Wang Y., Jergic S., Ma C., Lewis P. J., Dixon N. E., Beck J. L., Kelso M. J., Oakley A. J., J. Med. Chem. 2015, 58, 4693–4702. [DOI] [PubMed] [Google Scholar]
- 41. Yin Z., Wang Y., Whittell L. R., Jergic S., Liu M., Harry E., Dixon N. E., Kelso M. J., Beck J. L., Oakley A. J., Chem. Biol. 2014, 21, 481–487. [DOI] [PubMed] [Google Scholar]
- 42. Wijffels G., Johnson W. M., Oakley A. J., Turner K., Epa V. C., Briscoe S. J., Polley M., Liepa A. J., Hofmann A., Buchardt J., Christensen C., Prosselkov P., Dalrymple B. P., Alewood P. F., Jennings P. A., Dixon N. E., Winkler D. A., J. Med. Chem. 2011, 54, 4831–4838. [DOI] [PubMed] [Google Scholar]
- 43. Georgescu R. E., Yurieva O., Kim S.-S., Kuriyan J., Kong X.-P., O‘Donnell M., Proc. Natl. Acad. Sci. USA 2008, 105, 11116–11121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wijffels G., Dalrymple B. P., Prosselkov P., Kongsuwan K., Epa V. C., Lilley P. E., Jergic S., Buchardt J., Brown S. E., Alewood P. F., Jennings P. A., Dixon N. E., Biochemistry 2004, 43, 5661–5671. [DOI] [PubMed] [Google Scholar]
- 45. Kling A., Lukat P., Almeida D. V., Bauer A., Fontaine E., Sordello S., Zaburannyi N., Herrmann J., Wenzel S. C., König C., Ammerman N. C., Belén Barrio M., Borchers K., Bordon-Pallier F., Brönstrup M., Courtemanche G., Gerlitz M., Geslin M., Hammann P., Heinz D. W., Science 2015, 348, 1106–1112. [DOI] [PubMed] [Google Scholar]
- 46.Molecular Operating Environment (MOE), 2018.01; Chemical Computing Group ULC, 1010 Sherbooke St. West, Suite 910, Montreal, QC, Canada, H3A 2R7, 2019.
- 47. Whiting M., Muldoon J., Lin Y.-C., Silverman S. M., Lindstrom W., Olson A. J., Kolb H. C., Finn M. G., Angew. Chem. Int. Ed. 2006, 45, 1435–1439; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 1463–1467. [Google Scholar]
- 48. Weber L., Wallbaum S., Broger C., Gubernator K., Angew. Chem. Int. Ed. Engl. 1995, 34, 2280–2282; [Google Scholar]; Angew. Chem. 1995, 107, 2452–2454. [Google Scholar]
- 49. Cox E. D., Cook J. M., Chem. Rev. 1995, 95, 1797–1842. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary