

Abstract
The nature of cyclopropyl gold(I) carbene‐type intermediates has been reexamined as part of a mechanistic study on the formation of cis‐ or trans‐fused bicyclo[5.1.0]octanes in a gold(I)‐catalyzed cascade reaction. Benchmark of DFT methods together with QTAIM theory and NBO analysis confirms the formation of distinct intermediates with carbenic or carbocationic structures in the cycloisomerizations of enynes.
Keywords: cycloisomerization, cyclopropanation, DFT calculations, dienynes, gold(I) catalysis
Going for gold: The nature of cyclopropyl gold(I) carbene‐type intermediates has been explored as part of a mechanistic study on the gold(I)‐catalyzed formation of trans‐fused bicyclo[5.1.0]octanes. Complete computational analysis, that includes QTAIM theory and NBO analysis, confirms the formation of different species with carbenic or cationic nature as intermediates in the cycloisomerizations of enynes.
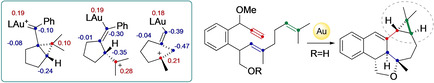
Introduction
Research in homogenous gold(I) catalysis has provided unique tools for the construction of molecular complexity. [1] Thus, fundamental knowledge gathered in the study of 1,n‐enyne cycloisomerizations [2] has led to many applications in total synthesis of complex natural products. [3] Although gold(I) carbenes have been proposed as key intermediates of many gold(I) catalyzed transformations, there is still some uncertainty regarding the structure of these species (carbenic or cationic character of C−Au bond), [4] especially considering their high reactivity, which makes their isolation very challenging. [5] In this context, our group recently reported the spectroscopic characterization of mesityl gold(I) carbenes in solution by NMR at low temperature, [6] which correspond to actual species present under catalytic conditions.
As part of our program on the total synthesis of jatrophalactone (1) [7] (Scheme 1), a cytotoxic diterpene isolated from the roots of Jatropha curcas, we observed the unexcepted formation of trans‐fused bicyclo[5.1.0]octanes (5) by a gold(I)‐catalyzed cyclization cascade. To date, no total synthesis of 1 has been reported. We envisioned that after the coordination of gold to the alkyne of dienyne 2, a 6‐exo‐dig cyclization would form cyclopropyl gold carbene A, which, after intramolecular nucleophilic attack of the OH moiety to the carbene, followed by OR elimination, would lead to the formation of intermediate B. Product 3 would finally be obtained by intramolecular cyclopropanation of the second alkene. In order to explore the feasibility of this transformation, simpler model substrate 4 was designed having a phenyl ring instead of the furan. However, to our surprise, product 5 was obtained in this reaction bearing a rare trans‐fused bicyclo[5.1.0]octane.
Scheme 1.
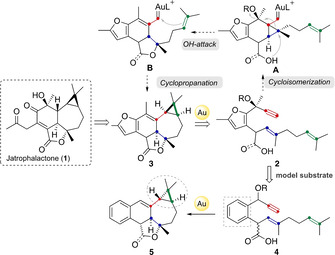
Proposed retrosynthesis of jatrophalactone (1) and model substrate 4 for the gold(I)‐catalyzed cycloisomerization key step.
Although, the trans‐bicyclo[5.1.0]‐octane motif is present in some natural products, [8] such as cneorubin B (6), emmottene (7), and hemerocallal A (8) (Figure 1), the formation of this ring system is rather unusual because it formally corresponds to the cyclopropanation of (E)‐cycloheptene, which is unstable at room temperature. [9] This type of trans‐fused bicyclo[5.1.0]octanes had only been obtained before as minor byproducts in cyclization cascade reactions catalyzed by gold(I). [10]
Figure 1.
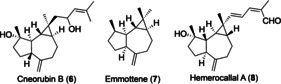
Natural products bearing a trans‐fused bicyclo[5.1.0]octane.
Herein, we present an experimental and computational study on the selective formation of cis‐ or trans‐fused bicyclo[5.1.0]octanes by a gold(I)‐catalyzed cascade. This investigation also led us to reconsider the puzzling structure of the cyclopropyl gold(I) carbene‐type intermediates. [11] In principle, upon coordination of gold(I) to the alkyne of the 1,6‐enyne in Int1, three different possible structures could be generated, Int2‐4. Whether or not structures Int2‐4 are resonance forms or distinct stationary points in the reaction coordinate is still an open question and the different interpretations coexist in the current literature [4] (Scheme 2). Thus, we performed DFT calculations (including benchmark of functionals, QTAIM theory, and NBO analysis) in order to further describe the structure of intermediates Int2–4 and how they are interconnected.
Scheme 2.
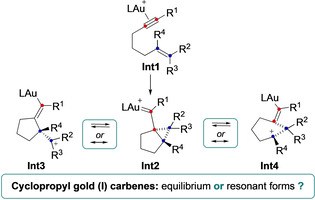
Different structures of cyclopropyl gold(I) carbene‐type intermediates Int2, Int3 and Int4.
Results and Discussion
Formation of trans‐fused bicyclo[5.1.0]octanes
We examined the cyclization of dienynes 4 a, 9 and 10, bearing OH or CO2H as intramolecular nucleophiles, as models for the key cascade cyclization for the synthesis of jatrophalactone (see Scheme 1). Dienynes 4 a, 9 and 10 reacted almost instantaneously with [(JohnPhos)Au(MeCN)]SbF6 as the catalyst at room temperature in CH2Cl2 (Scheme 3). Surprisingly, (E)‐configured enynes 9 a and 10 a (ca. 1:1 diastereomeric mixture at the benzylic positions), afforded 11 as a single diastereomer in 45–50 % yield, along with 2‐substituted naphthalene 12 (14 % yield). The presence of rare trans‐fused bicyclo[5.1.0]octane, along with a trans‐fused tetrahydronaphtho[1,2‐c]furan unit, in compound 11 was confirmed by X‐ray diffraction (Figure 2). Similarly, carboxylic acid 4 a yielded trans‐fused cyclopropane 5 a as a single diastereomer, albeit in lower yield. Again, naphthalene 12 was isolated in this reaction as a minor product. On the other hand, (Z)‐configured dienynes 9 b and 10 b gave an inseparable mixture of isomers 13 and 14 in moderate yield, together with traces of naphthalene (Z)‐12 (Scheme 3). Remarkably, when the two diastereomers of 9 b and 10 b were separated by chromatography and exposed to gold(I)‐catalysis, the same mixture of 13 and 14 was obtained, although (Z)‐12 was only formed from one of them. The relative configuration of pentacyclic products 13 and 14, which are clearly distinct from 11, was assigned by NOE NMR experiments. [12]
Scheme 3.
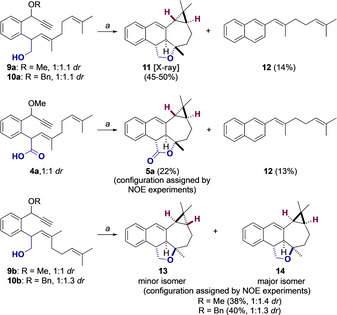
Gold(I) catalysis. a) Standard reaction conditions: [(JohnPhos)Au(MeCN)]SbF6 (2 mol %), CH2Cl2 (0.1 m), 23 °C, 10 min.
Figure 2.
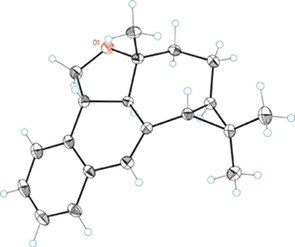
ORTEP structure of pentacyclic compound 11.
To understand the effect of substituents and alkene configuration in the stereoselectivity of these transformations, other dienynes were prepared and submitted to the gold(I)‐catalyzed cascade cyclization (Scheme 4). Therefore, simpler substrates 15 a,b and 16 missing the benzylic hydroxymethyl group gave products 17 a,b and 18 as single diastereomers in ca. 50 % yield. All of these products feature a cis‐fused bicyclo[5.1.0]octane system as confirmed by X‐ray diffraction analysis. The cascade reactions proved to be stereospecific with respect to the configuration of the alkene, as observed in many other gold(I)‐catalyzed transformations. Additionally, products 20 and 22 were obtained as single diastereomers by reaction of dienynes 19 a,b. Deprotection of TBS group of 20 and 22 gave rise to crystalline primary alcohols 21 and 23, whose relative configuration was confirmed by X‐ray diffraction.
Scheme 4.
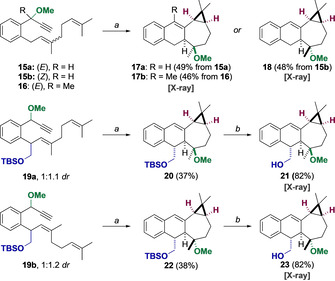
Gold(I) catalysis of modified dienynes. a) [(JohnPhos)Au(MeCN)]SbF6 (2 mol %), CH2Cl2, 23 °C, 10 min; b) THF/ 10 % HCl, 23 °C, 2 h.
In order to rationalize the formation of trans‐fused bicyclo[5.1.0]octanes and the different selectivity observed for (E)‐ and (Z)‐dienynes in the new gold(I)‐catalyzed cyclization cascade reactions we performed DFT calculations. Calculations were performed with B3LYP [13] ‐D3 [14] /6‐31G(d) [15] (C, H, O, P) and SDD [16] (Au) in CH2Cl2 (PCM) [17] and considering the model catalyst (PMe3)Au+. [12] We computed the system for (E)‐ and (Z)‐configured dienynes 9 a,b (Scheme 5). Four possible diastereomers 9 a (9 aa and 9 ab) and 9 b (9 ba and 9 bb) were studied. Thus, upon the activation of the alkyne moiety, three conformers of Int1 were located in all cases, differing on the orientation of the alkene (9 aa‐Int1 a–c, 9 ab‐Int1 a–c, 9 ba‐Int1 a–c, and 9 bb‐Int1 a–c). In the following mechanistic discussion, Int1–3 refer to structures similar to those depicted in Scheme 2.
Scheme 5.
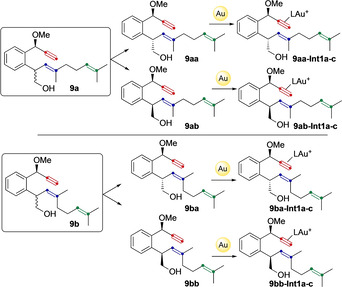
Gold(I)‐catalyzed formation of Int1 a–c of (E)‐configured 9 a and (Z)‐configured 9 b.
Firstly, we investigated the mixture of diastereomers 9 aa and 9 ab (Scheme 6). The six possible conformers of Int1 for enyne 9 a (9 aa‐Int1 a–c, 9 ab‐Int1 a–c) were found to have similar energies (within 0.9 kcal mol−1), although different reactivity. For 9 aa, the three conformers take different pathways generating different products. Transition states were located and energy barriers were found to be also similar. Interestingly, 9 aa‐Int1 a leads via 9 aa‐TS1 a‐5 directly, without the intermediacy of any cyclopropyl gold(I)‐type structure, to the formation of 9 aa‐Int5. This is a different type of intermediate (Int5) not depicted in Scheme 2 and corresponds to a structure similar to Int3, but with a fused tetrahydrofuran unit. Intermediates 9 aa‐Int1 b and 9 aa‐Int1 c lead to 9 aa‐Int3 and 9 aa‐Int2 respectively. It was also interesting to find that, in this system, open carbocation 9 aa‐Int3 is more stable than closed cyclopropyl gold(I) carbene 9 aa‐Int2 by 2.8 kcal mol−1, but 5.4 kcal mol−1 less than 9 aa‐Int5. On the other hand, for 9 ab, formation of cyclopropyl gold(I)carbene 9 ab‐Int2 is kinetically and thermodynamically more favored than 9 ab‐Int3 by 4.5 and 12.7 kcal mol−1, respectively. Intermediate 9 ab‐Int2 gives rise to 9 ab‐Int5 via 9 ab‐TS2 –5, which has a very low energy barrier (2.8 kcal mol−1). Furthermore, direct formation of 9 ab‐Int5 was also observed from 9 ab‐Int1 a through 9 ab‐TS1 a‐5. Therefore, we conclude that for 9 a, both isomers 9 aa and 9 ab give rise to Int5 type of products (9 aa‐Int5 and 9 ab‐Int5) although the reaction is more selective for isomer 9 ab giving only rise to 9 ab‐Int5.
Scheme 6.
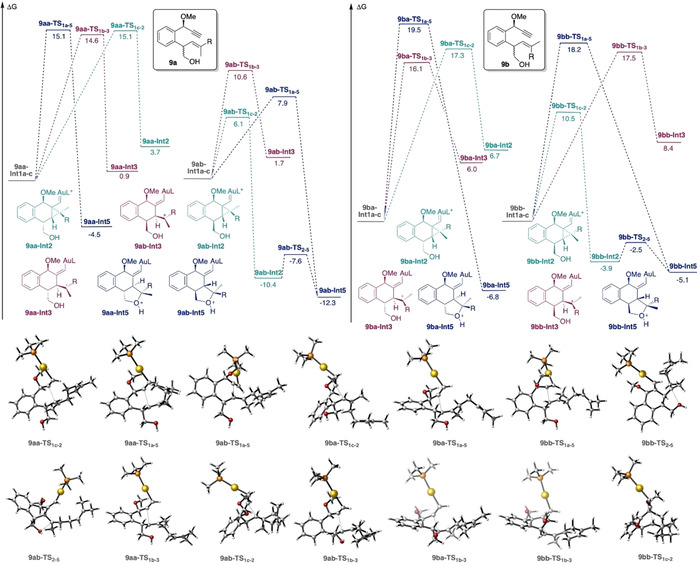
Computed formation of intermediates type of Int2, Int3 and Int5 for enynes 9 a and 9 b. L=PMe3. Free energies in kcal mol−1.
For those intermediates having the alcohol moiety in syn‐position with respect to the cyclopropyl (9 aa‐Int2) and to the carbocationic center (9 ab‐Int3), closure of the tetrahydrofuran ring was not observed. These intermediates could instead lead to the formation of naphthalene derivative side products 12 by loss of a molecule of formaldehyde in concomitance with a single cleavage rearrangement, connecting these intermediates (9 aa‐Int2 and 9 ab‐Int3) with 9 aa‐Int8 (Scheme 7). This mechanistic possibility was not computationally explored, but it is supported by the products observed experimentally.
Scheme 7.
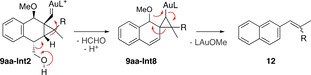
Proposed mechanism for the formation of naphthalene side product 12.
Similarly, computed pathways for diastereomer 9 ba have very similar energy barriers leading to 9 ba‐Int5. However, for 9 bb the predominant pathway by at least 7 kcal mol−1 is the formation of cyclopropyl gold(I) carbene 9 bb‐Int2 via 9 bb‐TS1 c‐2 , which would immediately give rise to 9 bb‐Int5 through 9 bb‐TS2 –5. Again, as it happened for 9 aa‐Int2, the formation of tetrahydrofuran product type Int5 was not observed from 9 ba‐Int2. We assumed that this intermediate would also lead to the formation of corresponding (Z)‐configured side product 12 (Scheme 7). Hence, 9 bb would selectively lead to the formation of 9 bb‐Int5, in good agreement with the experimental results, since no side‐product 12 was obtained for one of the diastereoisomers (presumably 9 bb, according to the presented calculations).
For both substrates 9 a,b the two pairs 9 aa‐Int5 and 9 ab‐Int5, and 9 ba‐Int5 and 9 bb‐Int5 would lead to the formation of same intermediates 9 a‐Int6 and 9 b‐Int6 by elimination of the methoxy group (Scheme 8). This explains why, when the reaction was attempted separately (9 ba and 9 bb), both diastereomers delivered the same products, arising from common intermediate 9 b‐Int6.
Scheme 8.
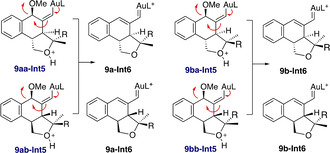
Formation of intermediates 9 a‐Int6 and 9 b‐Int6 for substrates 9 a and 9 b.
To understand the selective formation of trans‐fused rings 11 and 13 from 9 a‐b, the last cyclopropanation step was computed from intermediates 9 a‐Int6 and 9 b‐Int6 (Scheme 9). For substrate 9 a with an (E)‐configured alkene, intermediate 9 a‐Int6 can react further to form 9 a‐Int7 a via 9 a‐TS6 –7 a or 9 a‐Int7 b through 9 a‐TS6 –7 b, respectively. Remarkably, formation of 9 a‐Int7 a, which leads to trans‐fused cyclopropane 11, is 7.4 kcal mol−1 more favorable than formation of 9 a‐Int7 b, in agreement with our experimental results. However, for (Z)‐alkene 9 b, the energy difference for the two pathways is lower (3.6 kcal mol−1) and a mixture of cis/trans‐cyclopropane products could be formed, favoring cis‐14 (from 9 b‐Int7 b), as observed experimentally.
Scheme 9.
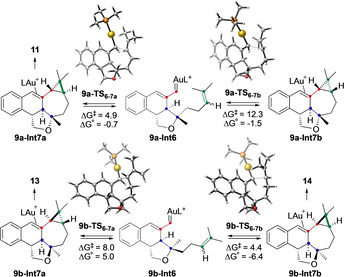
Different mechanistic pathways for cyclopropanation reactions of intermediates 9 a‐Int6 and 9 b‐Int6. L=PMe3. Free energies in kcal mol−1.
Structure of cyclopropyl gold(I) carbene intermediates
In the previous mechanistic study (Scheme 6) we found that the initial cyclization can lead to the formation of cyclopropyl gold carbenes (Int2) and open carbocations (Int3), such as the pairs 9 aa‐Int2 and 9 aa‐Int3, with very distinct energies and geometries. Therefore, to rigorously settle this issue computationally and to determine the dependence of the structure of the most stable intermediates on the different substituents, we studied the first step in the cyclization of several 1,6‐enynes.
First, we studied the behavior of 1‐phenyl‐7‐methyloct‐6‐en‐1‐yne (24) in the gold(I)‐catalyzed cyclization (Scheme 10). Initially, we performed a benchmark of DFT functionals using DLPNO‐CCSD(T)[ 18 , 19 ] as calibration method in order to discard any artifact associated to the chosen DFT method. [12]
Scheme 10.
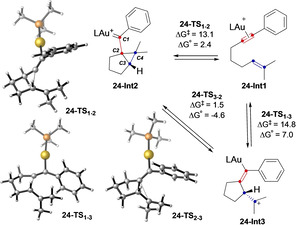
Formation of cyclopropyl gold(I) carbene 24‐Int2 and open carbocation 24‐Int3 from 24‐Int1, L=PMe3. Free energies in kcal mol−1.
We found that the most appropriate functionals for this system are M06 [20] ‐D3 [14] and B3LYP [13] ‐D3. [14] Hence, in this section, M06‐D3/6‐31G(d) [15] (C, H, P), SDD [16] (Au) level in CH2Cl2 (PCM) [17] level of theory was used to investigate the effect of substituents on the formation of different carbene‐type intermediates. Both cyclopropyl gold(I) carbene 24‐Int2 and open carbocation 24‐Int3 could be accessed from 24‐Int1, although the former intermediate was favored by 1.7 kcal mol−1. Interestingly, 24‐Int2 and 24‐Int3 are in equilibrium through 24‐TS2 –3. These intermediates show very different angles and bond lengths. [12] Thus, the C2‐C3‐C4 angle is 69.6° for 24‐Int2 and 115.9° for 24‐Int3. The C2−C4 distance in 24‐Int2 (1.734 Å) is significantly shorter than that for open carbocation 24‐Int3 (2.584 Å), whereas the C1−C2 distance shows the opposite trend (1.417 Å for 24‐Int2 vs. 1.351 Å for 24‐Int3).
We decided to investigate the effect of substituents on the initial formation of carbenic (Int2) or carbocationic (Int3 and/or Int4) intermediates with different enynes 24–31 (Scheme 11). In general, the three different intermediates Int2–4 were located having different angles and distances: cyclopropyl gold(I) carbene (Int2), open carbocation (Int3) and semi‐opened system (Int4) with a positive charge delocalized at C3 from enyne 24 that has a disubstituted alkene moiety. For 1,6‐enynes 24–26 with at least one methyl substituent at the terminal carbon of the alkene or a phenyl group at the alkyne, both Int2 and Int3 intermediates are formed, although in all cases the former is kinetically and thermodynamically favored. [21] On the other hand, for enynes 27–31 bearing terminal alkyne or alkene, formation of Int4 was observed. Similar results were observed when computing systems that contain aryl tethered 1,6‐enynes or alkyl tethered 1,7‐enynes. [12]
Scheme 11.
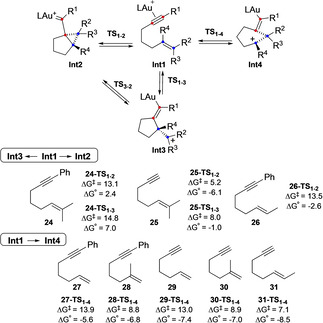
Possible pathways for the formation of carbene Int2, open carbocation Int3 and semi‐opened Int4. L=PMe3. Free energies in kcal mol−1.
The carbenic or carbocationic character of intermediates Int2, Int3, and Int4 was confirmed by a natural bond orbital (NBO) population analysis on representative structures (Figure 3).[ 12 , 22 ] The Au NPA charge was 0.193 for 24‐Int2, 0.189 for 24‐Int3 and 0.185 for 30‐Int4, being slightly higher for cyclopropyl gold(I) carbene species of type Int2, as expected. Moreover, NPA charges of the carbons connected to the gold atom are also slightly different for each system. For 24‐Int3 and 30‐Int4, positive charges are clearly delocalized and, for that reason charges on the carbon‐carbon double bond are more negative than for 24‐Int2.
Figure 3.
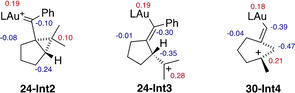
NPA charges by NBO analysis. Positive charges in red and negative charges in blue.
To further prove the structures of 25‐Int2, 25‐Int3, 30‐Int4, we carried out Quantum Theory Atoms in Molecules (QTAIM)[ 12 , 23 ] (Scheme 12). The bond critical points (BCPs) and the ring critical points (RCPs) were located and analyzed using Laplacian maps. Two RCP were located for intermediate 25‐Int2, whereas no RCP was observed between C8−C1 in the case of 25‐Int3, and the Laplacian clearly indicates the absence of this bond. Finally, only one ring critical point was observed in 30‐Int4 as a consequence of its semi‐opened ring system. Hence, QTAIM theory confirms that the molecular representation of these intermediates is accurate.
Scheme 12.
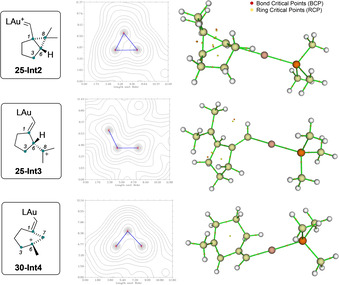
Molecular structure of 25‐Int2, 25‐Int3 and 30‐Int4. Laplacian map with electron density contour of atoms 1–6–8 and 1–6–7, bonds depicted in blue. QTAIM Topological Analysis graph with (BCPs) in red and (RCPs) in yellow.
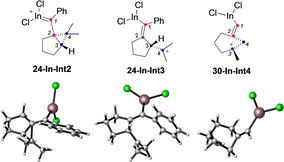
A mechanistic study on indium‐catalyzed cycloisomerizations of 1,6‐enynes found that the key intermediates for substrates of type 29 are also of type Int4. [24] For the sake of completeness, we also studied the possible involvement of intermediates related to Int2 and Int3 starting from enynes 24 and 30 using InCl3. We chose M06‐D3/6‐31G(d) (C, H) and LANL2DZ (In, Cl) level of theory in CH2Cl2 (PCM). We observed the formation of both intermediates 24‐In‐Int2 and 24‐In‐Int3 for enyne 24, whereas in the case of 30, only 30‐In‐Int4 was found (Table 1). Interestingly, 24‐In‐Int3 is the most favored intermediate species when using indium catalyst, whereas in the case of gold(I), cyclopropyl gold carbene 24‐Int2 is the preferred intermediate. Slight structural differences were observed when comparing these intermediates (24‐In‐Int2 and 24‐In‐Int3) with the ones found previously with gold(I) (24‐Int2, 24‐Int3). In the case of cyclopropyl carbene 24‐In‐Int2, longer distances were observed between C2−C4, indicating a less strained cyclopropane ring than for the gold(I) intermediate (Table 1, entry 2). In addition, the C2‐C3‐C4 angle was larger (72°) than for the gold(I)‐carbene 24‐Int2 (66.5°) (Table 1, entries 1–2). Intermediate 24‐In‐Int3 was found to be very similar to 24‐Int3 (Table 1, entries 3–4). On the other hand, for 30‐Int4 we observed the formation of a more opened system (longer distances between C2–C3), which presents the smallest angle observed in this study (Table 1, entries 5–6).
Table 1.
Optimized geometries and comparative list of calculated bond distances and angles of intermediates.[a]
|
| |||||
|---|---|---|---|---|---|
|
entry |
species |
d(C2‐C3) |
d(C2‐C4) |
∡(C2‐C3‐C4) |
Int2‐Int3[b] |
|
1 |
24‐Int2 |
1.536 |
1.652 |
66.5 |
0.0 |
|
2 |
24‐In‐Int2 |
1.542 |
1.768 |
72.0 |
0.0 |
|
3 |
24‐Int3 |
1.555 |
2.549 |
115.4 |
7.6 |
|
4 |
24‐In‐Int3 |
1.538 |
2.548 |
115.9 |
−4.4 |
|
5 |
30‐Int4 |
1.758 |
1.561 |
57.4 |
– |
|
6 |
30‐In‐Int4 |
2.213 |
1.566 |
44.9 |
– |
[a] DFT calculations performed using InCl3 as catalyst or L=PMe3 for Au. Free energies in kcal mol−1. Distances expressed in Å and angles in degrees. [b] Differences between free energies of (Int2, Int3) and (In‐Int2, In‐Int3).
Conclusions
In summary, we have uncovered a new gold(I)‐catalyzed cyclization cascade of substituted dienynes that can lead to selective formation of unexpected trans‐fused cyclopropanes within a trans‐bicyclo[5.1.0]octane framework, depending on the substrate geometry. DFT calculations and control experiments show that this specific selectivity is directed by the rigidity of the system. Likewise, computed pathways provide a rational for the role played by the fused tetrahydrofuran ring in the final cyclopropanation step. These new results expand the scope of these type of gold(I)‐catalyzed cyclizations for the formation of highly complex carbocyclic skeletons, in this case bearing trans‐fused cyclopropanes.
Our computational study included a reevaluation of the nature of the key intermediates in cycloisomerizations of enynes, providing evidence on the existence of three different types of cationic intermediates depending on the substitution of the initial substrate. The QTAIM theory confirms that the molecular representation of the different types of intermediates is accurate. Moreover, the metal carbenic or cationic character of these intermediates was confirmed by NBO analysis.
Experimental
Full details of all synthesis, characterization and DFT calculations can be found in the Supporting Information.
Deposition Numbers 1906609 (11), 1906610 (17 a), 1906613 (17 b), 1906611 (18), 1906612 (21), and 1906608 (23) contain the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service www.ccdc.cam.ac.uk/structures.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank the Ministerio de Ciencia e Innovación (PID2019‐104815GB‐I00 and FPU fellowship to H.A.‐R.), Severo Ochoa Excellence Accreditation 2020–2023 (CEX2019‐000925‐S), the European Research Council (Advanced Grant No. 835080), the Deutsche Forschungsgemeinschaft (postdoctoral fellowship to H.B.), the AGAUR (2017 SGR 1257), and CERCA Program/Generalitat de Catalunya for financial support. We also thank the ICIQ X‐ray diffraction unit for the X‐ray structures.
I. Escofet, H. Armengol-Relats, H. Bruss, M. Besora, A. M. Echavarren, Chem. Eur. J. 2020, 26, 15738.
References
- 1.
- 1a. Fürstner A., Chem. Soc. Rev. 2009, 38, 3208–3221; [DOI] [PubMed] [Google Scholar]
- 1b. Fensterbank L., Malacria M., Acc. Chem. Res. 2014, 47, 953–965; [DOI] [PubMed] [Google Scholar]
- 1c. Dorel R., Echavarren A. M., Chem. Rev. 2015, 115, 9028–9072; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1d. Day D. P., Chan P. W. H., Adv. Synth. Catal. 2016, 358, 1368–1384; [Google Scholar]
- 1e. Echavarren A. M., Muratore M. E., López-Carrillo V., Escribano-Cuesta A., Huguet N., Obradors C., Org. React. 2017, 92, 1–288; [Google Scholar]
- 1f. Boyle J. W., Zhao Y., Chan P. W. H., Synthesis 2018, 50, 1402–1416. [Google Scholar]
- 2.
- 2a. Jiménez-Núñez E., Echavarren A. M., Chem. Rev. 2008, 108, 3326–3350; [DOI] [PubMed] [Google Scholar]
- 2b. Obradors C., Echavarren A. M., Acc. Chem. Res. 2014, 47, 902–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.
- 3a. Pflästerer D., Hashmi A. S. K., Chem. Soc. Rev. 2016, 45, 1331–1367; [DOI] [PubMed] [Google Scholar]
- 3b. Stathakis C. I., Gkizis P. L., Zografos A. L., Nat. Prod. Rep. 2016, 33, 1093–1117; [DOI] [PubMed] [Google Scholar]
- 3c. Quach R., Furkert D. P., Brimble M. A., Org. Biomol. Chem. 2017, 15, 3098–3104; [DOI] [PubMed] [Google Scholar]
- 3d. Mayans J. G., Armengol-Relats H., Calleja P., Echavarren A. M., Isr. J. Chem. 2018, 58, 639–658. [Google Scholar]
- 4.
- 4a. Fürstner A., Morency L., Angew. Chem. Int. Ed. 2008, 47, 5030–5033; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 5108–5111; [Google Scholar]
- 4b. Hashmi A. S. K., Angew. Chem. Int. Ed. 2008, 47, 6754–6756; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 6856–6858; [Google Scholar]
- 4c. Seidel G., Mynott R., Fürstner A., Angew. Chem. Int. Ed. 2009, 48, 2510–2513; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 2548–2551; [Google Scholar]
- 4d. Benitez D., Shapiro N. D., Tkatchouk E., Wang Y., Goddard W. A., Toste F. D., Nat. Chem. 2009, 1, 482–486; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4e. Echavarren A. M., Nat. Chem. 2009, 1, 431–433; [DOI] [PubMed] [Google Scholar]
- 4f. Wang Y., Muratore M. E., Echavarren A. M., Chem. Eur. J. 2015, 21, 7332–7339; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4g. Harris R. J., Widenhoefer R. A., Chem. Soc. Rev. 2016, 45, 4533–4551. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Seidel G., Fürstner A., Angew. Chem. Int. Ed. 2014, 53, 4807–4811; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 4907–4911; [Google Scholar]
- 5b. Hussong M. W., Rominger F., Krämer P., Straub B. F., Angew. Chem. Int. Ed. 2014, 53, 9372–9375; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 9526–9529; [Google Scholar]
- 5c. Harris R. J., Widenhoefer R. A., Angew. Chem. Int. Ed. 2014, 53, 9369–9371; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 9523–9525; [Google Scholar]
- 5d. Joost M., Estévez L., Mallet-Ladeira S., Miqueu K., Amgoune A., Bourissou D., Angew. Chem. Int. Ed. 2014, 53, 14512–14516; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 14740–14744; [Google Scholar]
- 5e. Wang J., Cao X., Lv S., Zhang C., Xu S., Shi M., Zhang J., Nat. Comm. 2017, 8, 14625; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5f. Zeineddine A., Rekhroukh F., Carrizo E. D. S., Mallet-Ladeira S., Miqueu K., Amgoune A., Bourissou D., Angew. Chem. Int. Ed. 2018, 57, 1306–1310; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 1320–1324. [Google Scholar]
- 6. García-Morales C., Pei X.-L., Sarria Toro J. M., Echavarren A. M., Angew. Chem. Int. Ed. 2019, 58, 3957–3961; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 3997–4001. [Google Scholar]
- 7. Liu J.-Q., Yang Y.-F., Wang C.-F., Li Y., Qiu M.-H., Tetrahedron 2012, 68, 972–976. [Google Scholar]
- 8.
- 8a. J. M. Cronan Jr , Daviau T. R., Pannell L. K., J. H. Cardellina II , J. Org. Chem. 1995, 60, 6864–6865; [Google Scholar]
- 8b. Brochini C. B., Roque N. F., J. Braz. Chem. Soc. 2000, 11, 361–364; [Google Scholar]
- 8c. Yang Z., Chen H., Li Y., Helv. Chim. Acta 2003, 86, 3305–3309. [Google Scholar]
- 9.
- 9a. Gassman P., Williams F. J., Seter J., J. Am. Chem. Soc. 1968, 90, 6893–6895; [Google Scholar]
- 9b. Kirmse W., Hase C., Angew. Chem. Int. Ed. Engl. 1968, 7, 891–892; [Google Scholar]; Angew. Chem. 1968, 80, 914–914; [Google Scholar]
- 9c. Cain D., Pawar D. M., Noe E. A., J. Mol. Struct. Theochem. 2004, 674, 251–255; [Google Scholar]
- 9d. Squillacote M. E., DeFellipis J., Shu Q., J. Am. Chem. Soc. 2005, 127, 15983–15988. [DOI] [PubMed] [Google Scholar]
- 10. Jiménez-Núñez E., Molawi K., Echavarren A. M., Chem. Commun. 2009, 7327–7329. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Nieto-Oberhuber C., López S., Muñoz M. P., Cárdenas D. J., Buñuel E., Nevado C., Echavarren A. M., Angew. Chem. Int. Ed. 2005, 44, 6146–6148; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2005, 117, 6302–6304; [Google Scholar]
- 11b. Fürstner A., Davies P. W., Angew. Chem. Int. Ed. 2007, 46, 3410–3449; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 3478–3519; [Google Scholar]
- 11c. Cabello N., Jiménez-Núñez E., Buñuel E., Cárdenas D. J., Echavarren A. M., Eur. J. Org. Chem. 2007, 4217–4223; [Google Scholar]
- 11d. Jiménez-Núñez E., Claverie C. K., Bour C., Cárdenas D. J., Echavarren A. M., Angew. Chem. Int. Ed. 2008, 47, 7892–7895; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 8010–8013; [Google Scholar]
- 11e. Pérez-Galán P., Martin N. J. A., Campaña A. G., Cárdenas D. J., Echavarren A. M., Chem. Asian J. 2011, 6, 482–486. [DOI] [PubMed] [Google Scholar]
- 12.For further details see the Supporting Information. A dataset collection of computational results is available in the ioChem-BD repository and can be accessed via 10.19061/iochem-bd-1-189. Álvarez-Moreno M., de Graaf C., Lopez N., Maseras F., Poblet J. M., Bo C., J. Chem. Inf. Model. 2015, 55, 95–103. [DOI] [PubMed] [Google Scholar]
- 13. Becke A. D., J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar]
- 14. Grimme S., Antony J., Ehrlich S., Krieg H., J. Chem. Phys. 2010, 132, 154104. [DOI] [PubMed] [Google Scholar]
- 15. Hehre W. J., Ditchfield R., Pople J. A., J. Chem. Phys. 1972, 56, 2257–2261. [Google Scholar]
- 16. Andrae D., Haussermann U., Dolg M., Stoll H., Preuss H., Theor. Chim. Acta. 1990, 77, 123–141. [Google Scholar]
- 17. Cossi M., Barone V., Cammi R., Tomasi J., Chem. Phys. Lett. 1996, 255, 327–335. [Google Scholar]
- 18.
- 18a. Goerigk L., Grimme S., Phys. Chem. Chem. Phys. 2011, 13, 6670–6688; [DOI] [PubMed] [Google Scholar]
- 18b. Goerigk L., Kruse H., Grimme S., ChemPhysChem 2011, 12, 3421–3433; [DOI] [PubMed] [Google Scholar]
- 18c. Fischer M., Evers F. O., Formalik F., Olejniczak A., Theor. Chem. Acc. 2016, 135, 257; [Google Scholar]
- 18d. Dohm S., Hansen A., Steinmetz M., Grimme S., Checinski M. P., J. Chem. Theory Comput. 2018, 14, 2596–2608. [DOI] [PubMed] [Google Scholar]
- 19.
- 19a. Riplinger C., Sandhoefer B., Hansen A., Neese F., J. Chem. Phys. 2013, 139, 134101; [DOI] [PubMed] [Google Scholar]
- 19b. Iron M. A., Janes T., J. Phys. Chem. A 2019, 123, 3761–3781. [DOI] [PubMed] [Google Scholar]
- 20. Zhao Y., Truhlar D. G., Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- 21.The transition state between Int1 and Int3 was not found for enyne 26. In this case, an alternative pathway was found that leads directly to a product of formal [4+2] cycloaddition. For further details see the Supporting Information.
- 22.
- 22a. Weinhold F., Landis C. R., Valency and Bonding, Cambridge University Press, 2005; [Google Scholar]
- 22b. Landis C. R., Weinhold F., J. Am. Chem. Soc. 2006, 128, 7335–7345. [DOI] [PubMed] [Google Scholar]
- 23.
- 23a. Bader R. F. W., Atoms in Molecules, Clarendon Press, 1994; [Google Scholar]
- 23b. Bader R. F. W., Chem. Eur. J. 2006, 12, 7769–7772. [DOI] [PubMed] [Google Scholar]
- 24. Zhuo L.-G., Zhang J.-J., Yu Z.-Z., J. Org. Chem. 2014, 79, 3809–3820. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary