

Abstract
Epigenetic regulation is important in human health and disease, but the exact mechanisms remain largely enigmatic. DNA methylation represents one epigenetic aspect but is challenging to quantify. In this study, we introduce a digital approach for the quantification of the amount and density of DNA methylation. We designed an experimental setup combining efficient methylation‐sensitive restriction enzymes with digital polymerase chain reaction (PCR) to quantify a targeted density of DNA methylation independent of bisulfite conversion. By using a stable reference and comparing experiments treated and untreated with these enzymes, copy number instability could be properly normalized. In silico simulations demonstrated the mathematical validity of the setup and showed that the measurement precision depends on the amount of input DNA and the fraction methylated alleles. This uncertainty could be successfully estimated by the confidence intervals. Quantification of RASSF1 promoter methylation in a variety of healthy and malignant samples and in a calibration curve confirmed the high accuracy of our approach, even in minute amounts of DNA. Overall, our results indicate the possibility of quantifying DNA methylation with digital PCR, independent of bisulfite conversion. Moreover, as the context‐density of methylation can also be determined, biological mechanisms can now be quantitatively assessed.
Keywords: bisulfite conversion, digital PCR, DNA methylation, methylation‐sensitive restriction enzymes, quantification
In this study, we introduce a digital approach for the quantification of the amount and density of DNA methylation. We designed an experimental setup combining efficient methylation‐sensitive restriction enzymes with digital polymerase chain reaction (PCR), to accurately quantify DNA methylation independent of bisulfite conversion (a). The density of DNA methylation can be integrated into the analysis by the selection of PCR amplicon and methylation‐sensitive restriction enzyme (b).

1. INTRODUCTION
Epigenetic gene regulation is a complex process in which multiple mechanisms are involved. DNA methylation, typically referring to the reversible formation of 5‐methylcytosine in a CpG dinucleotide context (cytosine–phosphate–guanine: a cytosine directly followed by a guanine), represents one marker of epigenetic regulation with important roles in both human health and disease, including cancer. CpG‐rich genomic regions (“islands”) are frequently found in the promoter of a gene, and dense methylation of these islands is mostly associated with transcriptional silencing (Gardiner‐Garden & Frommer, 1987; Jones & Baylin, 2002). Aberrant DNA methylation of these islands may cause downregulation of tumor suppressor genes. In contrast, DNA methylation of specific CpGs may enhance the expression of some genes, thereby mimicking the effect of an oncogenic mutation (Barthel et al., 2017).
When studying DNA methylation, heterogeneous patterns are frequently observed (Figure 1). A particular DNA sample may comprise multiple alleles with varying DNA methylation: Alleles with methylation at all or none of the CpG dinucleotides (see Figure 1a) may be mixed with partially methylated alleles (methylation at a selection of the CpGs only, see Figure 1b). These mixtures are not always correctly interpreted, as most techniques measure the average methylation levels of the bulk or give a qualitative outcome, instead of providing a readout per individual CpG, per individual allele (Mikeska, Candiloro, & Dobrovic, 2010). Although the origin of heterogeneous DNA methylation remains incompletely understood, it may result from the presence of different cells with distinct methylation patterns, or the gradual accumulation of aberrant DNA methylation (Varley, Mutch, Edmonston, Goodfellow, & Mitra, 2009). To answer the questions surrounding DNA methylation and epigenetic regulation in general, accurate and quantitative assays are required to provide a detailed insight into the heterogeneity of DNA methylation.
Figure 1.

Heterogeneity in DNA methylation (addition of a methyl group to a cytosine nucleotide) can present in different ways. (a) A mixture of alleles with methylation at all cytosines, and alleles completely lacking DNA methylation. (b) A more complex mixture in which some alleles are partially methylated (i.e., methylation at a selection of the CpGs only)
Digital polymerase chain reaction (PCR) has proven to accurately address the genetic heterogeneity of (tumor‐derived) DNA samples. Mutations, copy number alterations, and cell type‐specific DNA markers have been quantified successfully to gain insight into the content and the evolutionary history of tumors (de Lange et al., 2015; Versluis et al., 2015; Zoutman et al., 2017). However, to measure DNA methylation in a similar quantitative fashion, new approaches are needed.
In this study, we present a digital PCR‐based approach to quantify a targeted density of DNA methylation by accurately measuring DNA digestion through a methylation‐sensitive restriction enzyme (MSRE). Whereas most analog methylation analysis methods depend on the chemical conversion of the input DNA with sodium bisulfite, MSREs can differentiate between methylated and unmethylated alleles independently of this conversion. Earlier and recent studies already showed the potential of combining MSREs with quantitative polymerase chain reaction or digital PCR, but were mainly focused on the detection of low fractions methylated DNA and/or benign conditions (Abe et al., 2019; Hashimoto, Kokubun, Itoi, & Roach, 2007; Hindson et al., 2011; Redshaw, Huggett, Taylor, Foy, & Devonshire, 2014; Tong et al., 2010). Instead, our experimental setup provides a complete quantification of DNA methylation with confidence intervals in the whole range from 0% to 100%, and can be applied in copy number unstable specimens, such as malignancies.
We evaluate our approach in the context of methylation in the RASSF1 transcript A promotor region. Ras association domain family member 1 (RASSF1) transcript A is an established tumor suppressor gene that is located on chromosome 3p21, and part of several tumorigenic molecular pathways (Donninger, Vos, & Clark, 2007). Epigenetic silencing in the form of DNA promoter methylation of this gene has been demonstrated in a wide variety of human malignancies. Most notably, frequencies of up to 88%–99% of affected cases have been reported in lung, prostate, and breast cancer (Donninger et al., 2007).
To validate our methodology, an in silico simulation of the digital PCR experiments is designed. A range of reference samples and an innovative dilution series are analyzed to investigate the sensitivity and dynamic range of the approach.
2. MATERIALS AND METHODS
2.1. Editorial policies and ethical considerations
All samples analyzed in this study were anonymized and obtained from healthy volunteers who provided informed consent. All specimens were handled in accordance with the institutional and national ethical guidelines and the Declaration of Helsinki.
2.2. Sample collection and DNA isolation
Five cancer cell lines derived from primary uveal melanomas (92.1, Mel‐202, Mel‐270, Mel‐285, and Mel‐290) were available in the Department of Ophthalmology, Leiden University Medical Center (LUMC), the Netherlands (De Waard‐Siebinga et al., 1995; Verbik, Murray, Tran, & Ksander, 1997).
Five male peripheral blood mononuclear cells (PBMC) and three fresh frozen placenta samples were a gift from the Department of Immunohematology and Blood Transfusion, LUMC. DNA was isolated using the QIAmp DNA Mini Kit, according to the instructions supplied by the manufacturer (Qiagen).
Three commercially available control DNA samples were purchased from Merck Millipore: CpGenome Universal Methylated DNA (enzymatically methylated human male genomic DNA), CpGenome Universal Unmethylated DNA vial A (human genomic DNA) and B (genomic DNA from a primary human fetal cell line).
2.3. Experimental design
MSRE BstUI (New England Biolabs) was selected based on its capacity to distinguish methylated from unmethylated DNA sequences. Incubation of 1 h at 60°C results in the digestion of unmethylated 5′‐CGCG‐3′ substrate sequences, whereas methylated 5′‐CGCG‐3′ sequences remain intact. To quantify DNA methylation, this digestion is measured in two separate duplex digital PCR experiments: one Baseline experiment measuring the initial presence of target of interest, and one MSRE+ experiment measuring the presence after MSRE digestion (Figure 2a). To correct for input differences, an independent and undigestible reference was measured simultaneously in both experiments.
Figure 2.
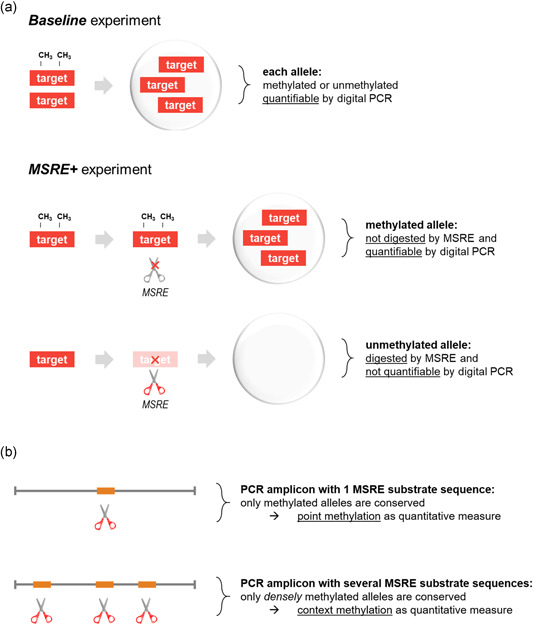
Concept of quantifying DNA methylation using methylation‐sensitive restriction enzyme (MSRE) and digital polymerase chain reaction (PCR). (a) An MSRE incubation of a DNA sample results in the digestion of unmethylated fragments by the MSRE, whereas methylated sequences remain intact. To calculate the fraction methylated alleles, the MSRE digestion is measured in two separate duplex digital PCR experiments: one Baseline experiment measuring the initial presence of target of interest, and one MSRE+ experiment measuring the presence after MSRE digestion. (b) The density of DNA methylation can be integrated into the analysis by the selection of PCR amplicon and MSRE. Densely methylated alleles can be quantified using a PCR target amplicon with multiple recognition substrates for the chosen MSRE, as the only methylation of all these restriction sites can prevent MSRE digestion. The absence of methylation at any of these sites will result in target fragmentation, impeding PCR, and thus, measurement of this allele. Alternatively, when only one recognition sequence is present in the PCR amplicon, the methylation state of that site specifically is measured, reflecting point methylation
To quantify RASSF1 methylation, an FAM digital PCR assay was designed (Sigma‐Aldrich) targeting the promoter region of RASSF1 transcript A and five 5′‐CGCG‐3′ BstUI recognition sequences (see Table S1 for exact genomic positions). Only alleles with consecutive methylation on these sequences are conserved by MSRE treatment and measurable in the MSRE+ experiment. In contrast, (partly) unmethylated alleles are digested by MSRE treatment. As an independent stable reference, an HEX digital PCR assay for TERT was used (Bio‐Rad), in which no BstUI recognition sequences were present.
The presence of RASSF1 is compared against reference TERT in each experiment by calculating the ratio RASSF1/TERT:
The methylation fraction (defined as the fraction RASSF1 alleles being methylated) is calculated by dividing the ratios RASSF1/TERT of the MSRE+ and Baseline experiment:
Note that, in copy number stable samples, the concentration of all RASSF1 alleles can be estimated by the concentration of TERT alleles. The ratio RASSF1/TERT of the MSRE+ experiment then already gives the methylation fraction. For consistency across all experiments, this approach was not applied in any of our samples.
In copy number unstable samples, TERT might be involved in genetic alterations. However, as it is only used to normalize input differences between the MSRE+ and Baseline experiments, its only requirement is being measurable without being affected by the MSRE. Therefore, a TERT copy number alteration would generally not influence the methylation fraction of RASSF1.
Confidence intervals on obtained concentration ratios can be calculated using the geometric interpretation of Fieller's theorem (Dube, Qin, & Ramakrishnan, 2008). This mathematical approach could easily be translated to our experimental design, as outlined in Supporting Information Data 1.
For the in silico simulations, R (version 3.6.0), RStudio (version 1.1.463), R packages rmarkdown (version 1.11), and digitalPCRsimulations (version 1.1.0) were used.
MIQE context sequences for both digital PCR assays are presented in Table S1.
2.4. MSRE incubation and digital PCR
Generally, 50 ng DNA was incubated for 1 h at 60°C with 1‐U BstUI and 0.5‐µl 10× CutSmart Buffer (New England Biolabs), in a total volume of 5.0 µl. For Baseline experiments, 50‐ng DNA was incubated for 1 h at 60°C with 0.5‐µl 10× CutSmart buffer in a total volume of 5.0 µl. All incubations took place in a T100 Thermal Cycler (Bio‐Rad).
Digital PCR experiments were performed using the QX200™ Droplet Digital™ PCR System (Bio‐Rad) following the general experimental guidelines as described earlier (Zoutman, Nell, & van der Velden, 2019). In short, 20‐ng incubated DNA was analyzed in a 22‐µl experiment, using 11‐µl ddPCR™ Supermix for Probes (No dUTP; Bio‐Rad) and primers and probes in a final concentration of 900 and 250 nM, respectively. PCR mixtures were partitioned into 20,000 droplets using the AutoDG™ System (Bio‐Rad). Subsequent PCR was performed in a T100 Thermal Cycler using the following protocol: 10 min at 95°C; 30 s at 94°C, and 1 min at 55°C for 40 cycles; 10 min at 98°C; cooling at 12°C for up to 48 h, until droplet reading. Ramp rate was set to 2°C/s for all steps. Droplet reading was performed in a QX200™ Droplet Reader (Bio‐Rad).
2.5. Data analysis
Raw digital PCR results were acquired using QuantaSoft (version 1.7.4; Bio‐Rad) and imported in online digital PCR management and analysis application Roodcom WebAnalysis (version 1.9.4; https://webanalysis.roodcom.nl/).
2.6. Methylation‐sensitive melting curve analysis (MS‐MCA) and bisulfite sequencing (BS)
To validate obtained methylation fractions by MS‐MCA and BS, a bisulfite conversion of MSRE+ and Baseline samples was carried out using the EZ DNA Methylation Kit (Zymo Research Corporation). Generally, 100‐ng DNA was converted according to the manufacturer's guidelines and eluted in 10‐µl M‐Elution Buffer. To remove possibly interfering salts, a DNA purification before the conversion was performed using DNA Clean & Concentrator (Zymo), following the manufacturer's instructions.
One microliter bisulfite‐converted DNA was analyzed in 7‐µl experiment, using 3.5‐µl iQ SYBR Green Supermix (Bio‐Rad) and primers in a final concentration of 700 nM. Consequent PCR was performed in a CFX384 Real‐Time PCR system (Bio‐Rad), using the following protocol: 3 min at 95°C; 10 s at 96°C, 10 s at 65°C, and 30 s at 72°C for 8 cycles; 10 s at 96°C, 10 s at 62°C, and 30 s at 72°C for 32 cycles; 10 s at 95°C; melting curve from 65°C to 95°C per 0.3°C.
MS‐MCA analysis was performed in CFX Manager (version 3.1; Bio‐Rad). Raw PCR products were purified and analyzed for “Quick Shot Short GC” Sanger Sequencing at BaseClear, Leiden, The Netherlands. AB1‐files were analyzed using Roodcom SangerSeq Analysis (version 1.0; https://sangerseq.roodcom.nl).
MIQE context sequence for the used primers is presented in Table S1.
3. RESULTS
3.1. Digital PCR experimental setup to quantify DNA methylation
To determine the fraction methylated alleles in a sample of interest, two separate duplex digital PCR experiments were carried out to quantify target and reference sequences before and after incubation with an MSRE. In the Baseline experiment, all target alleles (both methylated and unmethylated) were measured, while after digestion by the MSRE (i.e., the MSRE+ experiment), only the methylated target alleles were quantitated (Figure 2a). To correct for input differences between both experiments, the measured concentration target alleles were normalized to the concentration of a stable reference, which is indigestible by the MSRE. By comparing the results of the Baseline and MSRE+ experiments, the methylation fraction and its 95% confidence interval were determined.
We chose to evaluate our approach by quantifying dense methylation of the promoter region associated with RASSF1 transcript A. By selecting a PCR target amplicon containing multiple recognition sequences for the preferred MSRE, the only methylation of all these restriction sites (i.e., a densely methylated allele) can prevent MSRE digestion. Less than complete methylation will lead to fragmentation of the allele, hindering further PCR amplification, and thus, the measurement of this allele. Conceptually, this illustrates how the context of DNA methylation can be integrated in the quantification (Figure 2b). As the methylation of surrounding CpGs may be biologically relevant, a valuable aspect is included in the analysis. Alternatively, an MSRE may be chosen for which its substrate is only present once in the PCR amplicon. This will restrict the quantification to this site only, reflecting point methylation of a specific CpG.
3.2. In silico evaluation of experimental setup and mathematical rationale
The digital nature of our experimental setup enables an advanced in silico modeling of it (Supporting Information Data 1). For each of 50 different conditions (5, 10, 20, 50, and 100 ng DNA, with methylation fractions of 10%–100% per 10%), 10,000 digital PCR simulations were performed. This allowed an extensive evaluation of the accuracy and precision of our experimental approach.
Generally, the methylation fraction was correctly estimated across all simulated conditions (Figure 3a); though, the absolute uncertainty of the measurements becomes larger at lower input concentrations and higher methylation fractions (Figure 3b). Overall, in 95.1% of the total of 500,000 simulations, the true methylation fraction did lie in the calculated confidence interval. As this coverage is very close to our intended 95%, it shows the validity of the mathematical approach. The coverage per condition was very comparable between all input states (Figure 3c). In contrast, the absolute width of the calculated confidence intervals was highly variable among the amount of input DNA and input methylation fraction (Figure 3d). However, as the digital error was correctly estimated, broader confidence intervals were observed when a larger uncertainty was found, which was at lower input amounts and at higher methylation fractions.
Figure 3.
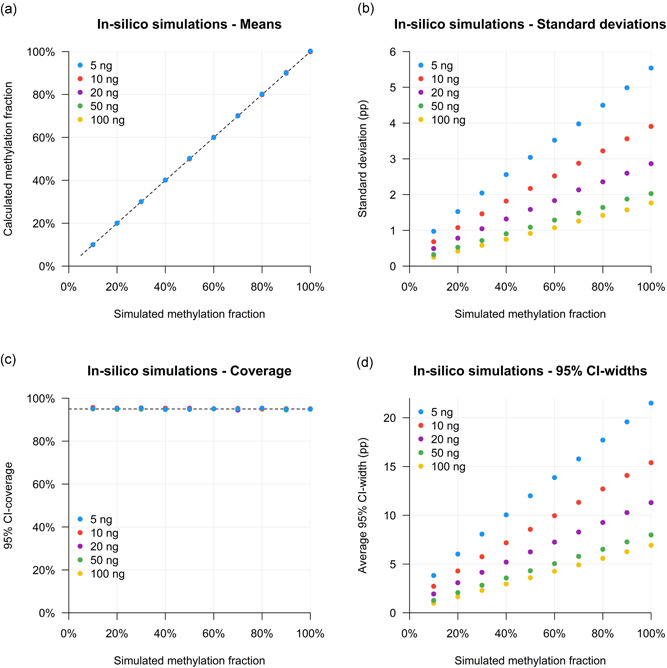
Validation of digital approach with methylation‐sensitive restriction enzyme sample digestion and digital polymerase chain reaction by in silico simulations for 20 methylation fractions and five input amount conditions. (a) Mean of calculated methylation fractions per input condition. (b) Standard deviation of calculated methylation fractions per input condition. (c) 95% confidence interval coverage per input condition. (d) Average absolute widths of 95% confidence intervals (CIs) per input condition
3.3. RASSF1 promoter methylation in references samples
A positive biological control for RASSF1 promoter methylation can be found in malignancies and in embryonic tissue (Chiu et al., 2007; Grawenda & O'Neill, 2015). Therefore, five uveal melanoma cell lines and three placental DNA samples were analyzed as reference samples. An illustration of the complete experimental and analytic workflow is given in Figure 4a.
Figure 4.

RASSF1 promotor methylation fractions in control samples. (a) Illustration of the experimental and analytic workflow to obtain the methylation fraction, for example, cell line Mel‐290. Summary of RASSF1 methylation fractions in (b) five uveal melanoma cell lines, (c) three placenta samples, (d) five healthy male peripheral blood mononuclear cell samples, and (e) three CpGenome control samples
In four out of the five uveal melanoma cell lines, in which previously extensive RASSF1 methylation was observed, only completely methylated alleles were detected (Figure 4b; Maat et al., 2007). The fifth cell line (Mel‐290), the only one that was previously typed as unmethylated, now presented with 6% methylation.
In placental tissue, both fetal and maternal cells are present, with hypo‐ and hypermethylated RASSF1 in distinct cell types (Chiu et al., 2007). We analyzed three DNA samples isolated from fresh frozen placenta tissues and found substantial RASSF1 methylation fractions of 70%, 77%, and 78% (Figure 4c). These high fractions validate the considerable presence of RASSF1‐methylated alleles in uncultured placentas (Paganini et al., 2015).
Although in healthy female blood, small amounts of methylated RASSF1 may be found (derived from current or prior pregnancy), healthy male blood only contains unmethylated RASSF1 alleles and can therefore be used as a negative control (van den Oever et al., 2013). Indeed, in five PBMC samples from healthy males, 0% RASSF1 methylation was detected (Figure 4d).
Unmethylated and methylated genomic DNA is commercially available and is an attractive source of control DNA for gene methylation studies. However, in two of the three available samples, heterogenous RASSF1 methylation was detected in our analyses. The methylated control demonstrated a 54% methylation fraction, whereas unmethylated controls A and B presented with fractions of 55% and 0% methylation, respectively (Figure 4e).
3.4. Validation by MS‐MCA and BS
To validate our findings, a selection of samples was treated with bisulfite and analyzed using MS‐MCA and BS (Figure 5 and Figure S1). After bisulfite conversion, the PCR products of methylated and unmethylated alleles have a different base composition: unmethylated cytosines have been converted to thymines, while methylated cytosines remain cytosines. As thymines bind to adenine via two hydrogen bonds, and cytosines to guanines via three hydrogen bonds, methylated and unmethylated alleles ultimately lead to distinct melting profiles when subjected to thermal denaturation (MS‐MCA; Wojdacz, Dobrovic, & Hansen, 2008). Moreover, these base differences are detectable using conventional Sanger sequencing (BS).
Figure 5.

Validation of RASSF1 methylation in reference samples with methylation‐sensitive melting curve analysis (MS‐MCA) and bisulfite sequencing (BS). (a) Fully methylated cell line Mel‐270 presents with methylated signals in the Baseline and MSRE+ experiments: methylated cytosines remain unaffected after bisulfite conversion, resulting in a relatively high melting peak around 85°C (due to the three hydrogen bonds between cytosine and guanine). The cytosines are also detectable by Sanger sequencing of the polymerase chain reaction (PCR) products. (b) Fully unmethylated blood sample PBMC 2 only shows unmethylated signals in the Baseline experiment: after bisulfite conversion and PCR amplification, unmethylated cytosines are converted to thymines, which results in a low melting peak around 80°C (due to the only two hydrogen bonds between thymines and adenines) and corresponding sequencing results. The unmethylated alleles are completely digested by the MSRE, leading to loss of all signals in the MSRE+ experiment. (c) Only unmethylated RASSF1 was detected in baseline MS‐MCA and BS analyses with Mel‐290, while after MSRE digestion, a methylated fraction emerged that was undetectable in the Baseline experiment
To validate that only methylated alleles resisted MSRE digestion, samples that were pre‐incubated with the restriction enzyme (MSRE+) were also analyzed.
The fully methylated cell lines (e.g., Mel‐270) were resistant to MSRE digestion and therefore showed similar results between the Baseline and MSRE+ experiments, all indicative of methylation (Figure 5a).
The unmethylated blood samples (e.g., PBMC 2) did not show signs of methylation and, as a consequence, only generated signals in the Baseline experiments. Typically, the lack of methylated alleles led to full sample digestion in the MSRE+ experiments, resulting in no amplification in the MS‐MCA and BS (Figure 5b).
Regarding the cell line Mel‐290 (6% methylation), no signs of methylation were observed in both the MS‐MCA and BS Baseline results. Only after the MSRE‐based enrichment for methylated alleles, the presence of a small number of methylated alleles was revealed by the combination of MS‐MCA or BS and MSRE enrichment for methylation (Figure 5c).
The placenta samples and two of the three commercially available methylation control samples (the methylated control and unmethylated control A) were all characterized by heterogeneous RASSF1 methylation, in which both methylated and unmethylated alleles were present. These findings were confirmed by the MS‐MCA and BS results, in which in the Baseline experiments, heterogenous peaks were found, and methylated peaks only in the MSRE+ experiments (Figure S1).
3.5. High accuracy and dynamic range
To evaluate the accuracy and dynamic range of the approach, a calibration curve was prepared. In different ratios, fully methylated target DNA (derived from cell line Mel‐270) was mixed with fully unmethylated DNA (derived from PBMC 2), maintaining a constant total DNA concentration. Subsequently, all mixtures were analyzed for RASSF1 methylation. In addition, the genotype status of single‐nucleotide polymorphism (SNP) rs1989839 was quantified in all mixtures. This SNP‐based approach allowed us to ascertain the actual diluted methylation fraction. As samples Mel‐270 and PBMC 2 can be distinguished with this SNP, mixtures of both samples can be reconstituted based on the exact presence of both alleles (Figure 6a).
Figure 6.
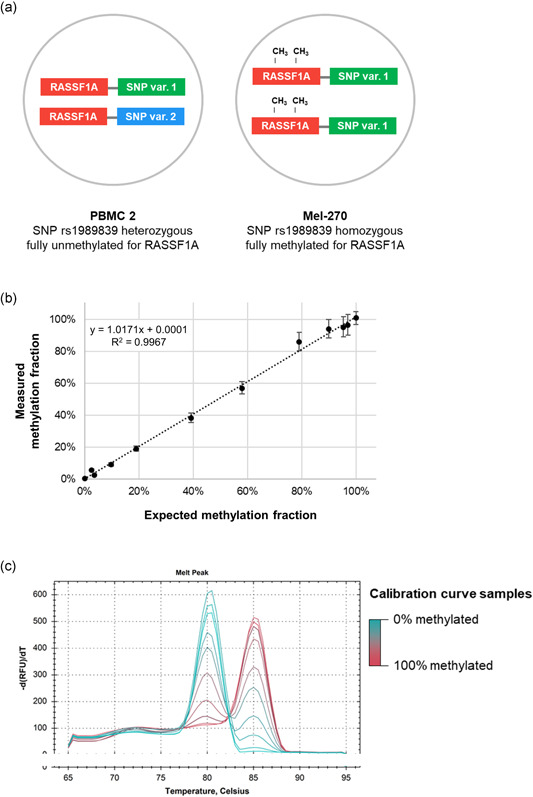
Accuracy and dynamic range determined with a calibration curve for RASSF1 methylation with diluted reference samples that includes a genetic calibrator by way of an SNP. (a) Equal concentrations of PBMC 2 (fully unmethylated for RASSF1 and heterozygous for SNP rs1989839) and Mel‐270 (fully methylated for RASSF1 and homozygous for SNP rs1989839) are mixed in different ratios, which are measured for RASSF1 methylation fraction (y axis) and SNP variant balance (x axis). This balance, calculated as % Mel‐270 = , validates the mixed ratio of both samples and is referred to as the expected methylation fraction. (b) A calibration curve comparing the expected and measured methylation fraction showed a high linear correlation (R 2 = .9967), indicating the quantitative nature of the approach. (c) Methylation‐sensitive melting curve analysis of all prepared ratios showed gradually increasing and decreasing peak heights at the methylated and unmethylated amplicon melting temperatures, but could not easily be translated into a quantitative measure
The results of both measurements are integrated, as shown in Figure 6b. A linear relationship between input ratio and calculated methylation fraction was observed (Pearson R 2 = .9967; slope = 1.0171), indicative of the high accuracy of the measurements.
The calibration curve samples were also treated with bisulfite and analyzed using MS‐MCA. As expected, gradually increasing and decreasing peak heights at the methylated and unmethylated amplicon melting temperatures were observed across the samples (Figure 6c). This peak distribution could, however, not easily be translated into a quantitative value, especially as small peaks were not consistently identifiable at the lowest and highest methylation fractions.
4. DISCUSSION
DNA methylation, one of the main markers of epigenetic regulation, is a fundamental process involved in human embryology, physiology, and pathology. Especially in cancer, aberrant methylation plays a role in the initiation and progression of the disease (Jones & Baylin, 2002, 2007).
Along with genetic alterations, also epigenetic changes diversify tumor cell populations (“intratumor heterogeneity”). Epigenetic subpopulations may arise during tumor evolution or due to epigenetically defined differentiation programs in normal cells (Ohgane, Yagi, & Shiota, 2008). Recent technological advances have enabled extensive, quantitative studies into the genetically determined tumor heterogeneity, which not only provided new insights into the content, but also an evolutionary history of tumors (de Lange et al., 2015; Oesper, Mahmoody, & Raphael, 2013). However, comparable quantitative analyses for epigenetic alterations are scarce, and quantifying epigenetic heterogeneity is still challenging (Mikeska et al., 2010).
Conventional methylation methods commonly do not recognize the presence of epigenetic heterogeneity and produce a dichotomous outcome, comparable to the traditional genetic analyses. In this way, the methylation state is interpreted as “yes” or “no” while the real dynamic range may be much wider (Figure 1). Hence, heterogeneous methylation patterns are often not correctly recognized (Mikeska et al., 2010).
Considering the wide variety of new applications with digital PCR‐based genetic analyses, we aimed to develop a comparable method to quantify epigenetic heterogeneity (de Lange et al., 2015; Versluis et al., 2015; Zoutman et al., 2017). In this study, we showed that digital PCR, in combination with an MSRE, allows quantification of a targeted density of DNA methylation accurately.
Widely used methods (methylation‐specific PCR, MS‐MCA, BS, pyrosequencing, bead microarray) require a bisulfite conversion of the DNA to make DNA methylation analyzable (Herman, Graff, Myöhänen, Nelkin, & Baylin, 1996). Ideally, this conversion deaminates unmethylated cytosines to uracils but does not change methylated cytosines. However, the experimental conditions for this conversion are critical. To avoid incomplete conversion and false‐positive results, an experiment is carried out that may result in extensive degradation (of up to 90%) of the input DNA (Grunau, Clark, & Rosenthal, 2001; Tanaka & Okamoto, 2007). Moreover, both consistent and inconsistent conversion introduces sequence differences and hence amplification biases are not uncommon, possibly leading to incorrect quantitative interpretations (Warnecke et al., 1997).
In this study, we prepared mixtures of fully methylated and fully unmethylated alleles and analyzed them by using our combination of MSRE and digital PCR (Figure 6b). Generally, these in vitro generated mixtures validated the quantitative nature of MSRE in combination with digital PCR. Validation with MS‐MCA showed patterns that were fully in line with the dilutions but nevertheless only allowed a qualitative interpretation. Sensitivity also turned out to be critical in MS‐MCA as the minor presence of methylated alleles in our 2.5% and 5% mixtures was undetectable, and could, therefore, not be quantified (Figure 6c). Our digital approach, in contrast, distinguished these minor fractions of methylated alleles and showed significant results of 2% and 5%. Similarly, uveal melanoma cell line Mel‐290 presented with a small fraction of 6% methylated alleles using digital PCR, which was again undetectable with MS‐MCA and BS (Figure 5c; Maat et al., 2007).
Whereas bisulfite conversion incorporates the original methylation state in the converted sequence, our approach relies on the specificity and sensitivity of the chosen MSRE to distinguish methylated from unmethylated sites. Consequently, when MSRE treatment leads to nonspecific or incomplete DNA digestion, under‐ or overestimated methylated fractions will be obtained (Hashimoto, Kokubun, Itoi, & Roach, 2014). However, a calibration curve made with validated control samples indicated a properly working and highly efficient MSRE, as the calculated methylation fractions accurately matched with the full range of prepared ratios (R 2 = .9967; Figure 6b). Importantly, as this result was obtained without a cumbersome bisulfite conversion, an accurate quantification can be obtained in a faster, easier, and less sample‐requiring experiment.
In a methodologically comparable approach, MSRE digestion was combined with real‐time PCR. Importantly, commercially available methylated and unmethylated control DNA samples were added as reference samples to the analysis to correct for nonspecific and incomplete DNA digestion (Hashimoto et al., 2014). However, here we showed that absolute quantification with digital PCR indicated the presence of both methylated and unmethylated RASSF1 alleles in two of these commercial control samples (Figures 4E and S1). Consequently, these samples cannot be used as absolute references in quantitative experiments. For MS‐MCA they may fit, as they provide control melting peaks that are useful for a qualitative characterization. Still, the use of these controls to correct for nonspecific and incomplete DNA digestion should be given careful consideration, as it may introduce a systematic mathematical error in the obtained quantifications.
Importantly, the limitations of our digital approach should also be considered. Although we confirmed theoretically that our mathematical rationale is valid and that methylation fraction can be correctly determined, our in silico simulations also indicated that the uncertainty of the results depends on the amount of input DNA and the methylation fraction itself (Supporting Information Data 1 and Figure 3). Whereas the input may be adjusted experimentally and its varying influence on result precision is a known characteristic of digital PCR, the methylation fraction is sample‐dependent and cannot be changed (Dube et al., 2008). Generally, lower input concentrations and higher methylation fractions are accompanied by higher absolute uncertainty. However, as the digital error of the experiments can be correctly estimated, this uncertainty is readily translated into broader confidence intervals (Figure 3c,d). These findings were confirmed and clearly illustrated by our calibration curve experiments (Figure 6b).
Moreover, the approach described in this study is not specifically designed for rare allele detection. Instead, we validated that our setup is providing accurate quantifications of methylation fractions across the whole range from 0% to 100% (Figure 6b). This is comparable to the application of digital PCR in DNA‐based T‐cell quantifications and covers the biological relevance of DNA methylation in most sample mixtures of hyper‐ or hypomethylated alleles (Zoutman et al., 2017).
To conclude, our results demonstrate the possibility of accurately quantifying DNA methylation with digital PCR, independent of bisulfite conversion. A superior measurement precision could be obtained compared with conventional techniques, and without the disadvantages of the conversion, more effective experiments may be carried out. Given the accuracy in quantifications, we propose that (sub)clone‐ or cell‐type‐specific DNA methylation markers, such as, but not limited to RASSF1, may be investigated accordingly in both benign and malignant samples, possibly providing new insights in both human health and disease. Moreover, as the context‐density of these DNA methylation markers can also be determined, important biological mechanisms can now be quantitatively assessed with digital PCR.
CONFLICT OF INTERESTS
The authors declare that there are no conflict of interests.
Supporting information
Supporting information.
Supporting information.
ACKNOWLEDGMENTS
We thank our colleagues from the Department of Immunohematology and Blood Transfusion, LUMC, for providing PBMC and placental reference samples, N. A. Gruis (Department of Dermatology, LUMC) for reviewing the manuscript and W. H. Zoutman (Department of Dermatology, LUMC) for helpful discussions and his expertise regarding bisulfite experiments. Rogier J. Nell is supported by the European Union's Horizon 2020 research and innovation program under Grant Agreement No. 667787 (UM Cure 2020 project).
Nell RJ, van Steenderen D, Menger NV, Weitering TJ, Versluis M, van der Velden PA. Quantification of DNA methylation independent of sodium bisulfite conversion using methylation‐sensitive restriction enzymes and digital PCR. Human Mutation. 2020;41:2205–2216. 10.1002/humu.24111
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- Abe, M. , Kagara, N. , Miyake, T. , Tanei, T. , Naoi, Y. , Shimoda, M. , … Noguchi, S. (2019). Highly sensitive detection of sentinel lymph node metastasis of breast cancer by digital PCR for RASSF1A methylation. Oncology Reports, 42(6), 2382–2389. 10.3892/or.2019.7363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barthel, F. P. , Wei, W. , Tang, M. , Martinez‐Ledesma, E. , Hu, X. , Amin, S. B. , … Verhaak, R. G. W. (2017). Systematic analysis of telomere length and somatic alterations in 31 cancer types. Nature Genetics, 49(3), 349–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu, R. W. K. , Chim, S. S. C. , Wong, I. H. N. , Wong, C. S. C. , Lee, W.‐S. , To, K. F. , … Lo, Y. M. D. (2007). Hypermethylation of RASSF1A in human and rhesus placentas. The American Journal of Pathology, 170(3), 941–950. 10.2353/ajpath.2007.060641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donninger, H. , Vos, M. D. , & Clark, G. J. (2007). The RASSF1A tumor suppressor. Journal of Cell Science, 120(Pt 18), 3163–3172. 10.1242/jcs.010389 [DOI] [PubMed] [Google Scholar]
- Dube, S. , Qin, J. , & Ramakrishnan, R. (2008). Mathematical analysis of copy number variation in a DNA sample using digital PCR on a nanofluidic device. PLOS One, 3(8), e2876 10.1371/journal.pone.0002876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardiner‐Garden, M. , & Frommer, M. (1987). CpG islands in vertebrate genomes. Journal of Molecular Biology, 196(2), 261–282. [DOI] [PubMed] [Google Scholar]
- Grawenda, A. M. , & O'Neill, E. (2015). Clinical utility of RASSF1A methylation in human malignancies. British Journal of Cancer, 113(3), 372–381. 10.1038/bjc.2015.221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grunau, C. , Clark, S. J. , & Rosenthal, A. (2001). Bisulfite genomic sequencing: Systematic investigation of critical experimental parameters. Nucleic Acids Research, 29(13), E65 10.1093/nar/29.13.e65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto, K. , Kokubun, S. , Itoi, E. , & Roach, H. I. (2007). Improved quantification of DNA methylation using methylation‐sensitive restriction enzymes and real‐time PCR. Epigenetics, 2(2), 86–91. 10.4161/epi.2.2.4203 [DOI] [PubMed] [Google Scholar]
- Hashimoto, K. , Kokubun, S. , Itoi, E. , & Roach, H. I. (2014). Improved quantification of DNA methylation using methylation‐sensitive restriction enzymes and real‐time PCR. Epigenetics, 2(2), 86–91. 10.4161/epi.2.2.4203 [DOI] [PubMed] [Google Scholar]
- Herman, J. G. , Graff, J. R. , Myöhänen, S. , Nelkin, B. D. , & Baylin, S. B. (1996). Methylation‐specific PCR: A novel PCR assay for methylation status of CpG islands. Proceedings of the National Academy of Sciences of the United States of America, 93(18), 9821–9826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hindson, B. J. , Ness, K. D. , Masquelier, D. A. , Belgrader, P. , Heredia, N. J. , Makarewicz, A. J. , … Colston, B. W. (2011). High‐throughput droplet digital PCR system for absolute quantitation of DNA copy number. Analytical Chemistry, 83(22), 8604–8610. 10.1021/ac202028g [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, P. A. , & Baylin, S. B. (2002). The fundamental role of epigenetic events in cancer. Nature Reviews Genetics, 3(6), 415–428. [DOI] [PubMed] [Google Scholar]
- Jones, P. A. , & Baylin, S. B. (2007). The epigenomics of cancer. Cell, 128(4), 683–692. 10.1016/j.cell.2007.01.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lange, M. J. , van Pelt, S. I. , Versluis, M. , Jordanova, E. S. , Kroes, W. G. , Ruivenkamp, C. , … van der Velden, P. A. (2015). Heterogeneity revealed by integrated genomic analysis uncovers a molecular switch in malignant uveal melanoma. Oncotarget, 6(35), 37824–37835. 10.18632/oncotarget.5637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maat, W. , van der Velden, P. A. , Out‐Luiting, C. , Plug, M. , Dirks‐Mulder, A. , Jager, M. J. , & Gruis, N. A. (2007). Epigenetic inactivation of RASSF1a in uveal melanoma. Investigative Ophthalmology and Visual Science, 48(2), 486–490. [DOI] [PubMed] [Google Scholar]
- Mikeska, T. , Candiloro, I. L. , & Dobrovic, A. (2010). The implications of heterogeneous DNA methylation for the accurate quantification of methylation. Epigenomics, 2(4), 561–573. 10.2217/epi.10.32 [DOI] [PubMed] [Google Scholar]
- Oesper, L. , Mahmoody, A. , & Raphael, B. J. (2013). THetA: Inferring intra‐tumor heterogeneity from high‐throughput DNA sequencing data. Genome Biology, 14(7), R80 10.1186/gb-2013-14-7-r80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Oever, J. M. E. , Balkassmi, S. , Segboer, T. , Verweij, E. J. , van der Velden, P. A. , Oepkes, D. , … Boon, E. M. J. (2013). Mrassf1a‐Pap, a novel methylation‐based assay for the detection of cell‐free fetal DNA in maternal plasma. PLOS One, 8(12), e84051 10.1371/journal.pone.0084051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohgane, J. , Yagi, S. , & Shiota, K. (2008). Epigenetics: The DNA methylation profile of tissue‐dependent and differentially methylated regions in cells. Placenta, 29(Suppl A), S29–S35. 10.1016/j.placenta.2007.09.011 [DOI] [PubMed] [Google Scholar]
- Paganini, L. , Carlessi, N. , Fontana, L. , Silipigni, R. , Motta, S. , Fiori, S. , … Tabano, S. (2015). Beckwith‐Wiedemann syndrome prenatal diagnosis by methylation analysis in chorionic villi. Epigenetics, 10(7), 643–649. 10.1080/15592294.2015.1057383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redshaw, N. , Huggett, J. F. , Taylor, M. S. , Foy, C. A. , & Devonshire, A. S. (2014). Quantification of epigenetic biomarkers: An evaluation of established and emerging methods for DNA methylation analysis. BMC Genomics, 15, 1174 10.1186/1471-2164-15-1174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka, K. , & Okamoto, A. (2007). Degradation of DNA by bisulfite treatment. Bioorganic & Medicinal Chemistry Letters, 17(7), 1912–1915. 10.1016/j.bmcl.2007.01.040 [DOI] [PubMed] [Google Scholar]
- Tong, Y. K. , Jin, S. , Chiu, R. W. , Ding, C. , Chan, K. C. , Leung, T. Y. , … Dennis Lo, Y. M. (2010). Noninvasive prenatal detection of trisomy 21 by an epigenetic‐genetic chromosome‐dosage approach. Clinical Chemistry, 56(1), 90–98. 10.1373/clinchem.2009.134114 [DOI] [PubMed] [Google Scholar]
- Varley, K. E. , Mutch, D. G. , Edmonston, T. B. , Goodfellow, P. J. , & Mitra, R. D. (2009). Intra‐tumor heterogeneity of MLH1 promoter methylation revealed by deep single molecule bisulfite sequencing. Nucleic Acids Research, 37(14), 4603–4612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verbik, D. J. , Murray, T. G. , Tran, J. M. , & Ksander, B. R. (1997). Melanomas that develop within the eye inhibit lymphocyte proliferation. International Journal of Cancer, 73(4), 470–478. [DOI] [PubMed] [Google Scholar]
- Versluis, M. , de Lange, M. J. , van Pelt, S. I. , Ruivenkamp, C. A. , Kroes, W. G. , Cao, J. , … van der Velden, P. A. (2015). Digital PCR validates 8q dosage as prognostic tool in uveal melanoma. PLOS One, 10(3), e0116371 10.1371/journal.pone.0116371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Waard‐Siebinga, I. , Blom, D. J. R. , Griffioen, M. , Schrier, P. I. , Hoogendoorn, E. , Beverstock, G. , … Jager, M. J. (1995). Establishment and characterization of an uveal‐melanoma cell line. International Journal of Cancer, 62(2), 155–161. [DOI] [PubMed] [Google Scholar]
- Warnecke, P. M. , Stirzaker, C. , Melki, J. R. , Millar, D. S. , Paul, C. L. , & Clark, S. J. (1997). Detection and measurement of PCR bias in quantitative methylation analysis of bisulphite‐treated DNA. Nucleic Acids Research, 25(21), 4422–4426. 10.1093/nar/25.21.4422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojdacz, T. K. , Dobrovic, A. , & Hansen, L. L. (2008). Methylation‐sensitive high‐resolution melting. Nature Protocols, 3(12), 1903–1908. [DOI] [PubMed] [Google Scholar]
- Zoutman, W. H. , Nell, R. J. , & van der Velden, P. A. (2019). Usage of droplet digital PCR (ddPCR) assays for T cell quantification in cancer. Methods in Molecular Biology, 1884, 1–14. 10.1007/978-1-4939-8885-3_1 [DOI] [PubMed] [Google Scholar]
- Zoutman, W. H. , Nell, R. J. , Versluis, M. , van Steenderen, D. , Lalai, R. N. , Out‐Luiting, J. J. , … van der Velden, P. A. (2017). Accurate quantification of T cells by measuring loss of germline T‐cell receptor loci with generic single duplex droplet digital PCR assays. Journal of Molecular Diagnostics, 19(2), 236–243. 10.1016/j.jmoldx.2016.10.006 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information.
Supporting information.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.