

Abstract
Objective
Parkinson disease (PD) has useful symptomatic treatments that do not slow the neurodegenerative process, and no significant disease‐modifying treatments are approved. A key therapeutic target in PD is α‐synuclein (αS), which is both genetically implicated and accumulates in Lewy bodies rich in vesicles and other lipid membranes. Reestablishing αS homeostasis is a central goal in PD. Based on previous lipidomic analyses, we conducted a mouse trial of a stearoyl–coenzyme A desaturase (SCD) inhibitor (“5b”) that prevented αS‐positive vesicular inclusions and cytotoxicity in cultured human neurons.
Methods
Oral dosing and brain activity of 5b were established in nontransgenic mice. 5b in drinking water was given to mice expressing wild‐type human αS (WT) or an amplified familial PD αS mutation (E35K + E46K + E61K ["3K"]) beginning near the onset of nigral and cortical neurodegeneration and the robust PD‐like motor syndrome in 3K. Motor phenotypes, brain cytopathology, and SCD‐related lipid changes were quantified in 5b‐ versus placebo‐treated mice. Outcomes were compared to effects of crossing 3K to SCD1−/− mice.
Results
5b treatment reduced αS hyperphosphorylation in E46K‐expressing human neurons, in 3K neural cultures, and in both WT and 3K αS mice. 5b prevented subtle gait deficits in WT αS mice and the PD‐like resting tremor and progressive motor decline of 3K αS mice. 5b also increased αS tetramers and reduced proteinase K‐resistant lipid‐rich aggregates. Similar benefits accrued from genetically deleting 1 SCD allele, providing target validation.
Interpretation
Prolonged reduction of brain SCD activity prevented PD‐like neuropathology in multiple PD models. Thus, an orally available SCD inhibitor potently ameliorates PD phenotypes, positioning this approach to treat human α‐synucleinopathies. ANN NEUROL 2021;89:74–90
Introduction
Parkinson disease (PD) is characterized pathologically by the deposition of insoluble and aggregated forms of α‐synuclein (αS) in hallmark Lewy bodies. Recent morphological analyses have revealed that Lewy‐type inclusions also contain substantial amounts of lipid membranes, 1 and this realization has relevance for understanding the pathogenesis of neuronal dysfunction. 2 Studies in cultured cells, including induced pluripotent stem cell (iPSC)‐derived human neurons, and in rodent models, provide compelling evidence that αS binds transiently to the external phospholipid bilayer of vesicles during normal neuronal secretory function. 3 , 4 , 5 Physiological αS tetramers in cells were first observed in 2011, 6 , 7 and the neuronal tetramer:monomer (T:M) equilibrium has been shown to be relevant to PD pathogenesis. 8 , 9 , 10 For example, mutations in glucocerebrosidase (GBA) result in dyshomeostasis of lysosomal lipids and can cause PD, in part by decreasing physiological αS tetramers and elevating free monomers in patient neurons. 11 Via autophagy, the lysosomal system is critical for removal of impaired organelles and lipid droplets (LDs), and it can also clear aggregated and toxic forms of αS. 12 Since the initial description of αS tetramers, certain tetramer‐stabilizing compounds have been identified through unbiased screens, candidate approaches, and quantitative lipidomics, and some hits have been found to affect αS‐vesicle association. 13 , 14 , 15 One such pharmacological approach is αS tetramer stabilization by downregulating stearoyl–coenzyme A desaturase (SCD), the rate‐limiting enzyme for monounsaturated fatty acid (MUFA) biosynthesis that is required for membrane phospholipid and triglyceride (TG) homeostasis. 16 SCD inhibitors have consistently decreased αS toxicity in yeast, and rodent and human neurons. 13 , 14 , 15 To advance SCD inhibition as a novel treatment for human testing, we set out to validate the target in a robust preclinical PD model, αS mutant mice, and conducted a controlled prevention trial.
Cellular αS normally undergoes a dynamic exchange of tetramers and monomers, 7 , 17 , 18 and the latter are more prone to post‐translational modifications 10 that may shift vesicle‐bound monomers into pathological aggregates. The abrogation of αS tetramers has been studied in vitro 19 and in mammalian cells, 10 , 13 , 20 for example, by introducing the E46K familial PD mutation and also amplifying it via analogous E➔K substitutions in the 2 adjacent KTKEGV repeat motifs (E35K + E46K + E61K ["3K"]). Both the E46K and 3K mutants have enhanced binding to negatively charged vesicle membranes, 21 leading to more sizeable vesicle‐ and lipid‐rich cytoplasmic αS inclusions relative to the fine granules seen in wild‐type (WT) αS mouse brain. 10 , 20
Culture studies alone cannot capture the complex neuronal regulation of the αS T:M equilibrium in the brain and the physiological and pathological significance of αS tetramerization. Therefore, we have generated 3K αS transgenic (tg) mice and found their tetramer abrogation to accentuate the neuropathological and biochemical phenotypes of the WT or the familial (f) PD E46K (1K) αS mutation in an age‐dependent manner. 10 , 22 The striking PD‐like motor syndrome of 3K mice includes resting tremor, bradykinesia, rigidity, and gait deficits that respond in part to L‐dopa treatment, while relatively subtle changes in forced gait patterns are observed in male WT αS‐overexpressing mice versus their female counterparts. 22 Similar to PD, 23 there is a male preponderance in 3K mice in that females show less lowering of the αS T:M ratio and increased autophagic turnover of αS+ lipid‐rich brain aggregates. 22 These multiple features make the 3K model highly suitable for the necessary preclinical testing of potentially disease‐modifying approaches to PD.
Here, we show that an orally available SCD inhibitor "5b" that is brain penetrant slows the gradually progressive motor syndrome of 3K mice. Moreover, 5b protects against the subtle gait imbalance caused by chronic overexpression of human WT αS in mice. The SCD inhibitor normalizes αS solubility and improves the T:M ratio in male 3K mice toward the higher level of 5b‐WT αS mice. 5b also lessened the proteinase K‐resistant, pS129+ fine granules seen in WT αS mice, and it reduced the large pS129+ and lipid‐rich foci seen in the 3K mice. In both mouse lines, 120‐day 5b treatment effectively reduced the brain C16:1 to C16:0 desaturation index (DI) and the levels of SCD‐modulated TGs. Deleting 1 SCD allele in 3K mice through genetic crossings to SCD knockout mice resulted in a similar rescue of the PD‐like phenotypes, providing target validation. Moreover, in E46K‐expressing neurogenin‐induced human neurons (induced neurons [iNs]) and in 3K‐expressing dopaminergic (DAergic) human neuroblastoma cells, the 5b SCD inhibitor diminished abnormal αS monomer association with vesicle membranes and corrected altered autophagic turnover of excess LDs. Our results document a fatty acid (FA)‐dependent regulation of brain αS homeostasis, including the T:M equilibrium. SCD inhibition mitigates multiple PD‐like motor phenotypes in vivo and PD‐relevant biochemical changes in cultured neurons.
Materials and Methods
Cell Culture Studies, Stable Cell Lines, and Induced Neurons
Human M17D neuroblastoma cells and stable cell pools were cultured, transfected, and imaged as previously described. 15 Briefly, inclusion formation was recorded in M17D‐TR/aS‐3K::YFP (=3KY) or M17D‐TR/aS‐3K::YFP//RFP (=3KYR) cells (96‐well plates) on an IncuCyte Zoom instrument (Essen Bioscience, Ann Arbor, MI). Constitutive mCherry signal (3KYR only) as well as total YFP integrated intensities and YFP inclusion integrated intensities were quantified using IncuCyte software and an algorithm as published. 15 Toxicity was assessed via lactate dehydrogenase release (G1780; Promega, Madison, WI) or cell titer blue (G8080; Promega) assay following the manufacturer's protocol. For inclusions, the graphs represent the integrated intensities over the mCherry signal to normalize the cell numbers. LDs were visualized on IncuCyte using the HCS LipidTOX deep red neutral lipid dye following the manufacturer's protocol. LD integrated intensities were quantified using the following algorithm: parameters: adaptive; threshold adjustment (red corrected unit): 2; edge split on; edge sensitivity: 0; hole fill: 0; adjust size (pixels): 0; filters: area, max 50, all other boxes unchecked. The overlap between αS::YFP and punctate deep red neutral lipid dye was determined using the Incucyte "Overlap" algorithm. For 5b treatment, cells were plated on day 1 and treated on day 2, induced with 1μg/ml final concentration of doxycycline HCL (Sigma, St Louis, MO) on day 3, and analyses were conducted 24 hours after induction. Intact‐cell crosslinking using 1mM disuccinimidyl glutarate (DSG), and sequential extractions of proteins using phosphate‐buffered saline (PBS) and 1% (vol/vol) Triton X‐100 detergent were performed as previously described. 15 For fPD E46K induced neurons, the induced human neurons were generated as described previously 24 and induction, harvesting of neurons, and intact‐cell crosslinking were performed as previously described. 13
Treatment of 3K Mutant Mice and Behavioral Testing
For the study, we used male 3K αS mutant mice (3K, line #3817) and expression‐matched human WT αS‐overexpressing mice (WT, line #3877) with 2–3‐fold overexpression of human αS controlled by the Thy1.2 promoter (generation and characterization described previously 10 ). All mice were bred and maintained at the Hale Building for Transformative Medicine facility in accordance with National Institutes of Health guidelines on use of laboratory animals and a protocol approved by the Brigham and Women's Hospital. Mice were kept in normal 12 hours light/12 hours dark cycles and had free access to food and water. For 5b treatment, 10‐week‐old nontransgenic (Ntg) C57Bl6 or 3K and WT αS male tg mice were administered 5b dissolved in dimethylsulfoxide (DMSO) in their drinking water, whereas the placebo cohort received drinking water with DMSO only (1.9% [vol/vol]), well below described toxic/side effects 25 ). One percent (vol/vol) sucrose was added to the drinking water to mask any bitter taste. All behavioral testing has been conducted as previously described. 10
Intact‐Cell Crosslinking, Sequential Extraction, and Western Blot Analysis
Intact‐cell crosslinking was then conducted on the washed brain bits as previously described. 10 , 26 Sequential extracts of dissected mouse brains were each run on 4 to 12% Bis‐Tris gels (Invitrogen, Carlsbad, CA) and electroblotted onto nitrocellulose membranes (Millipore, Bedford, MA). Blots were then incubated with human‐specific αS antibody (Ab; 15G7; Enzo, Farmingdale, NY; 1:500), or Abs that recognize both rodent and human αS (syn1, clone 42, 1:2,000; BD Biosciences, Franklin Lakes, NJ), or against serine 129 (pS129; ab51253; 1:1,000; Abcam, Cambridge, MA), lysosomal membrane protein 1 (LAMP1; ab25245, 1:4,000; Abcam), microtubule‐associated protein 1B‐light chain 3 LC3B (5F10, 1:500; NanoTools, Teningen, Germany), protein disulfide isomerase (PDI; 3501 T, 1:1,000; Abcam), binding immunoglobulin protein (BiP; 610978; 1:5,000, BD Biosciences), CTP:phosphocholine cytidylyltransferase α (CCTα; 4454S, 1:1,000; Cell Signaling Technology, Danvers, MA), transferrin receptor (TFR; 84036, 1:5,000, Abcam), or protein deglycase (DJ‐1) (3E8, 1:500, Stressgen, San Diego, CA), or loading controls beta‐actin (A5441, 1:3000; Sigma) or glyceraldehyde‐3‐phosphate dehydrogenase (G9545, 1:5,000, Sigma) in Phosphate‐buffered Saline with Tween‐20 (PBST) containing 5% bovine serum albumin overnight.
Lipid Kits and Enzyme‐Linked Immunosorbent Assay
Lipid Extraction Kit (ab211044, Abcam) and Triglyceride Quantification Assay Kit (colorimetric; ab65336, Abcam) were used to quantify TGs, and human αS concentrations were quantified using pieces of motor cortex per enzyme‐linked immunosorbent assay (ELISA) kit (#KHB0061; Invitrogen, Carlsbad, CA). All assays were conducted according to the manufacturer's protocol.
Lipid Analyses
Lipids were extracted by procedures similar to the Folch method. 27 For lipid assignment, aligned peaks were first searched against the LIPID MAPS Database of computationally generated “bulk” lipid species with mass tolerance of ±0.005Da 28 . We utilized the high‐resolution Fourier transform ion cyclotron resonance to accurately measure FA (16:0 and 16:1). At the liquid chromatography (LC) gradient of 99 to 95% mobile A in 7 minutes at 120μl/min and FA (16) eluted at 3.08 and 3.12 minutes. To increase anion sensitivity, free FAs were measured using continuous accumulation of selected ions (CASI) anion scan mode in a narrow mass range of 70Da. The DI was the ratio of the peak areas of FA (16:1)/FA(16:0).
RNA
Total RNA samples were isolated from brain cortices using mirVana miRNA Isolation Kit (AM1561), and RNA concentrations were determined by Nanodrop. The samples were converted into cDNA using a high‐capacity cDNA reverse transcription kit (#4368813; Applied Biosystems, Foster City, CA). Quantitative polymerase chain reaction (qPCR) was performed using the TaqMan Fast Universal PCR Mix (#4352042, Applied Biosystems) and the appropriate TaqMan primers for human SNCA (Hs00240906_m1, Applied Biosystems). qPCR reactions were run in an StepOnePlus Real‐Time PCR system, and the delta‐delta treshold cycles (ΔΔCt) calculations were made using StepOne software (Applied Biosystems).
High‐Performance LC
High‐performance LC (HPLC) was conducted as previously described 26 to estimate striatal monoamine levels at age 6 months in 3K αS tg (n = 5 per group) or Ntg control mice (n = 5–6 per group).
Immunohistochemistry
Diaminobenzidine and fluorescence labeling was performed as described. 10 Briefly, sections were blocked in 10% normal donkey serum and incubated overnight at 4°C with Abs to human αS (15G7, 1:1,000, Enzo), antiphosphorylated 129 αS (ab51253, 1:4,000, Abcam), LAMP1 (ab25245, 1:500, Abcam), LC3B (5F10, 1:500, NanoTools), perilipin (PLIN; G2 sc‐390169; 1:500; Santa Cruz Biotechnology, Santa Cruz, CA), and tyrosine hydroxylase (TH; ab152, 1:500, Abcam). This was followed by incubation with fluorescein isothiocyanate–conjugated secondary Abs (1:500 in PBS) for 3 hours at room temperature. Confocal microscopy was conducted with a Zeiss (Oberkochen, Germany) LSM 710 confocal scanning laser microscope. An ImageJ plug‐in called “colocalization highlighter” created a mask of either LAMP1 or PLIN pixels that overlapped with pS129‐αS pixels. The sizes of the colocalized pixels on the resultant 8‐bit images were quantified using the analyze particle function plug‐in of ImageJ. For striatal TH evaluation, 3 images within the dorsolateral striatum were acquired per section in 3 coronal sections (bregma 0.97–0.37mm), n = 5 mice per group. Fiji color thresholding was used to calculate the respective area fraction covered by TH staining; imaging was performed using a Zeiss LSM 780 confocal scanning laser microscope (×40 field planarity apochromate (PL APO) oil immersion objective).
Semithin Sections
Semithin (0.3μm) sections from TAAB Epon (TAAB Laboratories Equipment, Aldermaston, UK) embedded cryosections were cut with a diamond knife stained in 1% toluidine blue mounted in pure glycerol and viewed in a Zeiss AxioImager Z1 microscope with a ×100 oil immersion objective, using a Zeiss HR camera. For all light microscopic images, white balance was adjusted.
Experimental Treatment Design, Quantification, and Statistical Analysis
Behavioral testing was conducted prior to treatment and at 60 days and at 120 days of treatment as indicated. Experimental details specific for behavioral testing are included in behavior testing below. Details regarding each statistical test, biological sample size (n), and p value can be found in the corresponding figure legends. All data are represented as mean ± standard error of the mean (SEM). SEM represents variance within a group. In all experiments, the genotypes can be found in the corresponding legends. Data were collected and processed side by side in randomized order for all experiments. Behavioral and histological tests were routinely performed blind to the conditions of the experiments. Unpaired 2‐tailed t tests were used for comparison between 2 groups, with p ≤ 0.05 considered significant. For all comparisons involving multiple variables, analysis of variance (ANOVA) was performed followed by post hoc test for paired comparisons using p ≤ 0.05 for significance. For all experiments, between 4 to 8 (biochemistry, histology) and 5 to 12 (behavior) male mice per experiment were used. For HPLC, 5 to 8 mice per group were used, and 2 outliers (n = 1 3K‐placebo [Plb], n = 1 3K‐5b) were identified by the robust regression and outlier removal method (ROUT) (Q = 1%) and removed from the analyses. To analyze the differential abundance of each lipid belonging to TG(48–52), moderated t tests were applied using the LIMMA R package. 29 , 30 To control the false discovery rate, the p values were adjusted in each comparison using the Benjamini and Hochberg correction. Additional bar graphs have been included to show the appropriate statistical information (mean ± SEM). All statistical analyses were performed using (GraphPad Software, San Diego, CA).
Results
5b Is an Orally Available SCD Inhibitor Active in Brain
An iterative library design had previously identified 5b as an appropriate tool compound to inhibit SCD for potential use in models of obesity. 31 In rats, 5b achieved high exposure in vivo to effect dose‐dependent decreases of the plasma and liver DI (MUFA to saturated FA [SFA]; eg, 16:1 to 16:0 and 18:1 to 18:0). 31 To further evaluate the pharmacokinetic (PK) properties of 5b, we assessed the bioavailability of the drug in 3‐month‐old Ntg C57Bl6 mice after single doses of 3 mg/kg intravenous or 10 mg/kg per os (PO), using standard pharmacokinetic (PK) parameters (Fig 1). A Cmax of 4,413ng/ml in plasma was achieved after 60 minutes of PO dosing. The T1/2 was 1.4 hours in plasma. The 5b brain concentration 7 hours after a single PO dose averaged 120.9 ± 53.1, and the average brain:plasma ratio was 0.86 ± 0.12, demonstrating good brain penetration. This gave an area under the plasma concentration‐time curve from time zero to extrapolated infinity of 9,920ng/ml/h. The structure of 5b is shown in Figure 1D and the design of the mouse trial in Figure 1E.
FIGURE 1.
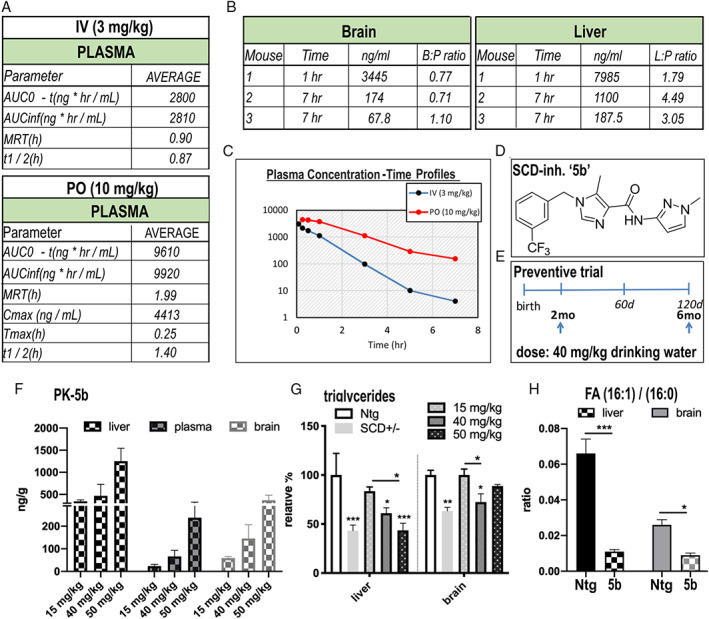
Pharmacokinetic (PK) and pharmacodynamic analyses after intravenous (IV) or oral (PO) dosing of the stearoyl–coenzyme A desaturase (SCD) inhibitor 5b in C57Bl6 (nontransgenic) mice. (A) PK analyses were performed after single dose of 3 or 10mg/kg body weight via IV injection or PO (gavage). AUC = area under the curve, MRT (mean residence time), Cmax (maximum concentration recorded and time (Tmax) to reach Cmax, t1/2 (elimination half‐life). (B) Calculated brain: plasma (B:P) ratio or liver:plasma (L:P) ratio. (C) Time profile after the single dose application. The PK parameters are means of 3 mice, and values at each time point are from different mice. The relative compound spread by oral dosing is shown in the graph (F) below. (D) Chemical structure of the SCD inhibitor 5b. (E) Trial design for the mouse study. (F) The spread of compound concentration in the mice is calculated from the mean values of n = 3 mice after oral dosing for 20 days at 3 different concentrations applied via the drinking water. (G) Triglyceride levels measured by enzyme‐linked immunosorbent assay in heterozygous SCD knockout (SCD+/−) and in sentinel (C57Bl6) mice treated at different doses of 5b for 20 days. (H) Liver and brain desaturation index showing lowering effect of SCD inhibitor 5b after 90 days of oral treatment. FA = fatty acid. Data are presented as mean value ± standard error of the mean. *p < 0.05, **p < 0.01, ***p < 0.001. [Color figure can be viewed at www.annalsofneurology.org]
We further quantified oral availability of 5b. Ten‐week‐old Ntg mice were dosed in the drinking water at 15, 40, or 50mg/kg daily for 10 days, and plasma, brain, and liver concentrations were recorded in a PK study. The compound rose dose‐dependently in the liver from 344 ± 31.34 to 1254 ± 290.8ng/g, in the plasma from 23.6 ± 7.2 to 237.7 ± 88.4ng/ml, and in the brain from 59.3 ± 6.0 to 368.0 ± 114.7ng/g (see Fig 1F). Two‐way ANOVA revealed a significant dose effect (F 2,18 = 9.815, p = 0.0013), and the calculated average brain:plasma ratio was ~1:1 (Fig 1F). Subsequently, we started long‐term dosing of C57Bl6 (sentinel mice) at 40mg/kg to further analyze the pharmacodynamic profile. Palmitoleic acid (16:1) is an endogenous SCD product, and enzymes that synthesize TGs require 16:1 as a substrate. The decrease in 16:1 and other MUFAs by SCD inhibitors or by genetic ablation of SCD causes hair follicle abnormalities, leading to sparse fur. 31 , 32 Homozygous SCD1 knockout (KO) mice (SCD−/−) are deficient in TGs, partly due to decreases in their endogenous 16:1 (by 55%), with intermediate levels found in SCD1+/− KO mice. 16 Accordingly, we observed the typical sparse fur in 90‐day 5b‐treated sentinel animals, resembling the appearance of SCD1−/− KO mice (data not shown) . We next analyzed TG levels in liver and brain of 5b‐treated mice and in SCD1+/− mice using a colorimetric TG assay. Ninety days of 5b treatment reduced liver and brain TGs to the approximate levels detected in heterozygous SCD+/− KO mice (Fig 1G). To specify SCD enzyme activity, we measured the DI by calculating the ratio of FA 16:1 to 16:0 using LC–mass spectrometry (MS). Dosing of 5b at 40mg/kg for 90 days revealed an 82% DI reduction in liver (p = 0.003) and a 63% reduction in brain (p = 0.006) (Fig 1H). Together, these results demonstrate that oral administration of 5b effectively inhibits SCD enzyme activity and significantly decreases the MUFA content in mouse brain.
5b Treatment Increases Soluble αS Tetramerization and Reduces pS129+ in fPD Culture
To test whether SCD inhibition by 5b restores αS homeostasis in neural cells (Fig 2), we next tested the 5b compound in our M17D αS‐3K cultures. 15 5b treatment produced a dose‐dependent reduction of αS inclusions (Fig 2A). This was a sensitive measure of activity in culture, as in either M17D human neuroblastoma cells or in primary rat neurons, all the SCD inhibitors we have tested have decreased αS inclusions to a similar extent. 15 , 33 To confirm relevance to the clinical fPD mutation, M17D cells stably expressing E46K αS were treated with 10μM 5b for 48 hours. Western blot (WB) analysis revealed a decrease in serine 129 αS phosphorylation (pS129+) immunoreactivity (p < 0.001) (Fig 2B), an increase in Tris‐buffered saline (TBS) solubility (cytosolic αS) versus TX‐100 membrane solubility (p = 0.017) (Fig 2C), and an increase in the αS 60:14 ratio upon intact‐cell DSG crosslinking 8 (p = 0.032) (Fig 2D). These changes are in the absence of cytotoxicity (Fig 2E, F).
FIGURE 2.
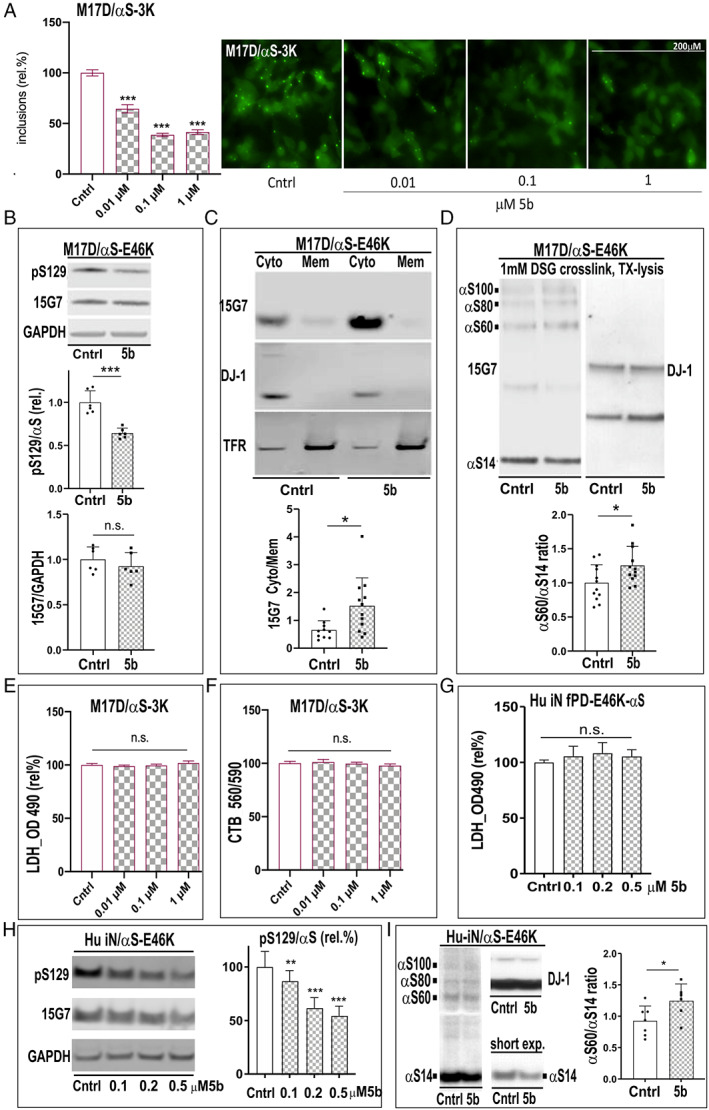
Stearoyl–coenzyme A desaturase inhibitor 5b reduces 3K αS inclusions and increases the soluble αS tetramer:monomer ratio in familial Parkinson disease E46K culture. (A) M17D cells (details of the M17D platform have been previously described 8 , 15 ) were treated for 24 hours with dimethylsulfoxide (DMSO‐control (Cntrl) or 5b, then transfected with YFP‐tagged 3K αS. Representative images of cells treated for 24 hours with 5b are shown. Graphs show quantitation of αS inclusions fold control, and representative images are shown on the right. (B) M17D/ αS‐E46K stable cell pools treated for 48 hours with 10μM 5b or DMSO alone. Western blots (WBs) for serine 129 (pS129) αS, total αS (15G7), and glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH; loading control) are shown. Quantification of pS129:15G7 αS ratio (normalized to DMSO; n = 6) is shown. (C) Sequential extraction and WB for αS. DJ‐1 and transferrin receptor (TFR) serve as controls for the extracts. Graph shows increase in Tris‐buffered saline (Cyto = cytosol) to TX (Mem = membrane) ratio. (D) WB of intact‐cell crosslinking of E46K αS using the cell‐penetrant crosslinker DSG and subsequent lysis in phosphate‐buffered saline/1% Triton‐X100; graph shows αS60:14 ratio by WB in n = 12. (E) Lactate dehydrogenase (LDH) release and (F) cell titer blue (CTB) in transient M17D/3K αS dopaminergic neuroblastoma cells and (G) LDH release in fPD E46K human (hu) induced neuron (iN) culture; n = 6 independent experiments were carried out twice. (H) Hu induced pluripotent stem cell E46K iNs were treated for 2 days with 5b at 3 different doses or DMSO (fresh media/5b after 2 days) and WB for pSer129, total αS, or GAPDH. (I) WB of intact‐cell crosslinking with DSG and graph. Data are expressed as means ± standard deviation; n.s. = nonsignificant; *p < 0.05, **p < 0.01, ***p < 0.001; 1‐way analysis of variance with Tukey post hoc test (A, E‐H) or unpaired 2‐tailed t test (B‐D, I). [Color figure can be viewed at www.annalsofneurology.org]
We then conducted quantitative analyses in human neurons differentiated from an iPSC line expressing the E46K fPD mutation. In absence of cytotoxicity (Fig 2G), 5b treatment produced a striking dose‐dependent decrease in pS129+ αS in the human neurons (1‐way ANOVA, F 3,75 = 73.45, p < 0.001; see Fig 2H. In addition, the αS 60:14 T:M ratio upon intact‐cell DSG crosslinking rose (p = 0.049) (Fig 2I).
Together, the above results validate the ability of 5b to decrease SCD activity in neuronal culture, to normalize αS solubility and tetramerization, and to reduce the abnormal pS129+ signal intensity. Based on these highly consistent in vitro results and our demonstration that 5b crosses the blood–brain barrier and efficiently lowers SCD activity in brain, we examined the phenotypic effects of 5b treatment in WT and 3K αS mice.
5b Treatment Produces On‐Target Effects Resembling SCD KO and Improves PD‐Type Motor Deficits
Because 5b decreased SCD activity in brain after 90 days at 40mg/kg in drinking water, we assessed whether prolonged oral treatment could ameliorate the motor impairments of the WT and 3K αS tg mice. The 3K mutation decreases the brain αS T:M ratio much more than overexpressing the human WT αS does, 10 and this is associated with gradually progressive motor deficits, including resting tremor and climbing, balance, and gait abnormalities, which typically begin at ~3 months in males and become notably worse by age 6 months, as demonstrated by several timed motor tests, including pole climbing, 4‐limb wire hanging, and rotarod walking. 10 , 22 We initially performed these tests at age 8 weeks to establish baseline motor performances, and we then randomly assigned WT and 3K αS tg mice matched for sex, age, and αS expression level to active 5b treatment (5b) or vehicle (Plb) groups (Fig 3).
We began continuous oral 5b application in the drinking water at age 10 weeks and monitored motor performance at 60 and 120 days after initiating treatment. By around 30 days of treatment, roughly 30% of the 5b‐treated 3K and WT αS tg mice had begun to display small patches (1–3mm) of hair loss and eye squinting (a sign of dry eye), which are well known effects of genetic or pharmacological SCD downregulation. 31 These changes thus provide a clear pharmacodynamic readout that the inhibitor was engaging its enzymatic target. By 120 days, all 5b‐treated mice had sparser and more grayish coats (Supplementary Videos SUPPLEMENTARY VIDEO M1–3, M5) that were similar to the described cutaneous phenotypes of SCD−/− mice. 32 This effect should represent at least a 50% loss of SCD activity, as such fur changes are minimal in SCD+/− mice at this age. 16 The fur loss ensuing from SCD inhibition has been specifically attributed to decreased palmitoleic acid (16:1), 32 which was significantly reduced by 5b treatment in our Ntg sentinel mice (see Fig 1H). No difference in weight was detected between genotypes in placebo‐treated (WT‐Plb, 33 ± 1g; 3K‐Plb, 32 ± 2g) and 5b‐treated mice (WT‐5b, 28 ± 1g; 3K‐5b, 27 ± 1g; 2‐way ANOVA, post hoc Bonferroni p > 0.05). However, there was a significant treatment effect showing overall lesser weight (not exceeding 15%) in the 5b cohort of both genotypes (data not shown). Veterinarians throughout the study monitored all mice, and no mouse required culling by euthanasia due to adverse effects.
Following treatment for 60 and 120 days, we analyzed treatment effects on the development of the relatively mild gait abnormalities in the WT αS tg mice and the progressive motor decline 10 , 22 in male 3K αS mutant mice (Fig 3). Two‐way ANOVA confirmed a significant interaction (F 2,33 = 3.27, p = 0.05) between 5b treatment and duration of the longitudinal pole climbing of 3K mice. Post hoc multiple comparisons revealed 5b to effectively reduce the time for 3K mice to climb down the pole (p = 0.03). In accord, we observed far fewer falls from the pole at 120 days in the 5b than placebo group, and the 2‐way ANOVA showed a highly significant interaction of pole duration and treatment (F 2,33 = 9.084, p = 0.0007) . This L‐dopa–sensitive pole climbing test 10 was not affected in the human αS–expressing WT line (see Fig 3B and also Supplementary Videos M1 and M2 showing simultaneously placed 3K‐Plb and 3K‐5b mice, and Supplementary Video M3 showing WT‐Plb vs WT‐5b mice). We next analyzed performance on the 4‐limb wire test , an established sensitive measure for muscle tone 34 that associates with the tremor unique for 3K mice (ie, not described for our or other WT αS tg mouse lines; see Table S2 in Nuber et al 10 ). Baseline analyses showed a decrease in wire‐hanging endurance as early as 3 months in 3K mice (see Fig 3C), and planned comparisons revealed that 120 days of 5b treatment improved the 4‐limb wire endurance in 3K mice (p = 0.02). Consistently, when placed on an elevated platform, 3K‐Plb mice had relatively more body tremor (Supplementary Video M4) than 3K‐5b mice (Supplementary Video M5). No changes in wire‐hanging were observed in the WT αS tg mice. We next challenged WT and 3K αS mice on the accelerating rotarod test. Two‐way ANOVA revealed significant treatment and duration effects in WT mice (F 2,30 = 25.15, p < 0.01) and an interaction in 3K mice (F 2,33 = 5.036, p = 0.0123). Post hoc comparisons showed 5b treatment protected against rotarod deficits seen in WT mice at 120 days and lessened the rotarod deficits at both 60 days (p = 0.01) and 120 days (p = 0.001) in 3K mice. To more finely analyze the underlying gait changes, we used high‐resolution photography of gait symmetry by placing mice on a transparent motorized belt. Two‐way ANOVA confirmed a significant 5b treatment interaction in WT mice at 120 days (F 2,30 = 7.612, p = 0.0021). 5b also improved the more severe gait deficits of the 3K mice at 120 days (placebo vs 120d 5b, p < 0.05). Thus, 5b treatment largely protected against the development of the PD‐relevant gait impairments of WT αS mice and significantly ameliorated the more marked motor decline in 3K αS mice.
FIGURE 3.
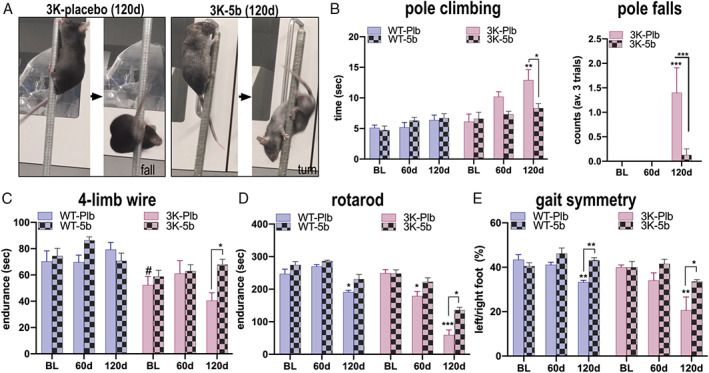
Pharmacological rescue of gait abnormalities by long‐term feeding of 5b to wild‐type (WT) αS and 3K αS transgenic mice. (A) Representative image of pole performance in placebo 3K (3K‐Plb) or 5b treated 3K (3K‐5b) after 120 days (120d) of treatment, showing abnormal climbing and subsequent fall in 3K‐Pbl versus normal turn and climbing down in 3K‐5b mice (see also representative Supplementary Videos M1–M3 of simultaneously placed placebo 3K and WT versus 3K‐5b or WT‐5b). (B) Graphs show quantitation of time to descend the pole and number of falls from the pole. (C) 5b treatment improves muscle tone–related endurance of 3K mice in the 4‐limb wire hanging test (see also Supplementary Videos M4 and M5 of placebo 3K and 5b‐treated 3K mice). (D) Quantitation of fine motor balancing skill in a 4 to 40rpm accelerating rotarod test shows 5b treatment hinders development of balancing deficits in WT and 3K αS mice. (E) 5b treatment prevents development of gait imbalance shown by harmonization of the left/right paw support in WT and ameliorates the more severe deficits in 3K αS mice. Data are mean ± standard error of the mean. *p < 0.05, **p < 0.01, ***p < 0.001; # indicates a significant difference at baseline between WT and 3K; 2‐way analysis of variance with Tukey post hoc test. BL = baseline performance at 8 weeks. [Color figure can be viewed at www.annalsofneurology.org]
SCD Inhibition Normalizes αS Solubility and T:M Ratio and Decreases Neuronal Aggregates in WT and 3K αS Mice
Having determined 5b to effectively decrease brain SCD activity and simultaneously increase the αS T:M ratio in E46K‐expressing iNs and 3K‐expressing M17D DAergic cells (see Fig 2), we next investigated the T:M ratio in the brains of 5b‐treated WT and 3K mice that had shown motor improvements over placebo at 120 days. First, we assessed αS solubility by sequential extraction of cortical homogenates (Fig 4). Using either conventional immunoblotting with the hu αS monoclonal Ab (mAb) 4B12 or a sensitive (lower limit of quantification = 0.2ng/ml) anti‐hu αS sandwich ELISA, we found that 5b treatment increased 3K αS levels in cytosol (TBS extract) and reduced the abnormally elevated level of membrane‐associated 3K (radioimmunoprecipitation assay extract; see Fig 4A,B). Post hoc analysis of the treatment effects calculated by 2‐way ANOVA (F 1,19 = 14.03, p = 0.0014) showed 5b treatment to significantly increase the low cytosol:membrane (cyt:mem) ratio in 3K brain (p = 0.046). No significant change was seen in cyt:mem ratio of WT tg mouse cortices, likely because the large majority of the human WT αS remains soluble (cytosolic). We then assessed whether the 5b‐induced shift of 3K αS protein from membrane to cytosol was associated with an improvement in the cytosolic αS T:M ratio. Intact‐cell crosslinking revealed a significant increase in 3K αS tetramers by 5b versus placebo (see Fig 4C,D). Pairwise comparison of the treatment effect (2‐way ANOVA, F 1,19 = 42.79, p < 0.0001) confirmed that 120 days of 5b treatment increased αS T:M ratio in 3K brain. The T:M ratio in WT αS did not reveal a significant change, but the tetramer level (αS60) showed a trend of rising (p = 0.07). Of note, 120 days of 5b treatment did not change the transcriptional (RNA) level of the human αS gene (see Fig 4D).
FIGURE 4.
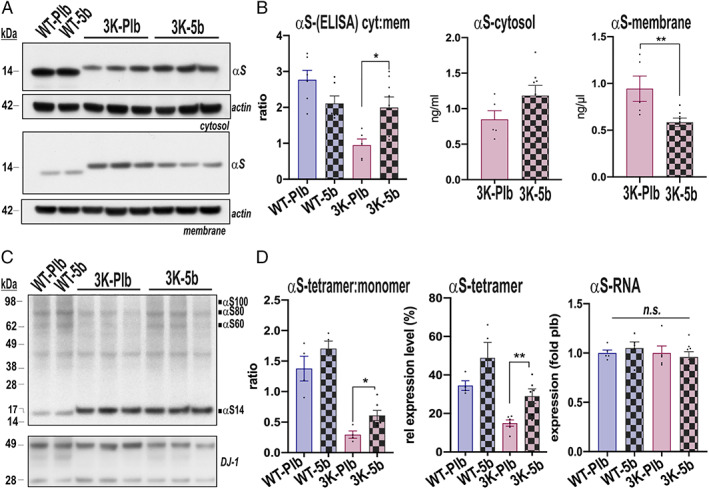
5b treatment for 120 days elevates αS solubility and tetramer:monomer (T:M) ratio. (A) Representative Western blot (WBs; noncrosslinked) of sequential extractions of Tris‐buffered saline–soluble (cytosolic), radioimmunoprecipitation assay–soluble (membrane) brain homogenates (15G7: human αS). (B) Human‐specific αS enzyme‐linked immunosorbent assay (ELISA) of the extracted homogenates shows increase in soluble (cytosolic:membrane [cyt:mem] ratio) αS by 5b treatment. (C) WB of intact‐cell crosslinking of αS in cortical brain bits from 3K mice. Syn1 antibody detects monomeric (αS14) and tetrameric (αS60) αS and probable conformers of the tetramer (αS80, αS100). 8 DJ‐1 serves as a cross‐linking control. (D) Quantitation of a representative WB of αS monomers (~14kDa) and tetramers (~60kDa) reveals an increased T:M ratio in treated 3K‐5b mice, due to the rise in tetramers. Right graph: Expression data for human αS gene. n = 4–6 per group. *p < 0.05; **p < 0.01; 2‐way analysis of variance with Tukey post hoc test or unpaired 2‐tailed t test. n.s. = non‐significant; Plb = placebo; WT = wild type. [Color figure can be viewed at www.annalsofneurology.org]
To assess whether 5b treatment lowers abnormal human αS accumulation and aggregation in the brain, we performed immunohistochemistry for total human αS using the 15G7 mAb, which targets an epitope at the C‐terminus (116–131), as well as a highly specific mAb for pS129 (EP1536Y 35 ; Fig 5). Because αS also acquires proteinase K‐digestion (PK‐digestion) resistance when aggregating into Lewy‐type lesions 30 that may include lipid membranes, 1 , 36 we additionally used PK‐digestion of the sections. Thus, correlating 5b treatment with PK resistance in pS129‐stained brain sections could indicate 5b efficacy on lipid‐rich aggregates. In general, PK‐digestion of total αS (15G7) and pS129+ stained sections revealed larger PK‐resistant immunoreactive foci in 3K mouse brain and finer PK‐resistant immunoreactive granules in WT mouse brain, observed in both cortex and striatum, and these were decreased in the brains of 5b‐ versus placebo‐treated mice. Two‐way ANOVA confirmed significant 5b treatment effects on the pS129+ pattern in WT (F 1,13 = 144, p < 0.001) and 3K cortical sections (F 1,15 = 13.88, p = 0.002) and a minor, nonsignificant reduction for the total (15G7+) αS signals. Post hoc comparisons revealed 5b decreased the total pS129+ puncta (p < 0.01) and also the pS129+ PK‐resistant granules (p < 0.001) in WT cortex. 5b further reduced the relatively large PK‐resistant inclusions in 3K cortex (p = 0.03). Analyzing the striatum, 2‐way ANOVAs showed significant 5b treatment effects (total αS: F 1,28 = 17.69, p = 0.0002; pS129 + αS: F 1,28 = 25.35, p < 0.0001) in WT striatal sections and a significant interaction (5b treatment × PK‐digestion) of pS129+ aggregates in the 3K sections (F 1,28 = 8.536, p = 0.0068). Paired comparisons validated 5b treatment to effectively reduce the striatal PK‐resistant granules of WT (p = 0.01) and of 3K brains (p = 0.003).
FIGURE 5.
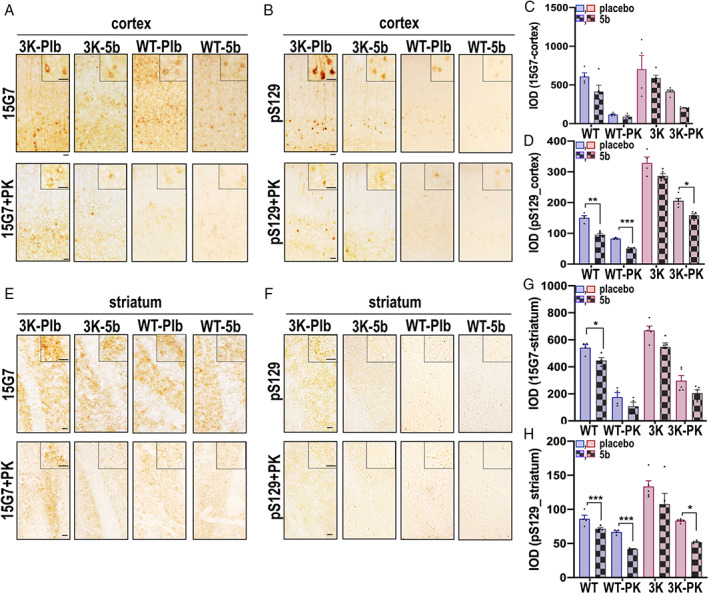
Stearoyl–coenzyme A desaturase inhibitor 5b ameliorates phosphorylated αS deposits in Parkinson disease–relevant brain regions of wild‐type (WT) and 3K αS mice. (A, E) Representative panels of cortical and striatal sections stained with the hu‐specific total αS (Ab 15G7) or (B, F) phosphorylated (Ab pS129+) αS after 120 days of 5b treatment without (upper panels) and with (lower panels) proteinase K pre‐treatment. Relative total and phosphorylated αS and their proteinase K (PK)‐resistant αS integrated optic densities were analyzed in the cortex (C,D) and the striatum (G,H). Data are mean ± standard error of the mean. *p < 0.05, **p < 0.01, ***p < 0.001; 2‐way analysis of variance with Tukey post hoc test. Scale bars = 25μm. [Color figure can be viewed at www.annalsofneurology.org]
SCD Inhibition Improves Striatal DAergic Integrity and Lessens Lysosomal and Endoplasmic Reticulum Dysregulation and Lipid Imbalance in Cortical Neurons
To investigate 5b treatment effects on the sizeable αS immunoreactive foci in 3K brain, we analyzed first the dorsal striatum, rich in DAergic nerve fibers, and probed for abnormal pS129+ immunoreactivity (see Fig 6). The quantification of colocalized pS129+/TH+ puncta revealed a significant decrease in the size of the DAergic nerve fiber foci in 3K‐5b versus 3K‐Plb mice (p = 0.013). We next analyzed cortical pyramidal neurons in 3K sections displaying relatively large somatic aggregates that colocalize with lysosomal vesicles. 10 Similar abnormalities are a common feature of PD. 37 Previous neuropathological analyses described accumulations of lysosomal vesicles close to 38 , 39 , 40 or as part of 41 human Lewy bodies (LBs), and their sizes have been postulated to increase in Lewy‐type pathology. 42 We quantified the pS129‐lysosomal aggregate sizes in cortical sections of the 5b‐treated 3K mice and observed that treatment with the SCD inhibitor significantly reduced pS129+/LAMP+ aggregate sizes in 3K cortical somata versus placebo (p = 0.0014).
FIGURE 6.
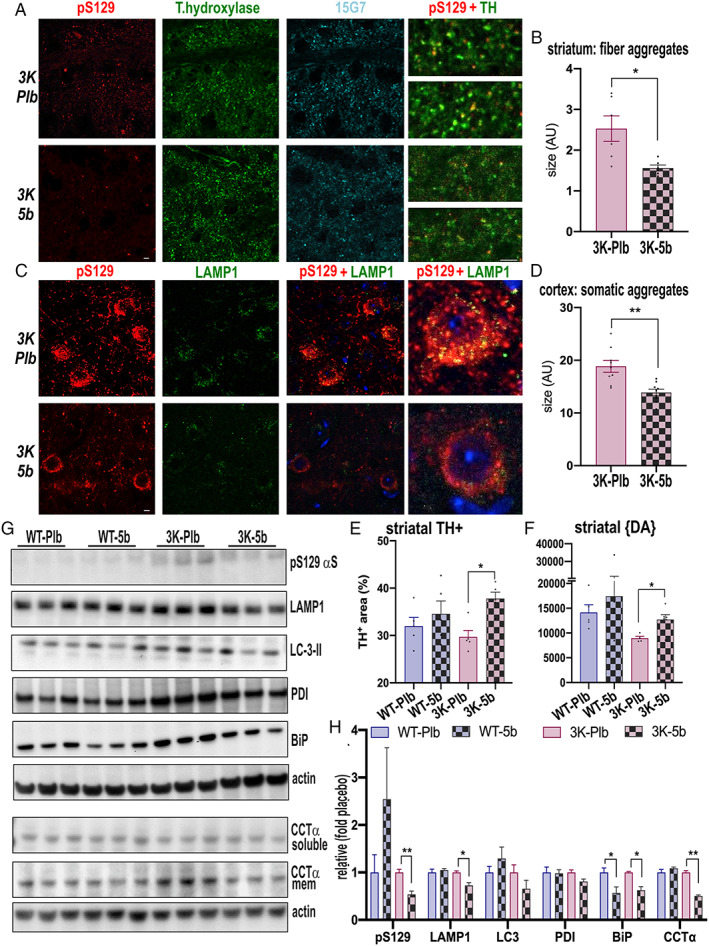
Stearoyl–coenzyme A desaturase inhibitor 5b reduces vesicle‐rich pS129+ aggregates, normalizes dopaminergic integrity and decreases the level of lipid droplet–associated proteins. (A) Confocal microscopy of dopaminergic striatal sections triple‐labeled pSer129 (red), tyrosine hydroxylase (TH; green), and total human αS (15G7; cyan), highlighting reduced pS129 human αS puncta within TH+ nerve fiber terminals by 5b. (B) Quantification of colocalized puncta of pSer129 and TH. (C) Confocal microscopy of cortical sections triple‐labeled pSer129 (red), lysosomal membrane protein 1 (LAMP1; green), and 4,6‐diamidino‐2‐phenylindole (blue), highlighting reduced pS129+ puncta in cortical neuron soma by 5b. (D) Quantification of pSer and LAMP1. (B, D) Colocalized points of the respective dual‐color confocal images were analyzed using ImageJ colocalization highlighter and then estimated for their size by the ImageJ particle analyzer plug‐in. Quantification for colocalized aggregate sizes (pS129 and the nerve fiber marker TH or the somatic marker LAMP1) in n = 10 fields each of sections from each genotype. (E) Estimating relative TH+ in striatal sections of placebo (Plb) wild‐type (WT) and 3K and 5b‐treated WT and 3K mice. (F) High‐performance liquid chromatography assay of striatal dopamine (DA) level in placebo WT and 3K, and 5b‐treated WT and 3K treated mice (n = 5–8 each group; duplicate sample measurements). (G) Quantitative western blots show pS129, LAMP1 accumulation, and lipid droplet marker binding immunoglobulin protein (BiP), protein disulfide isomerase (PDI), and CTP:phosphocholine cytidylyltransferase α (CCTα) by 5b treatment. (H) Quantification of (G). Data are mean ± standard error of the mean. *p < 0.05, **p < 0.01 placebo versus 5b, 2‐way analysis of variance with Tukey post hoc test or unpaired 2‐tailed t test. Scale bar: 20 µm. [Color figure can be viewed at www.annalsofneurology.org]
We next assessed whether SCD inhibition improves the DAergic nerve fiber integrity by quantifying TH+ immunoreactivity and dopamine (DA) levels in the dorsal striatum (see Fig 6E, F). Two‐way ANOVA showed significant treatment effect of 5b (F 1,16 = 11.38, p = 0.0039) but no significant interaction. The post hoc multiple comparisons revealed 5b significantly increased TH+ reactivity in 3K mice over that in placebo‐treated 3K mice. Accordingly, the low DA concentration in 3K striatum was increased by 5b treatment (44 ± 25%; p = 0.017).
WB analyses confirmed elevated protein levels of pS129 and LAMP1 in 3K cortex, suggesting impaired autophagy‐turnover of αS aggregates, as we previously described in 3K mice, 22 and these levels were reduced by 5b (p < 0.05; see Fig 6G, H). LDs and lipofuscin (end‐stage neuronal lysosomes) are an integral part of the 3K αS+ cytoplasmic aggregates, 10 , 22 and similar lesions are associated with and/or part of LBs in PD.1 Normally, most LDs bud from the endoplasmic reticulum (ER), cycle between the ER and lysosomes, and are subsequently degraded by autolipophagy for energy production and may serve as a sink for excess free FA to help prevent lipotoxicity. 43 , 44 Accordingly, we next probed ER chaperone proteins that are part of LD membranes, such as BiP 45 and PDI. 45 WBs of cortical extracts showed that 5b lowered BiP levels in both the WT and 3K models and showed a trend for reduced PDI level (p = 0.06) in 3K. CCTα, a key enzyme active on growing LD surfaces, 46 was decreased by 5b versus placebo in 3K brains (p < 0.01; see Fig 6H). Together, these data suggest a relatively pronounced dysregulation of LD formation in 3K mice and its mitigation by oral SCD inhibitor treatment.
In light of the 5b‐mediated decrease in size of abnormal lysosomal foci and lipid markers, we asked whether 5b diminishes the contribution of lipid droplets (LDs) in neurons harboring 3K αS aggregates (Fig 7). To quantify LDs, we used costaining of pS129 with the lipid‐dye BODIPY (493/503) (see Fig 7A) or the LD‐membrane–binding protein, PLIN on cortical sections (Fig 7C). BODIPY largely colabeled with pS129+ granules in cortical neuronal somata of the 3K‐Plb mice, as measured by particle sizes of the colocalized 8‐bit images (using ImageJ software), whereas much less colocalization was detected in 3K‐5b cortical neurons. To confirm the effect of reduced SCD activity by an independent approach, we also measured the pS129/LD coimmunoreactive aggregates in 3K mice bred on an SCD1 KO background (yielding heterozygous SCD+/− 3K mice; called "3K‐SKO"). Importantly, we observed a large reduction in the pS129‐LD inclusion sizes in 3K‐SKO cortex, similar to the effects of prolonged (120 days) 5b treatment (Fig 7C). To validate this finding of lesser LD signals, we conducted costaining of pS129 with the LD coating protein PLIN. Notably, PLIN+ LD are shuttled to lysosomes, where PLIN is extracted from the LD membranes, enabling autophagy lipolysis of the LD content. 47 We found PLIN to colocalize with pS129+ in 3K neuronal inclusions, and these were less apparent in both 3K‐5b and in 3K‐SKO brains (p < 0.01, 1‐way ANOVA, Tukey post hoc test) (Fig 7D). Light microscopic analyses showed a similar change in LD pattern that occasionally localized to dark degenerating neurons in toluidine blue–stained semithin cortical sections of 3K‐Plb mice (see Fig. 7E). To further confirm that SCD inhibition can downregulate the excess LD buildup with 3K αS, 13 we exposed M17D αS‐3K neural cultures to 5b in vitro and analyzed lipid dye (631/655) colabeling in response to increasing 5b concentrations. The SCD inhibitor produced a dose‐dependent reduction of LD‐rich αS inclusions (Fig 7F).
FIGURE 7.
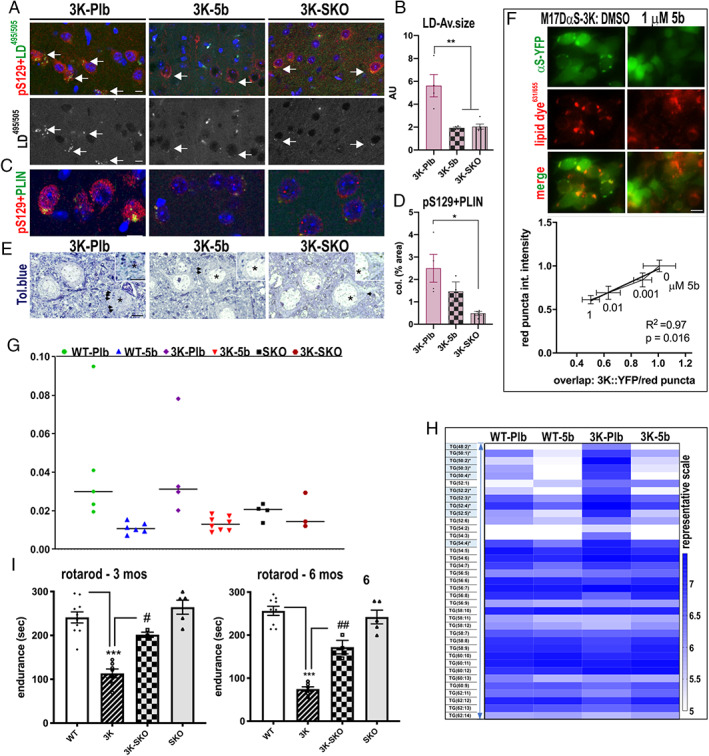
Reducing stearoyl–coenzyme A desaturase (SCD) lowers excess lipid droplets (LDs) associated with sizeable 3K‐αS aggregates, normalizes lipid profile, and rescues motor behavior in 3K mice. (A) Confocal microscopy of cortical sections labeled with pS129+ puncta (red) and an LD dye495/505 (green), quantified in B, or with (C) perilipin (PLIN; green) and (D) quantitation of colocalized particles using ImageJ particle size analyzer of the 8‐bit images. (E) Light microscopy of toluidine blue–stained semithin sections identifies LDs (arrows). LDs accumulate in a neuron with dark degenerative profile in 3K‐placebo (Plb; asterisk) versus normal neuron appearance in 3K‐5b and 3K‐SKO. (F) M17D cells were treated for 24 hours with dimethylsulfoxide or 5b, then transfected with YFP‐tagged 3K αS and stained with a lipid droplet dye631/655 (red). Representative images of cells treated for 24 hours with SCD inhibitor 5b are shown. Graph shows correlation between (fold) decrease in lipid colocalization with (fold) decrease in inclusions dependent on the 5b dose. (G) 16:1 to 16:0 fatty acid desaturation indices of placebo wild type (WT‐Pbl), 3K (3K‐Pbl), and treated WT‐5b, 3K‐5b versus 3K‐SKO, SKO in mouse cortex. (H) Triglyceride (TG) lipid profiles in 120‐day placebo 3K and WT and 3K‐5b. (I) Improved balancing skills by genetic deletion of 1 SCD allele in 3K (heterozygous crossing 3K to SKO) in the accelerating rotarod (4–40rpm). Data are mean ± standard error of the mean. 3K vs WT: *p < 0.05, **p < 0.01, ***p < 0.001; and 3K vs 3K‐SKO: # p < 0.05 and ## p < 0.01 in 3K vs 3K‐SKO; 2‐way analysis of variance with Tukey post hoc test. Scale bars = 20μm (A, C, F) or 10μm (E). [Color figure can be viewed at www.annalsofneurology.org]
Collectively, the above findings suggest that αS mutations that increase free monomers promote accumulation of LD, and this is significantly reduced by prolonged feeding of 5b. To attribute the changes by 5b treatment to SCD enzymatic activity, we measured the DI in WT‐5b and 3K‐5b versus their Plb controls, as well as in the heterozygous 3K‐SKO mice and SKO‐only mice by calculating the ratio of 16:1 to 16:0 FA in brain measured by LC‐MS. The dosing of 5b at 40mg/kg for 120 days led to a ~70% reduction in DI (p = 0.001, 2‐way ANOVA, Tukey post hoc test) in both the WT and the 3K tg mouse brain and ~53% DI reduction in 3K‐SKO brain (p = 0.032, 2‐way ANOVA, Tukey post hoc test; see Fig 7G).
To assess the changes in lipid classes in WT and 3K αS tg mice, we next measured the levels of lipids in the brain cortex through a discovery lipidomics approach. The most prominent changes were found in midchain glycerolipids (TGs) known to be regulated by SCD activity (see Figs 7H and 1G). 48 Paired comparisons revealed total abundance of these TGs increased in 3K‐Plb versus WT (p < 0.001; see Fig 7H). The 5b treatment decreased TGs in WT brain (p = 0.05) and also the elevated TG levels in 3K mouse brain (p < 0.001).
Finally, we investigated whether the decrease in lipid‐rich αS aggregates by genetic deletion of an SCD allele affected the motor performance of the 3K mice by measuring performance of 3‐ and 6‐month‐old 3K versus 3K‐SKO versus SKO mice on the challenging rotarod test (see Fig 7I). Post hoc comparisons of the differences by genotype revealed the 3K‐SKO crossing greatly benefitted the highly abnormal rotarod impairment of 3K mice at both 3 months (p = 0.05) and 6 months (p = 0.0002).
Discussion
Here, we assessed the ability of an orally available SCD inhibitor to prevent PD‐like cytopathology in iPSC‐derived human neurons expressing the fPD E46K mutation and in E46K‐expressing human DAergic M17D cells, as well as in "E46K‐amplified" (3K) neuronal cultures, and we examined this agent in vivo in 2 distinct PD‐relevant mouse models that transgenetically express either human WT αS (as occurs in almost all PD patients) or the E46K‐derived 3K mutant αS. Longitudinal assessment allowed evaluation of 5b efficacy against either the mild phenotypes observed in WT αS or the markedly progressive PD‐like motor syndrome of the 3K mice. We report that daily oral administration of a well‐characterized, brain‐penetrant SCD inhibitor significantly prevents these phenotypes, and the mechanistic target is fully supported by the genetic deletion of an SCD allele. Decreasing SCD enzymatic activity in 3K mice pharmacologically improved their more severe neuropathology, including αS insolubility and hyperphosphorylation, and ameliorated striatal DAergic fiber loss and the resultant motor decline. In WT mice, 5b prevented the buildup of pS129+ and PK‐resistant αS aggregates and subtle gait alterations. In cultured 3K neural cells and E46K‐expressing human neurons, a 5b‐mediated rescue of the αS T:M ratio was associated with reduced cytotoxic inclusions. The 5b‐mediated decrease in the 16:1/16:0 FA ratio (DI) was associated with improved partitioning of αS monomers from membranes toward principally cytosolic αS tetramers and with decreased lipid‐rich αS+ aggregates. Notably, we also found a striking dose‐dependent lowering of pS129+ signals in the E46K‐expressing human neurons. Importantly, genetic deletion of 1 SCD allele mitigated the progressive motor decline of 3K mice, supporting the specific role of the SCD enzyme in regulating human αS pathologies in multiple PD models. These findings, which represent the first report of the ability of SCD to regulate brain αS homeostasis in vivo, highlight the physiological importance of T:M equilibrium and FA metabolism as regulators of αS.
Increasing FA Saturation in vivo Enhances Physiological αS Tetramerization and Mitigates a PD‐like State
Consistent with previously reported effects of MUFAs 15 and polyunsaturated FAs 49 , 50 as contributing to pathological oligomerization of αS monomers in cultured neurons, the current results strongly support the hypothesis that decreasing the relative level of monounsaturated FAs protects against a PD‐like syndrome in mice. Supplementation with saturated FA can stimulate neuroprotective mechanisms in human cells from PD patients 15 or in PINK and PARKIN mutant PD fly models. 51 , 52 In addition, both single and repeated intragastric gavage of 8:0 SFA has been associated with increased DA signaling in a methylphenyltetrahydropyridine acute PD mouse model. 53 With direct clinical relevance, we observe here an increase in striatal DA levels in PD‐like 3K mice undergoing prolonged SCD inhibition.
How can changing the FA saturation state restore the physiological T:M ratio and mitigate the early onset and steadily progressive 3K neuropathological phenotype? Emerging evidence suggests an affinity of αS monomers for highly curved phospholipid membranes, 54 , 55 as consistently observed by αS overexpression across species. 5 , 56 , 57 , 58 , 59 Previous experimental studies reported that E46K αS has a higher affinity for binding to negatively charged vesicles than WT, 21 likely via abnormally increased membrane‐induced amphipathic helix formation, 60 and adding in‐register E➔K mutations into the adjacent KTKEGV motifs in the N‐terminal region further enhances this abnormality. Thus, at least 2 mechanistic explanations emerge for our therapeutic benefits of shifting the FA balance toward the saturated state: (1) direct stabilization of a proper amphipathic helix induced by the membrane binding of αS 55 and/or (2) a decrease of the abnormal αS membrane–lipid interaction of 3K αS by creating a more protein‐repellent vesicle surface. Notably, adding 16:0 SFA in the form of lyso‐phosphatidylcholine micelles to pure WT αS in vitro has been reported to directly stabilize the α‐helical conformation of human WT αS. 61 In addition, the αS N‐terminus harbors a motif homologous to a region in FA‐binding proteins, 62 which may thus facilitate the interaction of αS with FA on certain membrane lipids. Thus, in our 5b‐treated mice, the relative increase in 16:0 by chronic SCD inhibition may enhance physiological αS multimerization and the proper transient nature of αS vesicle‐binding, lessening the pathogenic consequences of the upstream tetramer‐abrogation by the 3K mutant.
Consistent with our previous report, 22 an increase in αS tetramer levels and consequent decrease in excessive binding of monomers to membranes may allow normalization of the trafficking of synaptic (and other) vesicles, including those containing DA at striatal nerve terminals and LAMP1+ endolysosomal vesicles within somata. We hypothesize that a physiologic αS T:M ratio may be required for normal vesicle flux, and this is seemingly reinstated in 3K mice by SCD inhibition or genetic deletion. Notably, the consistent increases in relative αS tetramer levels in 3K mice—either by SCD inhibition in the present study or by female sex in our previous study 22 —argue against the assumption that intact‐cell crosslinking increases and stabilizes tetramers by neutralizing the αS negative charges, 63 which charges would be unchanged by either of these conditions. A more plausible explanation for the beneficial results is that excess of human WT αS or mutant αS bearing E46K‐derived or other fPD mutations 9 may disturb the normal αS T:M equilibrium, and these effects are dynamic and reversible by relative increases in SFAs in membranes.
SCD Inhibition Reduces the Burden of Lipid‐Rich Aggregates
3K αS inclusions in yeast and neuron PD models are vesicle‐ and lipid‐rich. 13 , 20 We have shown previously 22 and confirm here that such aggregates in 3K mouse brain are regulated by autophagy turnover, reminiscent of evidence for aggregate development from autophagy impairment in PD. 37 Interestingly, histological analyses have revealed prominent lipid‐rich membranes in brainstem 64 and cortical LBs, 65 but their derivation remains unknown. Lipid dyshomeostasis is increasingly observed to concur with αS aggregation (recently reviewed 66 ), and several reports now exemplify how manipulations that alter lipid levels promote αS aggregate formation in vitro, 61 in yeast, 13 and in rodent and human neuronal cultures. 13 , 14 , 15 We found several LD membrane markers, such as PLIN and CCTα, the latter an enzyme detected on growing LD surfaces, 46 to associate with the αS aggregates in 3K mouse brain. PLINs protect against cytoplasmic lipolysis but are stripped off by lysosomal lipases, enabling metabolic turnover of the lipid content. 47 The accumulation of these LD markers further suggests impairments in lipophagy in the 3K αS mouse brain. Using an unbiased lipidomics approach, we found SCD inhibition decreased the mutant αS‐mediated upregulation of neutral lipids, namely TGs, a major LD component. Mechanistically, our results support the concept that accumulation of αS monomers may result in FA and other nutrient alterations and subsequently increase LD shuttling for lysosomal lipolysis. Thus, cytotoxicity induced by excess αS monomers may have different morphological correlates, including accumulation of LDs and autophagy stress, and these are associated with the accrual of abundant vesicle aggregates. Our results show that SCD downregulation efficiently stabilizes the αS T:M equilibrium and decreases the localization of αS on membranes measured biochemically, and with LDs in aggregates observed histologically, implying an enhancement of soluble αS homeostasis by saturated lipids.
The improvement of the αS T:M equilibrium in cytosol by SCD inhibition appears to be functionally significant, as evidenced by the striking mitigation of motor deficits in 3K mice: enhanced muscle tone 34 against tremor (as evidenced by the wire test) and improved gait fluidity against rigidity (as per the pole, rotarod, and gait tests). These results are consistent with our prior work showing less of motor phenotypes in 3K male mice by exposure to estrogen, which also increased the T:M ratio. Together, our previous and present findings highlight the significance of reestablishing normal tetramer levels, consistent with compelling evidence that changing lipid homeostasis, for example by miglustat treatment in GBA loss‐of‐function models, reduces the tetramer abrogation of Gaucher neurons. 11 , 67 Interestingly, CCTα activity is increased in neuronal models of Gaucher's, 68 further supporting that changes in lipid metabolism are associated with PD‐relevant phenotypes. In agreement with this observation, an excess of αS monomers, not only from αS mutations but also from loss‐of‐function mutations in the lysosomal lipid enzyme GBA, may be associated with more than one genetic form of PD.
Importantly, an SCD inhibitor aimed at enhancing soluble αS homeostasis in PD has recently entered a phase 1a human trial in healthy subjects (www.yumanity.com; press release, October 7, 2019). Our documentation of multiple benefits for PD‐like neuropathology and motor dysfunction provides the necessary preclinical animal data to advance this novel disease‐modifying approach to testing in PD patients.
Author Contributions
S.N., X.H., J.Wa., K.H., U.D., S.F., and D.J.S. contributed to the conception and design of the study. All authors contributed to the acquisition and analysis of the data. S.N., X.H., V.L., J.Wi., U.D., S.F., and D.J.S. contributed to drafting of the text and preparation of the figures.
Potential Conflicts of Interest
D.J.S. and U.D. are consultants to Yumanity.
Supporting information
SUPPLEMENTARY VIDEO M1: Video of pole performance of placebo 3K (right) and a 5b‐treated 3K mouse (left).
SUPPLEMENTARY VIDEO M2: Video of pole performance of another placebo 3K (left) and 5b‐treated 3K mouse (right).
SUPPLEMENTARY VIDEO M3: Video of pole performance of placebo wild‐type (WT; left) and 5b‐treated WT mouse (right).
SUPPLEMENTARY VIDEO M4: Video of body movement of a placebo 3K mouse on an elevated platform.
SUPPLEMENTARY VIDEO M5: Video of body movement of a 5b‐treated 3K mouse on an elevated platform.
Acknowledgment
Funded by NIH, NINDS R01 NS109510 (to S.N.) R01 NS099328 (to U.D.), R01 NS083845 (to D.J.S.), Michael J. Fox Foundation for Parkinson's Research grants (16296 to S.N. and 16261 to S.F.) and a gift of the Women's Brain Initiative (to SN).
We thank B. Caldarone for advice on mouse behavioral testing; P. Bechade for histological sectioning; V. Cardoso for help with mouse genotyping; I. Kandela (Northwestern University; supported by Integrated Molecular Structure Education and Research Center, 1‐S10‐OD021786‐01) and C. Rowbottom (Biogen) and D. Burdette (Biogen) for 5b bioavailability and pharmacokinetic studies; J. Ntambi, W. Hirst (Biogen) and the members of the Nuber, Dettmer, and Selkoe laboratories for helpful discussions; and R. Brathwaite and G. Dove for administrative assistance.
Contributor Information
Silke Nuber, Email: snuber@bwh.harvard.edu.
Dennis J. Selkoe, Email: dselkoe@bwh.harvard.edu.
References
- 1. Shahmoradian SH, Lewis AJ, Genoud C, et al. Lewy pathology in Parkinson's disease consists of crowded organelles and lipid membranes. Nat Neurosci 2019;22:1099–1109. [DOI] [PubMed] [Google Scholar]
- 2. Suzuki M, Sango K, Wada K, Nagai Y. Pathological role of lipid interaction with alpha‐synuclein in Parkinson's disease. Neurochem Int 2018;119:97–106. [DOI] [PubMed] [Google Scholar]
- 3. Diao J, Burre J, Vivona S, et al. Native alpha‐synuclein induces clustering of synaptic‐vesicle mimics via binding to phospholipids and synaptobrevin‐2/VAMP2. Elife 2013;2:e00592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Burre J, Vivona S, Diao J, et al. Properties of native brain alpha‐synuclein. Nature 2013;498:E4–E6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Logan T, Bendor J, Toupin C, et al. Alpha‐synuclein promotes dilation of the exocytotic fusion pore. Nat Neurosci 2017;20:681–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bartels T, Choi JG, Selkoe DJ. Alpha‐synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature 2011;477:107–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wang W, Perovic I, Chittuluru J, et al. A soluble alpha‐synuclein construct forms a dynamic tetramer. Proc Natl Acad Sci U S A 2011;108:17797–17802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dettmer U, Newman AJ, Luth ES, et al. In vivo cross‐linking reveals principally oligomeric forms of alpha‐synuclein and beta‐synuclein in neurons and non‐neural cells. J Biol Chem 2013;288:6371–6385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dettmer U, Newman AJ, Soldner F, et al. Parkinson‐causing alpha‐synuclein missense mutations shift native tetramers to monomers as a mechanism for disease initiation. Nat Commun 2015;6:7314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nuber S, Rajsombath M, Minakaki G, et al. Abrogating native alpha‐synuclein tetramers in mice causes a L‐DOPA‐responsive motor syndrome closely resembling Parkinson's disease. Neuron 2018;100:75–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kim S, Yun SP, Lee S, et al. GBA1 deficiency negatively affects physiological alpha‐synuclein tetramers and related multimers. Proc Natl Acad Sci U S A 2018;115:798–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Xilouri M, Brekk OR, Stefanis L. Autophagy and alpha‐synuclein: relevance to Parkinson's disease and related synucleopathies. Mov Disord 2016;31:178–192. [DOI] [PubMed] [Google Scholar]
- 13. Fanning S, Haque A, Imberdis T, et al. Lipidomic analysis of α‐synuclein neurotoxicity identifies stearoyl CoA desaturase as a target for Parkinson treatment. Mol Cell 2019;73:1001–1014.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Vincent BM, Tardiff DF, Piotrowski JS, et al. Inhibiting stearoyl‐CoA desaturase ameliorates alpha‐synuclein cytotoxicity. Cell Rep 2018;25:2742–2754. [DOI] [PubMed] [Google Scholar]
- 15. Imberdis T, Negri J, Ramalingam N, et al. Cell models of lipid‐rich alpha‐synuclein aggregation validate known modifiers of alpha‐synuclein biology and identify stearoyl‐CoA desaturase. Proc Natl Acad Sci U S A 2019;116:20760–20769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Miyazaki M, Man WC, Ntambi JM. Targeted disruption of stearoyl‐CoA desaturase1 gene in mice causes atrophy of sebaceous and meibomian glands and depletion of wax esters in the eyelid. J Nutr 2001;131:2260–2268. [DOI] [PubMed] [Google Scholar]
- 17. Trexler AJ, Rhoades E. N‐terminal acetylation is critical for forming alpha‐helical oligomer of alpha‐synuclein. Protein Sci 2012;21:601–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fernandez RD, Lucas HR. Isolation of recombinant tetrameric N‐acetylated alpha‐synuclein. Protein Expr Purif 2018;152:146–154. [DOI] [PubMed] [Google Scholar]
- 19. Rovere M, Powers AE, Jiang H, et al. E46K‐like alpha‐synuclein mutants increase lipid interactions and disrupt membrane selectivity. J Biol Chem 2019;294:9799–9812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dettmer U, Ramalingam N, von Saucken VE, et al. Loss of native alpha‐synuclein multimerization by strategically mutating its amphipathic helix causes abnormal vesicle interactions in neuronal cells. Hum Mol Genet 2017;26:3466–3481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Choi W, Zibaee S, Jakes R, et al. Mutation E46K increases phospholipid binding and assembly into filaments of human alpha‐synuclein. FEBS Lett 2004;576:363–368. [DOI] [PubMed] [Google Scholar]
- 22. Rajsombath MM, Nam AY, Ericsson M, Nuber S. Female sex and brain‐selective estrogen benefit alpha‐synuclein tetramerization and the PD‐like motor syndrome in 3K transgenic mice. J Neurosci 2019;39:7628–7640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bourque M, Morissette M, Di Paolo T. Repurposing sex steroids and related drugs as potential treatment for Parkinson's disease. Neuropharmacology 2019;147:37–54. [DOI] [PubMed] [Google Scholar]
- 24. Soldner F, Laganiere J, Cheng AW, et al. Generation of isogenic pluripotent stem cells differing exclusively at two early onset Parkinson point mutations. Cell 2011;146:318–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jacob SW, de la Torre JC. Pharmacology of dimethyl sulfoxide in cardiac and CNS damage. Pharmacol Rep 2009;61:225–235. [DOI] [PubMed] [Google Scholar]
- 26. Nuber S, Harmuth F, Kohl Z, et al. A progressive dopaminergic phenotype associated with neurotoxic conversion of alpha‐synuclein in BAC‐transgenic rats. Brain 2013;136:412–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Folch J, Lees M, Sloane Stanley GH. A simple method for the isolation and purification of total lipides from animal tissues. J Biol Chem 1957;226:497–509. [PubMed] [Google Scholar]
- 28. Fahy E, Sud M, Cotter D, Subramaniam S. LIPID MAPS online tools for lipid research. Nucleic Acids Res 2007;35:W606–W612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Raesch SS, Tenzer S, Storck W, et al. Proteomic and lipidomic analysis of nanoparticle corona upon contact with lung surfactant reveals differences in protein, but not lipid composition. ACS Nano 2015;9:11872–11885. [DOI] [PubMed] [Google Scholar]
- 30. Ritchie ME, Phipson B, Wu D, et al. Limma powers differential expression analyses for RNA‐sequencing and microarray studies. Nucleic Acids Res 2015;43:e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Atkinson KA, Beretta EE, Brown JA, et al. N‐benzylimidazole carboxamides as potent, orally active stearoylCoA desaturase‐1 inhibitors. Bioorg Med Chem Lett 2011;21:1621–1625. [DOI] [PubMed] [Google Scholar]
- 32. Stoffel W, Schmidt‐Soltau I, Jenke B, et al. Hair growth cycle is arrested in SCD1 deficiency by impaired Wnt3a‐palmitoleoylation and retrieved by the artificial lipid barrier. J Invest Dermatol 2017;137:1424–1433. [DOI] [PubMed] [Google Scholar]
- 33. Fanning S, Haque A, Imberdis T, et al. Lipidomic analysis of alpha‐synuclein neurotoxicity identifies stearoyl CoA desaturase as a target for Parkinson treatment. Mol Cell 2019;73:1001–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Crestani F, Low K, Keist R, et al. Molecular targets for the myorelaxant action of diazepam. Mol Pharmacol 2001;59:442–445. [DOI] [PubMed] [Google Scholar]
- 35. Delic V, Chandra S, Abdelmotilib H, et al. Sensitivity and specificity of phospho‐Ser129 alpha‐synuclein monoclonal antibodies. J Comp Neurol 2018;526:1978–1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Abd‐Elhadi S, Honig A, Simhi‐Haham D, et al. Total and proteinase K‐resistant alpha‐synuclein levels in erythrocytes, determined by their ability to bind phospholipids, associate with Parkinson's disease. Sci Rep 2015;5:11120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kett LR, Dauer WT. Endolysosomal dysfunction in Parkinson's disease: recent developments and future challenges. Mov Disord 2016;31:1433–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Watanabe I, Vachal E, Tomita T. Dense core vesicles around the Lewy body in incidental Parkinson's disease: an electron microscopic study. Acta Neuropathol 1977;39:173–175. [DOI] [PubMed] [Google Scholar]
- 39. Hayashida K, Oyanagi S, Mizutani Y, Yokochi M. An early cytoplasmic change before Lewy body maturation: an ultrastructural study of the substantia nigra from an autopsy case of juvenile parkinsonism. Acta Neuropathol 1993;85:445–448. [DOI] [PubMed] [Google Scholar]
- 40. Kosaka K. Lewy bodies in cerebral cortex. Report of three cases. Acta Neuropathol (Berl) 1978;42:127–134. [DOI] [PubMed] [Google Scholar]
- 41. Forno LS. Neuropathology of Parkinson's disease. J Neuropathol Exp Neurol 1996;55:259–272. [DOI] [PubMed] [Google Scholar]
- 42. Kuusisto E, Parkkinen L, Alafuzoff I. Morphogenesis of Lewy bodies: dissimilar incorporation of alpha‐synuclein, ubiquitin, and p62. J Neuropathol Exp Neurol 2003;62:1241–1253. [DOI] [PubMed] [Google Scholar]
- 43. Jacquier N, Choudhary V, Mari M, et al. Lipid droplets are functionally connected to the endoplasmic reticulum in Saccharomyces cerevisiae . J Cell Sci 2011;124:2424–2437. [DOI] [PubMed] [Google Scholar]
- 44. Henne M. And three's a party: lysosomes, lipid droplets, and the ER in lipid trafficking and cell homeostasis. Curr Opin Cell Biol 2019;59:40–49. [DOI] [PubMed] [Google Scholar]
- 45. Zhang S, Wang Y, Cui L, et al. Morphologically and functionally distinct lipid droplet subpopulations. Sci Rep 2016;6:29539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Krahmer N, Guo Y, Wilfling F, et al. Phosphatidylcholine synthesis for lipid droplet expansion is mediated by localized activation of CTP:phosphocholine cytidylyltransferase. Cell Metab 2011;14:504–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sztalryd C, Brasaemle DL. The perilipin family of lipid droplet proteins: gatekeepers of intracellular lipolysis. Biochim Biophys Acta 2017;1862:1221–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Attie AD, Krauss RM, Gray‐Keller MP, et al. Relationship between stearoyl‐CoA desaturase activity and plasma triglycerides in human and mouse hypertriglyceridemia. J Lipid Res 2002;43:1899–1907. [DOI] [PubMed] [Google Scholar]
- 49. Assayag K, Yakunin E, Loeb V, et al. Polyunsaturated fatty acids induce alpha‐synuclein‐related pathogenic changes in neuronal cells. Am J Pathol 2007;171:2000–2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sharon R, Bar‐Joseph I, Frosch MP, et al. The formation of highly soluble oligomers of alpha‐synuclein is regulated by fatty acids and enhanced in Parkinson's disease. Neuron 2003;37:583–595. [DOI] [PubMed] [Google Scholar]
- 51. Senyilmaz D, Virtue S, Xu X, et al. Regulation of mitochondrial morphology and function by stearoylation of TFR1. Nature 2015;525:124–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bajracharya R, Bustamante S, OBallard JW. Stearic acid supplementation in high protein to carbohydrate (P:C) ratio diet improves physiological and mitochondrial functions of Drosophila melanogaster parkin null mutants. J Gerontol A Biol Sci Med Sci 2019;74:1564–1572. [DOI] [PubMed] [Google Scholar]
- 53. Joniec‐Maciejak I, Wawer A, Turzynska D, et al. Octanoic acid prevents reduction of striatal dopamine in the MPTP mouse model of Parkinson's disease. Pharmacol Rep 2018;70:988–992. [DOI] [PubMed] [Google Scholar]
- 54. Westphal CH, Chandra SS. Monomeric synucleins generate membrane curvature. J Biol Chem 2013;288:1829–1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Dettmer U, Selkoe D, Bartels T. New insights into cellular alpha‐synuclein homeostasis in health and disease. Curr Opin Neurobiol 2015;36:15–22. [DOI] [PubMed] [Google Scholar]
- 56. Nemani VM, Lu W, Berge V, et al. Increased expression of alpha‐synuclein reduces neurotransmitter release by inhibiting synaptic vesicle reclustering after endocytosis. Neuron 2010;65:66–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Gitler AD, Bevis BJ, Shorter J, et al. The Parkinson's disease protein alpha‐synuclein disrupts cellular Rab homeostasis. Proc Natl Acad Sci U S A 2008;105:145–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wang L, Das U, Scott DA, et al. Alpha‐synuclein multimers cluster synaptic vesicles and attenuate recycling. Curr Biol 2014;24:2319–2326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Burre J, Sharma M, Tsetsenis T, et al. Alpha‐synuclein promotes SNARE‐complex assembly in vivo and in vitro. Science 2010;329:1663–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wise‐Scira O, Dunn A, Aloglu AK, et al. Structures of the E46K mutant‐type alpha‐synuclein protein and impact of E46K mutation on the structures of the wild‐type alpha‐synuclein protein. ACS Chem Nerosci 2013;4:498–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. O'Leary EI, Jiang Z, Strub MP, Lee JC. Effects of phosphatidylcholine membrane fluidity on the conformation and aggregation of N‐terminally acetylated alpha‐synuclein. J Biol Chem 2018;293:11195–11205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Sharon R, Goldberg MS, Bar‐Josef I, et al. Alpha‐synuclein occurs in lipid‐rich high molecular weight complexes, binds fatty acids, and shows homology to the fatty acid‐binding proteins. Proc Natl Acad Sci U S A 2001;98:9110–9115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Killinger BA, Melki R, Brundin P, Kordower JH. Endogenous alpha‐synuclein monomers, oligomers and resulting pathology: let's talk about the lipids in the room. NPJ Parkinsons Dis 2019;5:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. den Jager WA. Sphingomyelin in Lewy inclusion bodies in Parkinson's disease. Arch Neurol 1969;21:615–619. [DOI] [PubMed] [Google Scholar]
- 65. Gai WP, Yuan HX, Li XQ, et al. In situ and in vitro study of colocalization and segregation of alpha‐synuclein, ubiquitin, and lipids in Lewy bodies. Exp Neurol 2000;166:324–333. [DOI] [PubMed] [Google Scholar]
- 66. Fanning S, Selkoe D, Dettmer U. Parkinson's disease: proteinopathy or lipidopathy? NPJ Parkinsons Dis 2020;6:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kim YM, Shin DH, Park SB, et al. Case report of unexpected gastrointestinal involvement in type 1 Gaucher disease: comparison of eliglustat tartrate treatment and enzyme replacement therapy. BMC Med Genet 2017;18:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Bodennec J, Pelled D, Riebeling C, et al. Phosphatidylcholine synthesis is elevated in neuronal models of Gaucher disease due to direct activation of CTP:phosphocholine cytidylyltransferase by glucosylceramide. FASEB J 2002;16:1814–1816. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SUPPLEMENTARY VIDEO M1: Video of pole performance of placebo 3K (right) and a 5b‐treated 3K mouse (left).
SUPPLEMENTARY VIDEO M2: Video of pole performance of another placebo 3K (left) and 5b‐treated 3K mouse (right).
SUPPLEMENTARY VIDEO M3: Video of pole performance of placebo wild‐type (WT; left) and 5b‐treated WT mouse (right).
SUPPLEMENTARY VIDEO M4: Video of body movement of a placebo 3K mouse on an elevated platform.
SUPPLEMENTARY VIDEO M5: Video of body movement of a 5b‐treated 3K mouse on an elevated platform.