

Abstract
The photochemical deracemization of spiro[cyclopropane‐1,3′‐indolin]‐2′‐ones (spirocyclopropyl oxindoles) was studied. The corresponding 2,2‐dichloro compound is configurationally labile upon direct irradiation at λ=350 nm and upon irradiation at λ=405 nm in the presence of achiral thioxanthen‐9‐one as the sensitizer. The triplet 1,3‐diradical intermediate generated in the latter reaction was detected by transient absorption spectroscopy and its lifetime determined (τ=22 μs). Using a chiral thioxanthone or xanthone, with a lactam hydrogen bonding site as a photosensitizer, allowed the deracemization of differently substituted chiral spirocyclopropyl oxindoles with yields of 65–98 % and in 50–85 % ee (17 examples). Three mechanistic contributions were identified to co‐act favorably for high enantioselectivity: the difference in binding constants to the chiral thioxanthone, the smaller molecular distance in the complex of the minor enantiomer, and the lifetime of the intermediate 1,3‐diradical.
Keywords: enantioselectivity, hydrogen bonds, photochemistry, time-resolved spectroscopy, xanthones
A chiral thioxanthone‐ or xanthone‐based sensitizer allows for the differentiation of cyclopropane enantiomers and for a photochemical deracemization (17 examples, up to 85 % ee). The key intermediate of this entropically disfavored process was identified by transient absorption spectroscopy to be a 1,3‐ diradical with a lifetime of τ=22 μs.
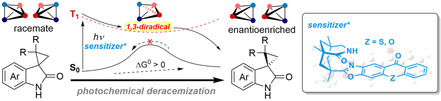
Introduction
Photochemistry offers access to reaction pathways which are not viable in the electronic ground state (thermal reactions). [1] In the context of enantioselective catalysis [2] it has been recognized that photoexcited chiral compounds can transfer their chirality to prochiral substrates and enable a selective isomerization. In particular, cyclic alkenes such as (Z)‐cycloheptene and (Z)‐cyclooctene have been frequently studied in the isomerization to their chiral (E)‐diastereoisomers. [3] Likewise, achiral cis‐1,2‐diphenylcyclopropane has been employed as a precursor to chiral trans‐1,2‐diphenylcyclopropane. [4] While the latter method has never led to useful enantioselectivities, the isomerization of cycloalkenes has provided access to enantiomerically enriched products in up to 87 % enantiomeric excess (ee). [3f] However, the reaction suffers from the fact that significant amounts of the achiral (Z)‐isomer remain unconverted. A conceptually different approach which avoids stable achiral intermediates aims at the deracemization [5] of a chiral compound via a transient achiral intermediate which decays to either one of the two enantiomers. The achiral intermediate does not accumulate but serves only to racemize the compound. An achiral catalyst thus enables in combination with light a conversion of an enantiopure compound to a racemic product mixture (Scheme 1). This entropically favored process has ample precedents in thermal chemistry and is frequently a nuisance but rarely synthetically desirable.
Scheme 1.
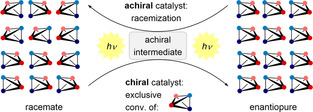
Photochemical deracemization of a racemate by a chiral catalyst vs. racemization of an enantiopure compound by an achiral catalyst.
The reverse process, that is, a photochemical deracemization, [6] however, is synthetically very useful. Indeed, a large number of chiral compounds are prepared in racemic form and need to be separated at a later stage by sophisticated methods. [7] A deracemization enables a complete conversion of the racemate to a single enantiomer. A key element of the pathway shown below is to find a chiral catalyst which processes only one enantiomer of a given substrate to provide the putative achiral intermediate. Since the achiral intermediate decays to both enantiomers [8] enrichment of the other enantiomer is ensured by repetitive racemization cycles. Although photochemical deracemization methods have been suggested for some time, [2h] it was not until very recently that the first highly enantioselective method was reported. Based on the use of chiral thioxanthone 1 [9] it was found that 3‐(1′‐alkenylidene)‐piperidin‐2‐ones (3) could be deracemized almost quantitatively and with high ee. [10] In subsequent studies, the thioxanthone was used in both enantiomeric forms (1 and ent‐1) as was the related compound 2 [11] and its enantiomer ent‐2. Work with cyclopropanes 4 [12] was performed employing mainly catalyst ent‐1 and chiral sulfoxides 5 [13] were deracemized with catalyst ent‐2. Allenes have remained so far the best suited substrate class and 3‐(1′‐alkenylidene)‐pyrrolidin‐2‐ones (6) have recently been employed in the context of synthetic work which aimed at a subsequent conversion of axial into point chirality. [14] In the same study, catalyst 1 was also found to successfully deracemize azepan‐2‐one 7 albeit in lower ee.
In all cases depicted in Figure 1, it had been postulated that the achiral intermediates involved in the photochemical processes are triplet intermediates. [15] Indeed, a typical workflow for the discovery of a sensitized deracemization reaction aims to find suitable conditions for a racemization of enantiopure compounds induced by an achiral triplet sensitizer, for example, by the parent compounds thioxanthen‐9‐one (TX) with a reported ET=268 kJ mol−1 (77 K, EtOH) [16] or by xanthen‐9‐one [ET=308 kJ mol−1, 77 K, ethanol– ether–isopentane (EPA)]. [17] Once it is confirmed that the catalyst is—in concert with light—solely responsible for the racemization, the deracemization with chiral catalysts can be studied and optimized. Although some circumstantial evidence was collected to support the hypothesis of triplet sensitization as the operating mechanistic pathway in the above‐mentioned deracemization reactions, the postulated radical intermediate has not yet been detected spectroscopically.
Figure 1.
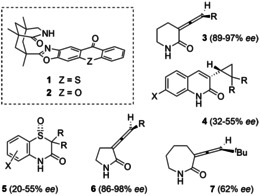
Structure of deracemization catalysts 1, 2 and of products 3–7 obtained enantioselectively by a photochemical deracemization reaction.
In the present study, we have investigated the deracemization of biologically and synthetically relevant [18] spirocyclopropyl oxindoles (spiro[cyclopropane‐1,3′‐indolin]‐2′‐ones) in the presence of catalysts 1 and 2. Our results demonstrate an efficient deracemization for this compound class to be possible. Furthermore, we investigated the mechanism of the process by transient absorption spectroscopy and computational studies and identified the diradical intermediate previously proposed. The results are discussed in the context of different selectivity parameters generally relevant for deracemization reactions.
Results and Discussion
Racemization and Detection of the 1,3‐Diradical Intermediate. Spirocyclopropyl oxindoles with the general structure I (Scheme 2) exhibit a lactam motif which enables hydrogen bonding to sensitizers 1 and 2. It was hypothesized that the compounds might undergo ring opening upon triplet sensitization and form 1,3‐diradical II as an achiral intermediate. If so, a deracemization as generally described in Scheme 1 was conceivable for this substrate class. Although the ring opening of cyclopropanes upon triplet sensitization has some precedents, [19] there is also evidence that the compounds can undergo ring opening by a single electron transfer process. [20] Both reductive and oxidative quenching of a photoexcited chromophore are possible and a racemization/deracemization could also occur for example via putative radical cation III.
Scheme 2.
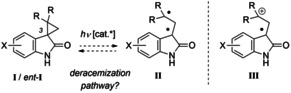
Putative formation of 1,3‐diradical II from spirocyclopropyl oxindoles I as a possible sensitized (energy transfer) pathway for racemization/deracemization and structure of an alternative intermediate III accessible via electron transfer.
The synthesis of racemic spirocyclopropyl oxindoles was readily achieved from 3‐diazo‐indolin‐2‐ones which served as precursors for a carbene addition to the respective olefin. [21] The studies commenced with the dichloro‐substituted substrate rac‐8 a which was available from 1,1‐dichloroethene (Figure 2). The two enantiomers 8 a and ent‐8 a were separable by chiral HPLC which enabled us to assess the integrity of the stereogenic C3 carbon center upon irradiation. The first electronic absorption band ranges from ca. 335 nm to 275 nm peaking at λ=295 nm (MeCN, ϵ=1690 m −1 cm−1). The triplet energy of the oxindole 8 a was determined as ET=298 kJ mol−1 from its phosphorescence spectrum (77 K, EtOH). Upon extended irradiation at λ=405 nm (Figure 2, left; for spectral data of all light sources, see the Supporting Information) enantiopure oxindole 8 a remained configurationally stable. Surprisingly, achiral TX induced a rapid racemization despite the fact that its triplet energy is significantly lower (ΔE≈30 kJ mol−1) than the triplet energy of compound 8 a. The racemization experiment was also performed at low temperature (−25 °C; Supporting Information) under otherwise identical conditions. After 10 hours of irradiation, compound 8 a retained its configuration (90 % ee) indicating that the energy transfer (ET) requires a thermal activation to be successful, either because it is endergonic (as likely the case here) and/or a steric hindrance has to be overcome. Irradiation with light of shorter wavelength (λ=350 nm) led to a racemization which was complete after 14 hours (Figure 2, right). The racemization was notably decelerated by addition of piperylene. Since piperylene is an established triplet quencher [22] the latter result suggested that the racemization occurs via a triplet intermediate. The former result indicated that sensitizer 1 might be a suitable deracemization catalyst because its triplet energy is similar (ET=263 kJ mol−1, 77 K, trifluorotoluene) [10] to that of TX. Further, it was of importance to choose an irradiation wavelength which results only in sensitized excitation via the chiral catalyst and avoids any direct excitation of the substrate.
Figure 2.
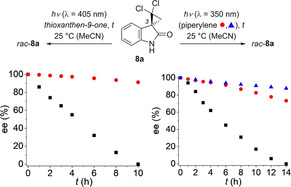
Racemization (▪) of compound 8 a to rac‐8 a in MeCN as the solvent: Time profile (c=1.5 mm) for λ exc=405 nm in the absence (•) and in the presence (▪) of 0.15 mm thioxanthen‐9‐one (left); time profile for λ exc=350 nm in the absence (▪) and in the presence of piperylene (right, 15 mm: •; 30 mm: ▴).
In the next step, we investigated the mechanism of the racemization reaction by transient absorption (TA) spectroscopy using an advanced set‐up [23] allowing for simultaneous recording of temporal and spectral information on the reaction dynamics (for a detailed description of the used set‐up and data analysis, see the Supporting Information). To this end, the triplet sensitizer TX which is achiral and lacks a binding site was employed. The dynamics of TX in various solvents are well studied. Intersystem crossing (ISC) occurs within a few ps [24] and leads to the formation of a characteristic triplet‐triplet (Tn←T1) absorption band that decays with a lifetime of 45 μs in argon‐saturated MeCN at room temperature. [16] In our TA experiments with TX in MeCN under ambient conditions, the data showed two temporal components (Figure 3 a).
Figure 3.
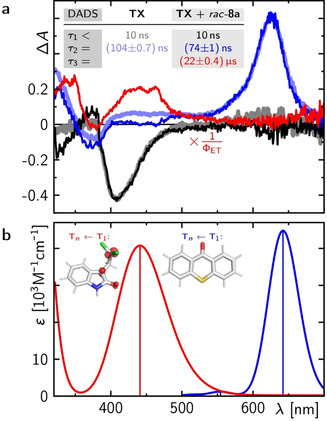
a) DADS of TA studies with TX in the absence (gray and light blue) and presence (black, dark blue, red) of rac‐8 a (12.3 mm) in MeCN, excited at λ exc=355 nm. b) Electronic transitions (sticks) for 3 TX and 1,3‐diradical 3 8 a convoluted with gaussians (the peak widths are taken from a fit of the corresponding experimental absorption bands) calculated at the RHF‐CPCM(MeCN)‐TD‐DFT‐B3LYP//DEF2‐TZVP level of theory (Supporting Information). The inset shows geometrically optimized structures.
The faster component was limited by the duration of the excitation pulse (ca. 10 ns) and comprised the characteristic spontaneous emission from 1 TX around 410 nm, as evidenced by the gray decay‐associated difference spectrum (DADS) in Figure 3 a. The slower component with a decay time of 104 ns (=k TX −1=(k bisc + k q [O2])−1, light blue DADS) represents the Tn←T1 signal and ground‐state bleach, in full agreement with the literature[ 16 , 24a ] and corroborated by a quantum chemical calculation using the extended multi‐configuration quasi‐degenerate perturbation theory (XMCQDPT) on top of the complete active space self‐consistent field (CASSCF) method (Figure 3 b, blue line). No transient signals remained on longer timescales, indicating that 3 TX decays entirely back to the ground state, with the reduced lifetime due to quenching by 3O2. In the presence of 12.3 mm rac‐8 a, the 3 TX decay time was reduced to 74 ns (dark blue DADS in Figure 3 a), and again 3 TX completely returned to the ground state. However, the DADS describing the triplet decay showed a lower amplitude in the spectral region between 400 and 500 nm than without a quencher (compare light and dark blue DADS, respectively), because the decay of 3 TX was accompanied by the formation of a new transient component (red DADS). The latter spectrum revealed two distinct absorption bands peaking at ca. 350 nm and 450 nm. The new species, thus, had a rise time of 74 ns and decayed with a time constant of 22 μs.
A comparison of the spectral position and shape of the spectrum with the calculated triplet absorption spectrum of 3 8 a (red line in Figure 3 b) corroborates a successful ET from 3 TX to rac‐8 a. The quantum yield Φ for ET from the triplet can be determined from the corresponding decay rates to ΦET=1−k TX/k TX+rac‐8 a=(29±2)%. The red DADS in Figure 3 a was rescaled accordingly by a factor of 1/ΦET in order to emphasize the good agreement between experiment and theory for both involved triplet species. Considering the concentration of rac‐8 a, the bimolecular ET rate is (3.2±0.2)×108 m −1 s−1 and thus the reaction is rather efficient but still slower than theoretically expected for a fully diffusion limited reaction (1.86×1010 m −1 s−1; Supporting Information). The calculations further show that the geometrically optimized structure of 3 8 a is a 1,3‐diradical resulting from opening of the cyclopropane ring (see inset of Figure 3 b). The TA experiments support the mechanism depicted in Scheme 2 for the sensitized reaction and allowed us to determine the lifetime of the 1,3‐diradical as τ=22 μs in non‐degassed MeCN at room temperature. [25]
Since the reduced radical anion TX.− of TX was reported to also exhibit an absorption between 400–500 nm, [26] we recorded its absorption spectrum upon electron transfer from N,N‐diisopropylethylamine to 3 TX. The lifetime and its spectrum (Supporting Information) were completely different from the lifetime and spectrum of the intermediate observed for the reaction between 3 TX with rac‐8 a (Figure 3 a). Furthermore, the kinetics of TX.− are accompanied by the persistence of the TX ground‐state bleach signal, in contrast to the case of the 1,3‐diradical 3 8 a. A single electron transfer as predominant pathway can consequently be ruled out.
Photochemical Deracemization. An initial screening of the reaction conditions for the desired deracemization (rac‐8 a → 8 a) was performed with fluorescent lamps emitting at λ=420 nm (Table 1). This set‐up had been previously used for reactions catalyzed by thioxanthone 1 and its enantiomer ent‐1.[ 10 , 12 ] Based on the racemization data and supported by time‐dependent measurement of the ee an irradiation time of eight hours was considered sufficient to reach the photostationary state and was selected to evaluate possible solvents for the reaction. Benzene and trifluorotoluene gave the best results (entries 3 and 4) while dichloromethane was slightly (entry 1) and toluene (entry 2) significantly inferior, at least in terms of enantioselectivity. It was gratifying to note, though, that even at this stage the ee was higher than the ee achieved with quinolone‐derived cyclopropanes 4 (Figure 1). [12] The originally chosen concentration of c=2.5 mm was based on precedent from previous work[ 10 , 12 ] and it was found that in the present case a further decrease of the concentration led to an increased enantioselectivity (entry 5). An increase in the catalyst concentration led to a slight ee improvement (entry 6). Further variations did not lead to better results under the chosen irradiation conditions (for further optimization attempts, see the Supporting Information).
Table 1.
Reaction optimization of the photochemical deracemization rac‐8 a→8 a catalyzed by chiral thioxanthone 1.
|
Entry[a] |
c [mm] |
Solvent |
1 [mol %] |
Light source[a] |
t [b] [h] |
ee [c] [%] |
|---|---|---|---|---|---|---|
|
1 |
2.5 |
CH2Cl2 |
10 |
FL |
8 |
55 |
|
2 |
2.5 |
PhMe |
10 |
FL |
8 |
31 |
|
3 |
2.5 |
PhH |
10 |
FL |
8 |
60 |
|
4 |
2.5 |
PhCF3 |
10 |
FL |
8 |
59 |
|
5 |
1.25 |
PhCF3 |
10 |
FL |
8 |
65 |
|
6 |
2.5 |
PhCF3 |
15 |
FL |
8 |
62 |
|
7 |
1.5 |
PhCF3 |
10 |
LED |
4 |
85 |
|
8 |
1.5 |
MeCN |
10 |
LED |
4 |
35 |
|
9 |
1.5 |
CH2Cl2 |
10 |
LED |
4 |
22 |
|
10 |
3.0 |
PhCF3 |
10 |
LED |
4 |
80 |
|
11 |
1.5 |
PhCF3 |
5 |
LED |
4 |
55 |
|
12[d] |
1.5 |
PhCF3 |
10 |
LED |
4 |
64 |
|
13[e] |
1.5 |
PhCF3 |
10 |
LED |
5 |
70 |
[a] Reactions were carried out in the given solvent at room temperature. Irradiation was performed at λ exc=420 nm either with a fluorescent lamp (FL) or with a light‐emitting diode (LED). Spectral data of the light sources are provided in the Supporting Information. [b] irradiation time. [c] The enantiomeric excess was calculated from the enantiomeric ratio (8 a/ent‐8 a) as determined by chiral HPLC analysis. [d] The reaction was performed at 0 °C. [e] The reaction was performed at λ exc=405 nm (LED).
Since the fluorescent lamps show a broad emission band that stretches into the UV region, we speculated that the intrinsic non‐catalyzed racemization (Figure 2) might deteriorate the outcome of the reaction (vide supra). A light‐emitting diode (LED) with a narrower emission spectrum was consequently chosen. The photostationary state was reached under these conditions already after four hours and the use of the LED resulted indeed in an improved selectivity (entry 7). It was confirmed that trifluorotoluene remained the preferred solvent under these conditions when compared to acetonitrile and dichloromethane (entries 8 and 9) and it was also confirmed that a low substrate concentration remains preferable (entry 10). Lowering the catalyst loading led to a decrease in enantioselectivity (entry 11). Exemplarily, entries 12 and 13 depict additional changes which were tried (lower reaction temperature, different light source) but did not further improve the selectivity. The ideal conditions for achieving a high enantioselectivity for the deracemization rac‐8 a → 8 a (Scheme 3) included an excitation at 420 nm, a substrate concentration of 1.5 mm in trifluorotoluene, and a catalyst loading of 10 mol %. The yield of isolated product was high (85 %) confirming the stability of the compound under the reaction conditions. Despite the optimization, the enantioselectivity was not fully consistent (80–85 % ee) due to minimal variations in the content of water and air. In the specific case of product 8 a, the routinely performed analogous reaction at −25 °C did not improve the enantioselectivity. In some other cases (products 8 b and 8 e) a minimal improvement was detected at lower temperature. In general, the degree of enantioselectivity for substituted spirocyclopropyl oxindoles 8 b–8 h was slightly lower as compared to the parent compound 8 a but it should also be noted that the conditions (concentration, solvent, light source) were not individually adapted to each substrate. The extensive functional group tolerance of sensitizer 1 had been demonstrated in previous studies.[ 9 , 10 , 12 ] The absolute configuration of compound 8 a was determined by anomalous X‐ray diffraction and the assignment for the other cyclopropanes was based on analogy. [27]
Scheme 3.
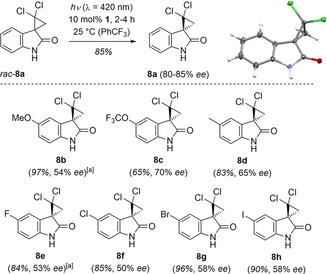
Deracemization of 2,2‐dichlorospiro[cyclopropane‐1,3′‐indoline]‐2′‐ones (rac‐8) catalyzed by chiral thioxanthone 1. [a] The reaction was performed at −25 °C.
The second substrate class which we investigated carried a dialkyl‐substituted carbon atom in the cyclopropane ring and optimization was performed with 2,2‐diethyl‐spiro[cyclopropane‐1,3′‐indoline]‐2′‐one (rac‐9 a). Although compound 9 a has a similar triplet energy (ET=298 kJ mol−1, 77 K, EtOH) as 8 a, an attempted deracemization with thioxanthone 1 failed. It was found that xanthone 2 with a higher triplet energy facilitates the desired reaction when the irradiation was performed at λ exc=366 nm. In a control experiment, we secured that irradiation with the chosen light source in the absence of a sensitizer does not lead to a racemization. The subsequently optimized reaction conditions (Supporting Information) could be transferred to several other substrates rac‐9 (Scheme 4) and the desired enantioenriched products were isolated in 82–98 % yield with 65–82 % ee. The high yield of the photochemical deracemization is notable given the high triplet energy of the sensitizer and its notorious tendency for C−H abstraction in the excited state.[ 11 , 28 ] Xanthone 2 was shown to be compatible with chloro, alkoxy and protected amino groups. From prior work, [13] the compatibility of the catalyst with bromo, sulfoxy, and cyano groups had been established.
Scheme 4.
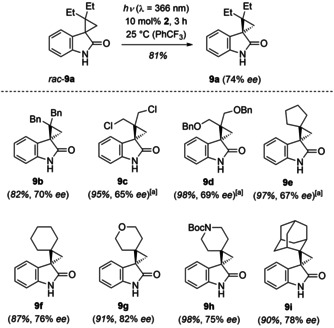
Deracemization of 2,2‐dialkyl‐substituted spiro[cyclopropane‐1,3′‐indoline]‐2′‐ones (rac‐9) catalyzed by chiral xanthone 2. [a] The reaction was performed at −25 °C.
Mechanistic Analysis and Discussion. Among the critical parameters that have been identified for the deracemization to be successful,[ 10 , 12 , 13 , 14 ] the most obvious parameter is the association of the two oxindole enantiomers I and ent‐I to the sensitizer. In an ideal scenario, the major enantiomer I does not bind to the sensitizer while enantiomer ent‐I shows efficient binding. We determined the specific binding constants for the two enantiomers ent‐8 a and 8 a to sensitizer 1 by NMR titration [29] and identified a better binding of the former enantiomer (Scheme 5). However, the values were unexpectedly low and indicated only a minor binding preference for ent‐8 a. Based on these data alone the degree of deracemization and the observed enantioselectivity cannot be accounted for.
Scheme 5.
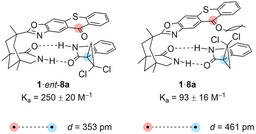
Binding constants (K a) of complexes 1⋅ent‐8 a and 1⋅8 a as determined by 1H NMR titration at 25 °C in [D6]benzene. Distances between the marked carbon atoms were calculated by DFT methods (Supporting Information).
A second parameter which influences the enantioselectivity is the rate of sensitization within the complexes. We have previously proposed [10] that the known distance dependence of energy transfer [30] plays a major role in the deracemization process. Indeed, DFT studies (Supporting Information) support the notion that the distance d between the two chromophores in 1⋅ent‐8 a and 1⋅8 a is significantly different with Δd in the range of ca. 100 pm. According to the theory of energy transfer,[ 30 , 31 ] the rate difference correlates to the distance by e−βΔd with the critical parameter β representing the sensitivity of the electron transfer to the distance of separation. Although there are no data available for the energy transfer from 1 to a given substrate, typical values for β are in the order of 10−2 pm−1.[ 1b , 30c , 30d ] Very crudely, the increased distance between the energy donor and the energy acceptor in complex 1⋅8 a should consequently lead to a decrease of the rate for electron transfer by at least 1/e, that is, by ca. 0.37, relative to 1⋅ent‐8 a. If one takes into account the above‐mentioned preference for the formation of 1⋅ent‐8 a vs. 1⋅8 a, the two factors co‐act towards the observed enantioselectivity.
For a mechanistic understanding, a third important contribution for the deracemization process has now been obtained for 8 a, that is, the lifetime of the triplet intermediate 3 8 a. Given that typical lifetimes for complexes between sensitizers such as 1 and 2 and hydrogen bonding substrates are below 1 μs, [28] it can be safely assumed that the cyclization will not occur within complex 1⋅3 8 a, as substantiated by the 22 μs lifetime determined for 3 8 a (equilibrium in Scheme 6). Since the steric bias exerted by the thioxanthone skeleton would in this case favor a cyclization to ent‐8 a (bottom left reaction in light gray in Scheme 6), a cyclization in the absence of any steric bias is desirable leading to a racemic mixture of 8 a and ent‐8 a (reaction on the right in Scheme 6). Support for this assumption stems from the enantioselective cyclization of 1,4‐ and 1,5‐diradicals mediated by a chiral template closely related to compound 1. [32]
Scheme 6.
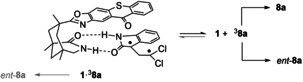
Dissociation of triplet 1,3‐diradical 3 8 a from sensitizer 1 and its decay to spirocyclopropyl oxindoles 8 a and ent‐8 a.
Based on the above‐mentioned data a preliminary kinetic scheme can be drawn (Scheme 7) for the sensitized deracemization of spirocyclopropyl oxindoles I which helps to visualize the key data. The pathway on the right (in black) is preferred over the pathway on the left (in gray). Both the higher association constant of ent‐I [K a (ent‐I)>K a(I)] to the sensitizer (Sens) and the more rapid sensitization followed by ring opening to the 1,3‐diradical (k′1>k 1) guarantee that the minor enantiomer is processed more rapidly. However, it is also crucial that the dissociation of the intermediate II is rapid because the cyclization within the complex would favor the production of ent‐I (k′2>k 2). Cyclization of the free 1,3‐diradical II leads to the racemic mixture with a rate constant k II the upper limit of which can be determined by its lifetime τ.
Scheme 7.
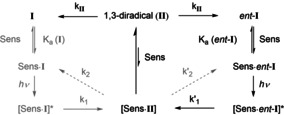
General scheme for the deracemization of spirocyclopropyl oxindoles (Ι) catalyzed by a chiral sensitizer (Sens) via a 1,3‐diradical ΙΙ.
As a consequence of the mechanistic picture it is important to note that the association itself is not the only relevant factor for a successful photochemical deracemization by sensitization. This statement is supported by comparing our current results with previous data obtained for a 3‐cyclopropyl‐substituted quinolone (4, Figure 1, R=Me, X=H). [12] The binding constants (25 °C, [D6]benzene) in this case were 2300±150 m −1 for one and 253±14 m −1 for the other enantiomer. While these data seem to suggest a much better differentiation in the deracemization reaction, the enantioselectivity (≤55 % ee) was lower than for most substrates 8 and 9 employed in the current study. Indeed, preliminary DFT calculations had suggested for the 3‐cyclopropylquinolones an almost identical distance (Δd≈0) between the chromophores in the complex to the sensitizer. In addition, it was speculated that the lifetime of the intermediate was low. Against this background the current study clearly emphasizes the importance of the energy transfer and the cyclization step.
Conclusion
In summary, the catalytic deracemization of spirocyclopropyl oxindoles has been achieved to a degree (up to 85 % ee) which has not been reached previously. The key components of the sensitized reaction pathway were elucidated by NMR titration experiments, transient absorption spectroscopy and computational studies. The postulated triplet intermediate of the reaction could be identified and its lifetime was determined. In addition, it was found that the association behavior of the cyclopropanes to the catalyst shows only little preference for one enantiomer and thus cannot solely explain the observed high enantioselectivity. Rather, the latter is achieved by an interplay with two additional aspects, namely an enantiosensitive distance in the complex formation with the chiral sensitizer, rendering energy transfer more likely in the desired substrate‐sensitizer combination, and a lifetime of the achiral intermediate that is longer than the mean complex lifetime, so that relaxation to the substrate's ground state is a racemic process. Future studies will explore the individual roles of these mechanisms with regard to the demonstrated deracemization reactions.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
Financial support by the Deutsche Forschungsgemeinschaft (Ba 1372/24‐1) is gratefully acknowledged. XL thanks the Alexander von Humboldt foundation for a research fellowship. O. Ackermann, F. Pecho, and J. Kudermann (all TU München) are acknowledged for their help with HPLC and GLC analyses. We are grateful to J. Großkopf (TU München) for his assistance in recording the luminescence spectra. Dr. G. Storch (TU München) is acknowledged for discussions regarding the DFT calculations. Open access funding enabled and organized by Projekt DEAL.
X. Li, R. J. Kutta, C. Jandl, A. Bauer, P. Nuernberger, T. Bach, Angew. Chem. Int. Ed. 2020, 59, 21640.
Contributor Information
Dr. Xinyao Li, http://www.oc1.ch.tum.de/home_en/
Prof. Dr. Thorsten Bach, Email: thorsten.bach@ch.tum.de.
References
- 1.
- 1a. Klán P., Wirz J., Photochemistry of Organic Compounds, Wiley, Chichester, 2009; [Google Scholar]
- 1b. Turro N. J., Ramamurthy V., Scaiano J., Modern Molecular Photochemistry of Organic Molecules, University Science Books, Sausalito, 2010. [Google Scholar]
- 2.For reviews covering enantioselective photochemical reactions, see:
- 2a. Zhou Q.-Q., Zou Y.-Q., Lu L.-Q., Xiao W.-J., Angew. Chem. Int. Ed. 2019, 58, 1586–1604; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 1600–1619; [Google Scholar]
- 2b. Sherbrook E. M., Yoon T. P. in Specialist Periodical Reports: Photochemistry, Vol. 46 (Eds.: Albini A., Protti S.), Royal Society of Chemistry, Croydon, 2019; [Google Scholar]
- 2c. Brenninger C., Jolliffe J. D., Bach T., Angew. Chem. Int. Ed. 2018, 57, 14338–14349; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 14536–14547; [Google Scholar]
- 2d. Garrido-Castro A. F., Maestro M. C., Alemán J., Tetrahedron Lett. 2018, 59, 1286–1294; [Google Scholar]
- 2e. Silvi M., Melchiorre P., Nature 2018, 554, 41–49; [DOI] [PubMed] [Google Scholar]
- 2f. Brimioulle R., Lenhart D., Maturi M. M., Bach T., Angew. Chem. Int. Ed. 2015, 54, 3872–3890; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 3944–3963; [Google Scholar]
- 2g. Inoue Y., Chem. Rev. 1992, 92, 741–770; [Google Scholar]
- 2h. Rau H., Chem. Rev. 1983, 83, 535–547. [Google Scholar]
- 3.
- 3a. Inoue Y., Yokoyama T., Yamasaki N., Tai A., Nature 1989, 341, 225–226; [Google Scholar]
- 3b. Inoue Y., Matsushima E., Wada T., J. Am. Chem. Soc. 1998, 120, 10687–10696; [Google Scholar]
- 3c. Hoffmann R., Inoue Y., J. Am. Chem. Soc. 1999, 121, 10702–10710; [Google Scholar]
- 3d. Inoue Y., Ikeda H., Kaneda M., Sumimura T., Everitt S. R. L., Wada T., J. Am. Chem. Soc. 2000, 122, 406–407; [Google Scholar]
- 3e. Lu R., Yang C., Cao Y., Tong L., Jiao W., Wada T., Wang Z., Mori T., Inoue Y., J. Org. Chem. 2008, 73, 7695–7701; [DOI] [PubMed] [Google Scholar]
- 3f. Maeda R., Wada T., Mori T., Kono S., Kanomata N., Inoue Y., J. Am. Chem. Soc. 2011, 133, 10379–10381. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Hammond G. S., Cole R. S., J. Am. Chem. Soc. 1965, 87, 3256–3257; [Google Scholar]
- 4b. Murov S. L., Cole R. S., Hammond G. S., J. Am. Chem. Soc. 1968, 90, 2957–2958. [Google Scholar]
- 5.For general reviews on the topic, see:
- 5a. Palmans A. R. A., Mol. Syst. Des. Eng. 2017, 2, 34–46; [Google Scholar]
- 5b. Rachwalski M., Vermue N., Rutjes F. P. J. T., Chem. Soc. Rev. 2013, 42, 9268–9282; [DOI] [PubMed] [Google Scholar]
- 5c. Servi S., Tessaro D., Pedrocchi-Fantoni G., Coord. Chem. Rev. 2008, 252, 715–726; [Google Scholar]
- 5d. Gruber C., Lavandera I., Faber K., Kroutil W., Adv. Synth. Catal. 2006, 348, 1789–1805. [Google Scholar]
- 6.Recent reviews:
- 6a. Shi Q., Ye J., Angew. Chem. Int. Ed. 2020, 59, 4998–5001; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 5030–5033; [Google Scholar]
- 6b. Yang C., Inoue Y., Nature 2018, 564, 197–199. [DOI] [PubMed] [Google Scholar]
- 7. Lorenz H., Seidel-Morgenstern A., Angew. Chem. Int. Ed. 2014, 53, 1218–1250; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 1240–1274. [Google Scholar]
- 8.For chiral induction in a planar photochemical intermediate by a memory effect, see:
- 8a. Jesuraj J. L., Sivaguru J., Chem. Commun. 2010, 46, 4791–4793; [DOI] [PubMed] [Google Scholar]
- 8b. Ayitou A. J.-L., Jesuraj J. L., Barooah N., Ugrinov A., Sivaguru J., J. Am. Chem. Soc. 2009, 131, 11314–11315. [DOI] [PubMed] [Google Scholar]
- 9. Alonso R., Bach T., Angew. Chem. Int. Ed. 2014, 53, 4368–4371; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 4457–4460. [Google Scholar]
- 10. Hölzl-Hobmeier A., Bauer A., Silva A. V., Huber S. M., Bannwarth C., Bach T., Nature 2018, 564, 240–243. [DOI] [PubMed] [Google Scholar]
- 11. Müller C., Bauer A., Bach T., Angew. Chem. Int. Ed. 2009, 48, 6640–6642; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 6767–6769. [Google Scholar]
- 12. Tröster A., Bauer A., Jandl C., Bach T., Angew. Chem. Int. Ed. 2019, 58, 3538–3541; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 3576–3579. [Google Scholar]
- 13. Wimberger L., Kratz T., Bach T., Synthesis 2019, 51, 4417–4424. [Google Scholar]
- 14. Plaza M., Jandl C., Bach T., Angew. Chem. Int. Ed. 2020, 59, 12785–12788; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 12885–12888. [Google Scholar]
- 15.For a recent report on a mechanistically different photochemical deracemization, see: Shin N. Y., Ryss J. M., Zhang X., Miller S. J., Knowles R. R., Science 2019, 366, 364–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Iyer A., Clay A., Jockusch S., Sivaguru J., J. Phys. Org. Chem. 2017, 30, e3738. [Google Scholar]
- 17. Pownall H. J., Mantulin W. M., Mol. Phys. 1976, 31, 1393–1406. [Google Scholar]
- 18.Reviews:
- 18a. Bacher E. P., Ashfeld B. L., Tetrahedron 2020, 76, 130692; [Google Scholar]
- 18b. Cao Z.-Y., Zhou F., Zhou J., Acc. Chem. Res. 2018, 51, 1443–1454; [DOI] [PubMed] [Google Scholar]
- 18c. Mei G.-J., Shi F., Chem. Commun. 2018, 54, 6607–6621; [DOI] [PubMed] [Google Scholar]
- 18d. Dalpozzo R., Adv. Synth. Catal. 2017, 359, 1772–1810; [Google Scholar]
- 18e. Dalpozzo R., Org. Chem. Front. 2017, 4, 2063–2078; [Google Scholar]
- 18f. Cao Z.-Y., Zhou J., Org. Chem. Front. 2015, 2, 849–858; [Google Scholar]
- 18g. Hong L., Wang R., Adv. Synth. Catal. 2013, 355, 1023–1052; [Google Scholar]
- 18h. Singh G. S., Desta Z. Y., Chem. Rev. 2012, 112, 6104–6155; [DOI] [PubMed] [Google Scholar]
- 18i. Dalpozzo R., Bartoli G., Bencivenni G., Chem. Soc. Rev. 2012, 41, 7247–7290. [DOI] [PubMed] [Google Scholar]
- 19.
- 19a. Zimmerman H. E., Flechtner W. T., J. Am. Chem. Soc. 1970, 92, 6931–6935; [Google Scholar]
- 19b. Becker R. S., Edwards L., Bost R., Elam M., Griffin G. J., J. Am. Chem. Soc. 1972, 94, 6584–6592; [Google Scholar]
- 19c. Paquette L. A., Meehan G. V., Henzel R. P., Eizember R. F., J. Org. Chem. 1973, 38, 3250–3256; [Google Scholar]
- 19d. Sivaguru J., Sunoj R. B., Wada T., Origane Y., Inoue Y., Ramamurthy V., J. Org. Chem. 2004, 69, 5528–5536; [DOI] [PubMed] [Google Scholar]
- 19e. Sivaguru J., Sunoj R. B., Wada T., Origane Y., Inoue Y., Ramamurthy V., J. Org. Chem. 2004, 69, 6533–6547; [DOI] [PubMed] [Google Scholar]
- 19f. Sivaguru J., Wada T., Origane Y., Inoue Y., Ramamurthy V., Photochem. Photobiol. Sci. 2005, 4, 119–127. [DOI] [PubMed] [Google Scholar]
- 20.
- 20a. Wong P. C., Arnold D. R., Tetrahedron Lett. 1979, 20, 2101–2104; [Google Scholar]
- 20b. Roth H. D., Schilling M. L. M., J. Am. Chem. Soc. 1980, 102, 7956–7958; [Google Scholar]
- 20c. Roth H. D., Sauers R. R., Photochem. Photobiol. Sci. 2012, 11, 931–937; [DOI] [PubMed] [Google Scholar]
- 20d. Woźniak Ł., Magagnano G., Melchiorre P., Angew. Chem. Int. Ed. 2018, 57, 1068–1072; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 1080–1084; [Google Scholar]
- 20e. Liu Z.-P., Li J.-L., Cheng X.-L., Cui J.-G., Huang Y.-M., Gan C.-F., Su W., Xiao J.-A., Eur. J. Org. Chem. 2019, 4085–4088. [Google Scholar]
- 21.For selected reviews, see references [18a,f]. For selected examples, see:
- 21a. Schwarzer D. D., Gritsch P. J., Gaich T., Angew. Chem. Int. Ed. 2012, 51, 11514–11516; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 11682–11684; [Google Scholar]
- 21b. Awata A., Arai T., Synlett 2013, 24, 29–32; [Google Scholar]
- 21c. Cao Z.-Y., Zhou F., Yu Y.-H., Zhou J., Org. Lett. 2013, 15, 42–45; [DOI] [PubMed] [Google Scholar]
- 21d. Cao Z.-Y., Wang X., Tan C., Zhao X.-L., Zhou J., Ding K., J. Am. Chem. Soc. 2013, 135, 8197–8200; [DOI] [PubMed] [Google Scholar]
- 21e. Muthusamy S., Ramkumar R., Tetrahedron Lett. 2014, 55, 6389–6393; [Google Scholar]
- 21f. Karthik G., Rajasekaran T., Sridhar B., Reddy B. V. S., Tetrahedron Lett. 2014, 55, 7064–7067. [Google Scholar]
- 22.
- 22a. Cozzens R. F., Fox R. B., J. Chem. Phys. 1969, 50, 1532–1535; [Google Scholar]
- 22b. Guillory J. P., Cook C. F., J. Am. Chem. Soc. 1973, 95, 4885–4891; [Google Scholar]
- 22c. Rosenfeld R., Alchalel A., Ottolenghi M., J. Phys. Chem. 1974, 78, 336–341. [Google Scholar]
- 23. Kutta R.-J., Langenbacher T., Kensy U., Dick B., Appl. Phys. B 2013, 111, 203–216. [Google Scholar]
- 24.
- 24a. Angulo G., Grilj J., Vauthey E., Serra-Andrés L., Rubio-Pons Ò., Jacques P., ChemPhysChem 2010, 11, 480–488; [DOI] [PubMed] [Google Scholar]
- 24b. Rodrigues J. F., de Assis da Silva F., Netto-Ferreira J. C., J. Braz. Chem. Soc. 2010, 21, 960–965; [Google Scholar]
- 24c. Villnow T., Ryseck G., Rai-Constapel V., Marian C. M., Gilch P., J. Phys. Chem. A 2014, 118, 11696–11707; [DOI] [PubMed] [Google Scholar]
- 24d. Rai-Constapel V., Villnow T., Ryseck G., Gilch P., Marian C. M., J. Phys. Chem. A 2014, 118, 11708–11717; [DOI] [PubMed] [Google Scholar]
- 24e. Mundt R., Villnow T., Ziegenbein C. T., Gilch P., Marian C., Rai-Constapel V., Phys. Chem. Chem. Phys. 2016, 18, 6637–6647; [DOI] [PubMed] [Google Scholar]
- 24f. Kayal S., Roy K., Adithya Lakshmanna Y., Umapathy S., J. Phys. Chem. A 2018, 122, 6048–6054. [DOI] [PubMed] [Google Scholar]
- 25.Lifetimes of triplet 1,3-diradicals are variable and span a range from ca. 100 ps to 100 μs:
- 25a. Mizuno K., Ichinose N., Otsuji Y., Caldwell R. A., J. Am. Chem. Soc. 1985, 107, 5797–5798; [Google Scholar]
- 25b. Adam W., Grabowski S., Platsch H., Hannemann K., Wirz J., Wilson R. M., J. Am. Chem. Soc. 1989, 111, 751–753; [Google Scholar]
- 25c. Engel P. S., Lowe K. L., Tetrahedron Lett. 1994, 35, 2267–2270; [Google Scholar]
- 25d. Karki S. B., Dinnocenzo J. P., Farid S., Goodman J. L., Gould I. R., Zona T. A., J. Am. Chem. Soc. 1997, 119, 431–432; [Google Scholar]
- 25e. Muray E., Illa O., Castillo J. A., Álvarez-Larena Á., Bourdelande J. L., Branchadell V., Ortuño R. M., J. Org. Chem. 2003, 68, 4906–4911; [DOI] [PubMed] [Google Scholar]
- 25f. Abe M., Chem. Rev. 2013, 113, 7011–7088; [DOI] [PubMed] [Google Scholar]
- 25g. Matsui Y., Oishi T., Ohta E., Ikeda H., J. Phys. Org. Chem. 2017, 30, e3636. [Google Scholar]
- 26. Yates S. F., Schuster G. B., J. Org. Chem. 1984, 49, 3349–3356. [Google Scholar]
- 27. Deposition Number 1991020 (for 8a) contains the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service www.ccdc.cam.ac.uk/structures.
- 28. Müller C., Bauer A., Maturi M. M., Cuquerella M. C., Miranda M. A., Bach T., J. Am. Chem. Soc. 2011, 133, 16689–16697. [DOI] [PubMed] [Google Scholar]
- 29.
- 29a. Fielding L., Tetrahedron 2000, 56, 6151–6170; [Google Scholar]
- 29b. Bakowski A., Dressel M., Bauer A., Bach T., Org. Biomol. Chem. 2011, 9, 3516–3529. [DOI] [PubMed] [Google Scholar]
- 30.
- 30a. Dexter D. L., J. Chem. Phys. 1953, 21, 836–850; [Google Scholar]
- 30b. Brown A., Wilkinson F., J. Chem. Soc. Faraday Trans. 2 1979, 75, 880–895; [Google Scholar]
- 30c. Closs G. L., Piotrowiak P., MacInnis J. M., Fleming G. R., J. Am. Chem. Soc. 1988, 110, 2652–2653; [Google Scholar]
- 30d. Albinsson B., Mårtensson J., Phys. Chem. Chem. Phys. 2010, 12, 7338–7351; [DOI] [PubMed] [Google Scholar]
- 30e. Curutchet C., Voityuk A. A., J. Phys. Chem. C 2012, 116, 22179–22185. [Google Scholar]
- 31. Speiser S., Chem. Rev. 1996, 96, 1953–1976. [DOI] [PubMed] [Google Scholar]
- 32. Bach T., Aechtner T., Neumüller B., Chem. Eur. J. 2002, 8, 2464–2475. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary