

Abstract
Divalent lanthanide complexes of Eu (1) and Yb (2) coordinated by a chelating pyridine‐based bis(silylene) ligand were isolated and fully characterized. Compared to the EuII complex 1, the YbII complex 2 presents a lower thermal stability, resulting in the activation of one SiII−N bond and formation of an YbIII complex (3), which features a unique silylene‐pyridyl‐amido ligand. The different thermal stability of 1 and 2 points towards reduction‐induced cleavage of one SiII−N bond of the bis(silylene) ligand. Successful isolation of the corresponding redox‐inert bis(silylene) CaII complex (5) was achieved at low temperature and thermal decomposition into a CaII complex (4) bearing the same silylene‐pyridyl‐amido ligand was identified. In this case, the thermolysis reaction proceeds through another, non‐redox induced, mechanism. An alternative higher yielding route to 4 was developed through an in situ generated silylene‐pyridyl‐amine proligand.
Keywords: amidinate ligands, calcium, lanthanides, silicon, silylenes
Divalent Eu, Yb and Ca complexes coordinated by a chelating bis(silylene) ligand were synthesized and their reactivity was studied.
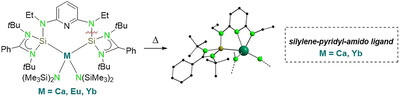
Introduction
N‐heterocyclic silylenes (NHSis) have evolved rapidly from laboratory curiosities to popular reagents in organometallic chemistry. [1] They have been widely used as ligands in diverse homogenous catalytic systems, [2] and have also played a dominant role in main group chemistry, especially to promote small molecule activation. [3] Although numerous transition metal silylene complexes can be found in the literature, [4] only a handful of f‐block silylene complexes has been reported to date (Figure 1). [5] In 2000, the group of Lappert isolated the first group 3 and lanthanide silylene complexes A by reacting the silylene Si[{N(CH2 tBu)}2‐C6H4] with [YCp3] or [YbCp3] in non‐polar solvents. [5a] The only other examples featuring Ln‐SiII (Ln=lanthanide) bonds are the divalent SmII complexes B and C reported in 2003 and in 2015, respectively.[ 5b , 5c ] Very recently, the group of Arnold described the synthesis of the first actinide NHSi complexes D. [5d] The paucity of f‐element silylene complexes compared to their transition metal analogues might be due to the hard‐soft mismatch between the hard f‐block metal center and the soft silicon donor, [6] which disfavors bonding interaction between lanthanides or actinides and silylene ligands. Compared with LnII ions, a similar coordination behavior is expected for CaII metal centers because of their similar ionic radii and electropositive nature. [7] Not surprisingly, only one report describing the coordination of NHSis to calcium has appeared in the last few years. [8]
Figure 1.

NHSi complexes of group 3 and f elements. [5]
In the past few years, a series of NHSi pincer‐type ligands containing two amidinate‐stabilized NHSi donors and a bridging spacer has been developed. [2d] The well‐established high yield access to the parent chlorosilylene LPhSiCl [9] (LPh=PhC(NtBu)2) enabled the formation of the desired bis(silylene) ligands through a straightforward salt metathesis route. The coordination chemistry of these multidentate bis(silylene) ligands is still relatively unknown, only documented for a few transition metals (Mn, Fe, Co, Ni, Cu, Pd, Ir) especially relevant for catalytic applications.[ 2d , 3d , 4k , 4m , 10 ] It is worth noting that, in comparison with analogous bis(phosphine) ligands, bis(silylene) ligands exhibit a significantly better σ‐donor ability. [4k]
In earlier work, we and others have shown that low valent main group compounds can be coordinated to LnII complexes. [11] Following our recent report on the reactivity of N‐heterocyclic carbene (NHC) LnII complexes towards CO2, [12] we were interested in the synthesis and study of the heavier NHSi analogues to investigate how the reactivity of the lanthanide metal centers would be modulated. We anticipated that formation and stabilization of such complexes could be facilitated by taking advantage of the chelating effect of the bis(silylene) ligand despite the soft nature of the neutral silicon donor. Similar strategies have already been used to achieve coordination of soft tertiary phosphine donors to YbII and EuII centers, by using bidentate bis(phosphine) ligands. [13]
Herein, we report the synthesis, structural features and different thermal stability of the first divalent EuII and YbII bis(silylene) complexes. The redox‐inert CaII analogue was also prepared in order to compare and rationalize the unusual reactivity.
Results and Discussion
Formation of EuII and YbII complexes bearing the reported pyridine‐based bis(silylene) ligand [SiNSi] [10b] (Scheme 1) was first targeted. A synthetic approach to such complexes requires lanthanide precursors soluble in non‐polar solvents and featuring a vacant coordination site or a loosely bound ligand, which can readily be displaced. Indeed, polar oxygen‐donor solvents (such as THF, Et2O or DME) should be avoided since they might compete with and suppress coordination of the bis(silylene) ligand. [5b] Equimolar reaction of the divalent lanthanide precursors [Ln{N(SiMe3)2}2(THF)2] (Ln=Eu, Yb) [14] and [SiNSi] in toluene at room temperature resulted in the formation of the corresponding bis(silylene) complexes [SiNSi‐Eu] (1) and [SiNSi‐Yb] (2), respectively (Scheme 1). Analytically pure products could be obtained in high yield (91 % for 1 and 89 % for 2) after removal of the solvent in vacuo and washing of the residue with a small amount of cold n‐pentane. The molecular structures were confirmed by means of multinuclear NMR spectroscopy (in case of the diamagnetic complex 2), elemental analyses and single crystal X‐ray diffraction studies. Both complexes are isostructural but not isomorphous, as complex 1 crystallizes in the triclinic space group P and 2 in the monoclinic space group P21/n. Both structures are displayed in Figure 2.
Scheme 1.

Synthesis of [SiNSi‐Eu] (1) and [SiNSi‐Yb] (2).
Figure 2.

Molecular structures of 1 and 2 in the solid state. H atoms and the SiMe3 groups are omitted for clarity. Selected bond lengths [Å] and angles [°] in 1: Eu−Si1 3.2810(11), Eu−Si2 3.2875(11); Si1‐Eu‐Si2 95.25(3); in 2: Yb−Si1 3.1999(11), Yb−Si2 3.1505(11); Si1‐Yb‐Si2 95.45(3).
The metal center in each complex is lying in a distorted tetrahedral geometry, coordinated by two nitrogen atoms of the amide ligands and two silicon atoms of the NHSi ligand. The distances between the Ln center and the central pyridine N atom (Eu⋅⋅⋅N4=3.728(3) and Yb⋅⋅⋅N4=3.668(3) Å) are too long to represent significant bonding interaction. [15] As expected, the Ln−Si bonds in 1 (3.2810(11) and 3.2875(11) Å) are slightly longer than those in 2 (3.1505(11) and 3.1999(11) Å) due to the larger ionic radius of EuII compared with that of YbII. [7] In addition, the Yb−Si bond distances are similar to those reported in YbII silyl complexes, [16] and somewhat longer than the Yb‐Si separation in the YbIII silylene complex A (2.984(2) Å), [5a] which is consistent with the smaller ionic radius of YbIII as compared to YbII. [7] Surprisingly, the metal centers are lying away from the mean plane of the pyridine ligand (2.397 and 2.326 Å for 1 and 2, respectively), which may be the result of the large ionic radii of the LnII metal centers and the relative rigidity of the [SiNSi] ligand.
The 1H NMR spectrum of 1 in C6D6 only revealed very broad unresolved features in the range −4 to +10 ppm due to the highly paramagnetic nature of the EuII ion. In contrast, the diamagnetic complex 2 could be fully characterized by multinuclear (1H, 13C, 29Si and 171Yb) NMR spectroscopy. The 1H NMR spectrum of 2 exhibits one single resonance for the four tBu groups at δ 1.24 ppm and another singlet for the four SiMe3 moieties at δ 0.62 ppm, thus revealing a C 2v symmetry of 2 in solution. Such a high symmetry in solution is not consistent with the fixed conformation observed in the solid state and indicates a fluxional behavior in solution at room temperature. In the 29Si{1H} NMR spectrum, two singlets were detected at δ 18.6 ppm (NSiN) and δ−14.0 ppm (SiMe3), the assignment being confirmed by 29Si INEPT and 1H‐29Si HMBC experiments. Coordination of the bis(silylene) ligand to the Yb atom results in a significant downfield shift for the SiII signal (resonances in the range δ −13.8 to −17.1 ppm for the free [SiNSi] ligand). [10b] Moreover, a singlet was detected at δ 810 ppm in the 171Yb NMR spectrum, downfield shifted compared with the signal of the precursor, [Yb{N(SiMe3)2}2(THF)2] (δ 680 ppm). [17] No 171Yb satellites could be clearly identified in the 29Si NMR spectrum,[ 16b , 17 ] which may point to a relatively labile coordination.
We further studied the lability of the bis(silylene) ligand in complex 2 by NMR‐scale reactions using THF and different NHCs (Scheme 2). Dissolution of crystalline 2 in [D8]THF resulted in complete decoordination of the [SiNSi] ligand along with formation of [Yb{N(SiMe3)2}2(THF)2], as evidenced by 1H NMR analysis (see Figure S16). Such a competition reaction is not surprising taking into account the hard and oxophilic nature of the YbII center. A similar ligand dissociation induced by THF was reported for the monodentate silylene adduct of samarocene, B (Figure 1). [5b] Reactivity towards different NHCs was also investigated. Reaction of 2 with ITMe (1,3,4,5‐tetramethylimidazol‐2‐ylidene) in C6D6 in a 1:2 molar ratio led to substitution of the coordinated bis(silylene) by two carbene molecules. The resulting [(ITMe)2Yb{N(SiMe3)2}2] complex was identified in the 1H and 13C{1H} NMR spectra by comparison with the published NMR spectroscopic data (Figures S17 and S18). [18] In contrast, the [SiNSi] bis(silylene) ligand in 2 was not be displaced by the bulkier IPr (1,3‐bis(2,6‐diisopropylphenyl)imidazole‐2‐ylidene) free carbene, probably due to the lower nucleophilicity of IPr compared to ITMe. [19]
Scheme 2.

Reaction of [SiNSi‐Yb] (2) with THF and ITMe.
A sealed NMR sample of 2 in C6D6 was stored at room temperature and the 1H NMR spectrum was recorded again after one week. Surprisingly, traces of several additional paramagnetically shifted signals were detected. The intensity of the new signals increased faster by heating the NMR tube at 80 °C. To investigate the thermal decomposition of 2, a toluene solution of the complex was heated at 80 °C for 48 h, which led to total conversion of 2 into the trivalent YbIII complex 3 (Scheme 3). Single crystals of 3 suitable for X‐ray diffraction analysis were obtained from a concentrated n‐pentane solution at room temperature in 63 % yield. The asymmetric unit of the molecular structure of 3 contains two independent molecules with similar metrical data. Therefore, the following discussion will be restricted to only one of the two molecules, which is displayed in Figure 3. The Yb atom in complex 3 is in a distorted square pyramidal geometry (τ<0.13), [20] chelated by a newly formed anionic silylene‐pyridyl‐amido ligand ([SiNN]). Only a few examples of multidentate anionic silylene ligands have been reported in the last 10 years, most of them being accessed by intramolecular C−H or Si−H bond activation when using low‐valent transition metal precursors.[ 2c , 3d , 4k , 21 ] The distance between the Yb atom and the pyridine nitrogen atom in 3 (2.314(4) Å) is slightly longer than the distance with the amide nitrogen donor (2.256(4) Å), suggesting that the anionic charge is localized at the amide N‐atom and supporting an amidopyridine (rather than aminopyridinato) [22] coordination mode of the ligand. A similar distribution of bond lengths (Yb−Npyr>Yb−Namido) was found in related YbIII amidopyridine complexes. [23] In addition, the neutral NHSi moiety is coordinated to the Yb atom with a significantly shortened Yb−Si bond (3.0426(15) Å) compared to the divalent Yb complex 2 (3.1505(11) and 3.1999(11) Å). Similarly, the Yb−Nsilylamide separations in complex 3 (2.214(4) and 2.226(3) Å) are ca. 0.15 Å shorter than the corresponding bond distances in 2 (2.363(3) and 2.370(3) Å), supporting the oxidation of the Yb center in its trivalent state. Monitoring the thermolysis process by 29Si{1H} NMR spectroscopy revealed the formation of a singlet at δ 75.8 ppm (see Figure S10), which could be unambiguously assigned to the SiI compound LPhSi‐SiLPh. [24] Therefore, thermal decomposition of 2 consists in the cleavage of one of the SiII−N bonds with formation of a SiI fragment and concomitant oxidation of the YbII metal center to YbIII. Analogous reduction of SiII to SiI has already been observed in the reaction of the chlorosilylene LPhSiCl with highly reducing f‐element complexes, such as [Cp*2Sm(OEt2)] [5c] and [(CpSiMe3)3U]. [5d] In the 1H NMR spectrum of 3, signals were detected between δ +81 and −13 ppm due to its paramagnetic nature. Nonetheless, evaluation of the peak intensities and 2D correlation spectra (1H‐1H COSY and 1H‐13C HMQC) allowed full assignment of all signals (see Figures S5–S9). In contrast to the thermal instability of 2, no decomposition of 1 in solution was detected over several weeks at room temperature. Paramagnetic 1H NMR analyses revealed that thermolysis of 1 can be achieved at higher temperatures upon extensive heating (>80 °C for more than one month), which precludes a convenient large‐scale synthesis. The difference in the thermal stability of 1 and 2 can be explained by the lower reducing ability of EuII compared to YbII (Ln3+/Ln2+ redox potentials of −0.35 V and −1.15 V vs. NHE, respectively, NHE=normal hydrogen electrode). [25]
Scheme 3.

Thermolysis of complex 2 leading to 3 and LPhSi‐SiLPh.
Figure 3.
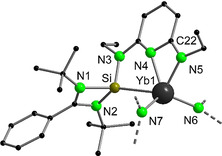
Molecular structure of 3 in the solid state. H atoms and the SiMe3 groups are omitted for clarity. Selected bond lengths [Å] and angles [°]: Yb1−Si1 3.0426(15), Yb1−N4 2.314(4), Yb1−N5 2.256(4); N4‐Yb1‐N5 58.93(13), N4‐C22‐N5 109.9(4).
The coordination behavior of divalent lanthanides has often been compared to that of CaII because of their similar ionic radii and high electropositive character. [26] These features, together with the redox‐inert nature of CaII, prompted us to investigate the formation of the CaII analogue of 1 and 2. Surprisingly, the equimolar reaction of [SiNSi] and [Ca{N(SiMe3)2}2(THF)2] in toluene either at room temperature or upon heating to 80 °C only gave an unidentifiable mixture, from which a few crystals of 4 could be isolated as a minor product (Scheme 4). Despite several attempts, no other side products could be isolated for the reaction at high temperature. Complex 4 consists of a CaII complex bearing the same [SiNN] silylene‐pyridyl‐amido ligand as obtained in the thermolysis product 3. In this case, it results from the formal cleavage of one Si−N bond of the [SiNSi] ligand, and elimination of one coordinated N(SiMe3)2 moiety with possible formation of LPhSi{N(SiMe3)2} [27] as a by‐product. In contrast to the formation of complex 3, which is formed by a redox reaction, initiated by the oxidation of YbII to YbIII, the formation of 4 is purely thermally induced. This different mechanism may explain the low yield of 4 and the formation of significant amounts of undefined by‐products. It should be noted that related ligand exchange reactions have already been reported on attempted syntheses of Ca and Zn silylene complexes.[ 4n , 8 ] The solid‐state structure of 4 features a dimeric arrangement with bridging amide ligands (Figure 4). The five‐coordinate Ca centers adopt highly distorted square pyramidal geometries (τ<0.14), [20] coordinated by two N donors of the chelating pyridyl‐amido moiety, one bridging Namido donor, one N of the silylamido ligand and one Si atom of the silylene. The Ca−Si distances in the dimer (3.0511(9) and 3.0654(9) Å) are shorter than those in the two other existing calcium silylene complexes, [Cp*2CaSi(O‐C6H4‐tBu){(NtBu)2CPh}] (3.2732(5) Å) and [Cp*2CaSi(NtBuCH)2] (3.090 Å in the calculated optimized structure), [8] indicating a stronger coordination of the silylene ligand towards Ca in 4. To the best of our knowledge, no other Ca silylene complex has been reported to date.
Scheme 4.

Different synthetic routes to access complexes 4 and 5.
Figure 4.
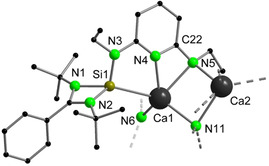
Molecular structure of 4 in the solid state (simplified view). H atoms, the SiMe3 groups and non‐coordinating solvent molecules are omitted for clarity. Selected bond lengths [Å] and angles [°]: Ca1−Si1 3.0426(15), Ca1−N4 2.314(4), Ca1−N5 2.524(2); N4‐Ca1‐N5 54.55(6), N4‐C22‐C5 113.9(2).
The bonding situation of Ca with the pyridyl‐amido moiety is different from that in the YbIII complex 3. The Ca−Namido bonds (Ca1−N5 2.524(2) and Ca2−N11 2.532(2) Å) are slightly longer than the Ca−Npyr bonds (Ca1−N4 2.468(2) and Ca2−N10 2.451(2) Å), suggesting a higher weight of the aminopyridinato resonance form. [22] Despite the popularity and versatility of amidinate‐coordinated Ca complexes, only a handful of Ca complexes bearing pyridyl‐amido ligands has been reported. [28] The very poor yield of 4 in this procedure precluded further spectroscopic studies. We therefore sought a more rational and higher yielding synthetic approach to complex 4. The NHSi‐containing proligand [SiNNH] was synthesized by mono‐deprotonation of the diamine [NNNH2] followed by reaction with LPhSiCl, and was further engaged in situ in a transamination reaction with [Ca{N(SiMe3)2}2(THF)2] (Scheme 4). Using this procedure, 4 could be isolated in a yield up to 38 % after crystallization and fully characterized.
1H NMR studies revealed different types of signals depending on the solvent used ([D8]THF or C6D6, see Figures S11 and S14), which can be explained by the dimeric nature of the complex in non‐coordinating solvents. In C6D6, a complex spectrum with broad resonances was observed at room temperature but sharper signals could be identified and assigned upon heating to 70 °C. A variable temperature 1H NMR analysis from 25 °C to 70 °C revealed the decoalescence of several signals (see Figure S15), which gives further evidence that the dimeric arrangement of 4 is retained is C6D6 solution at room temperature. In [D8]THF, well‐defined resonances for the different proton signals were observed, consistent with a C s symmetry in solution. Two singlet signals were detected in the 29Si{H} NMR spectrum at δ −9.0 and −15.9 ppm for the silylene and silylamide resonances, respectively. The silylene resonance of 4 in [D8]THF is slightly shifted upfield compared to that in C6D6 at room temperature (δ−4.4 ppm), which may indicate THF‐induced decoordination of the silylene donor, as observed in case of the YbII complex 2.
To investigate whether formation of 4 results from the thermal decomposition of the bis(silylene) calcium analogue of 1 and 2, the reaction between [SiNSi] and [Ca{N(SiMe3)2}2(THF)2] was performed under strict temperature control at −30 °C in toluene (Scheme 4). Single crystals of the Ca‐coordinated complex 5 were obtained at low temperature and the molecular structure is isomorphous with that of the YbII analogue 2 (Figure 5). The Ca−Si bond distances (3.1646(8) and 3.2080(9) Å) are slightly longer than those in 4. Complex 5 is highly temperature sensitive and slowly decomposes in solution at above −30 °C, as evidenced by a gradual color change of the solution from light yellow to dark orange. However, thermal decomposition of 5 at room temperature does not lead to a clean conversion into 4 but instead results in the formation of a complex mixture of products as evidenced by 1H NMR analyses. Thus, although it seems logical, we cannot claim without doubt that 4 is directly formed from 5.
Figure 5.

Molecular structure of 5 in the solid state. H atoms and the SiMe3 groups are omitted for clarity. Selected bond lengths [Å] and angles [°]: Ca−Si1 3.1646(8), Ca−Si2 3.2080(9); Si1‐Ca‐Si2 92.82(3).
We used the TURBOMOLE program package [29] to calculate the reaction energies for the thermolysis of 1 and 2, as well as for the thermolysis of 5 to 4. Structure parameters have been optimized and reaction energies have been calculated at the DFT level, employing the PBE0 hybrid‐functional [30] and the def2‐TZVP basis sets. [31] The reaction in Scheme 3 is possible rather for the Yb complex 2 (only slightly endothermic: +17 kJ mol−1) than for the Eu complex 1 (endothermic: +169 kJ mol−1). Furthermore, the reaction energy for the thermolysis of 5 to 4 is −113 kJ mol−1, thus supporting the assumption that 4 might be formed directly from 5. For reasons we do not see from the calculations, this reaction does not happen for the Eu or Yb complexes, although the calculated reaction energies are also exothermic and similar in size (Eu: −94 kJ mol−1, Yb: −107 kJ mol−1). Employment of the D4 dispersion correction [32] shifts reaction energies by ca. +60 kJ mol−1 throughout. When accounting for temperature effects from partition sums within the standard harmonic oscillator approximation for molecules in the gas phase, [33] calculated with the combination PBE/def2‐TZVP, reactions become more exothermic by 10 to 30 kJ mol−1, but the qualitative picture is unchanged. Results obtained with other functionals can be found in the ESI and are overall similar.
Conclusions
In conclusion, the first bis(silylene)‐coordinated divalent lanthanide complexes 1 (Eu) and 2 (Yb) have been isolated and fully characterized. Despite chelation of the metal center, the bis(silylene) ligand in these complexes is very labile and undergoes substitution by THF or small carbene (ITMe) ligands. The more reducing YbII complex 2 presents a lower thermal stability, resulting in the easy activation of one SiII−N bond and formation of the YbIII complex 3 which features a unique silylene‐pyridyl‐amido ligand. Successful isolation of the corresponding redox‐inert CaII complex 5 could only be achieved at low temperature and thermal decomposition into the CaII silylene‐pyridyl‐amido complex 4 was identified. In this case, formation of the silylene‐pyridyl‐amido ligand proceeds through another mechanism probably involving a ligand exchange reaction. A higher yielding route to 4 was developed using an in situ generated [SiNNH] proligand. Such a route opens new perspectives for the formation of silylene‐pyridyl‐amido complexes and more generally of complexes bearing silylene ligands functionalized by anionic donors. The reactivity described in this manuscript also sheds light on possible decomposition reactions that can occurs when functionalized silylene ligands are associated with highly reducing metal centers.
Experimental Section
See Supporting Information for general procedures.
Synthesis of [SiNSi‐Eu] (1): To a mixture of [SiNSi] (0.145 g, 0.213 mmol) and [Eu{N(SiMe3)2}2(THF)2] (0.131 g, 0.213 mmol) was condensed toluene (10 mL) at −78 °C. The reaction mixture was allowed to warm to room temperature, resulting in an orange solution. After stirring at room temperature for 2 h, all volatiles were removed under reduced pressure. The resulting solid was washed with cold n‐pentane (2 mL) and dried in vacuo for 20 min affording the title product as an orange solid. Yield: 0.223 g (0.193 mmol), 91 %. Crystals suitable for X‐ray diffraction analysis were grown by slow evaporation of the toluene solution at 20 °C. Anal. Calcd for C51H95EuN9Si6 (1154.86): C, 53.04; H, 8.29; N, 10.92. Found: C, 53.06; H, 8.18; N, 10.77. The 1H NMR spectrum of [SiNSi‐Eu] (1) in C6D6 only revealed very broad unresolved features in the range −4 to 10 ppm due to the highly paramagnetic nature of the complex. IR (ATR): (cm−1)=3435 (w), 3059 (w), 2963 (vs), 2928 (s), 2899 (m), 2868 (m), 2163 (w), 1647 (m), 1589 (vs), 1553 (m), 1521 (w, sh), 1488 (s, sh), 1475 (s), 1445 (vs), 1386 (m), 1355 (m), 1337 (w), 1313 (m), 1254 (s), 1239 (s), 1203 (m), 1157 (s), 1114 (w), 1066 (w), 1025 (w), 980 (m, sh), 955 (s), 929 (m), 881 (m), 864 (m), 842 (s, sh), 821 (s), 774 (m, sh), 747 (m), 721 (w), 707 (m), 697 (m, sh), 662 (m), 611 (m), 532 (w), 488 (w), 427 (w).
Synthesis of [SiNSi‐Yb] (2): To a mixture of [SiNSi] (0.150 g, 0.220 mmol) and [Yb{N(SiMe3)2}2(THF)2] (0.140 g, 0.220 mmol) was condensed toluene (10 mL) at −78 °C. The reaction mixture was allowed to warm to room temperature, resulting in a dark‐red solution. After stirring at room temperature for 2 h, all volatiles were removed under reduced pressure. The resulting solid was washed with cold n‐pentane (2 mL) and dried in vacuo for 20 min affording the title product as a purple solid. Crystals suitable for X‐ray diffraction analysis were grown by slow evaporation of the toluene solution at 20 °C. Yield: 0.230 g (0.196 mmol), 89 %. 1H NMR (400.30 MHz, C6D6): δ [ppm]: 7.59 (br s, 2 H, CH Ph), 7.32 (t, 3 J HH=7.9 Hz, 1 H, p‐CH pyr), 7.07–6.93 (m, 8 H, CH Ph), 6.21–6.20 (br d, 2 H, m‐CH pyr), 3.40–3.38 (br m, 4 H, CH 2CH3), 1.24 (s, 36 H, C(CH 3)3), 1.16 (br t, 6 H, CH2CH 3), 0.62 (s, 36 H, Si(CH 3)3). 13C{1H} NMR (100.66 MHz, C6D6): δ [ppm]: 168.7 (NCN), 161.0 (o‐C pyr), 138.8 (p‐C pyr), 133.0 (C quat,Ph), 130.2 (Ph), 129.9 (Ph), 128.8 (Ph), 128.4 (Ph), 127.7 (Ph), 102.8 (m‐C pyr), 53.4 (C(CH3)3), 39.0 (CH2CH3), 32.2 (C(CH3)3), 15.0 (CH2 CH3), 7.2 (Si(CH3)3). 29Si{1H} NMR (79.53 MHz, C6D6): δ [ppm]: 18.6 (NSiN), −13.9 (SiMe3) (assignment confirmed by 29Si‐INEPT and 1H‐29Si HMBC experiments) 171Yb NMR (52.52 MHz, C6D6): δ [ppm]: 810 (br s, Δν1/2 ≈140 Hz). IR (ATR): (cm−1)=3436 (w), 3083 (w), 3061 (w), 2991 (m, sh), 2964 (vs), 2929 (s), 2901 (s), 2868 (s), 2165 (w), 1647 (s), 1614 (s, sh), 1597 (s, sh), 1576 (vs), 1519 (w), 1488 (s, sh), 1475 (s), 1459 (s, sh), 1441 (vs), 1410 (vs), 1392 (s, sh), 1359 (m), 1328 (w), 1306 (w), 1286 (w), 1269 (m), 1242 (s), 1203 (s), 1154 (s), 1108 (w), 1099 (w, sh), 1078 (m), 1065 (m), 1035 (s), 1010 (vs), 970 (m, sh), 928 (m), 886 (m), 869 (s), 821 (vs), 809 (s, sh), 783 (m), 769 (m), 748 (s), 722 (m), 705 (s), 672 (m, sh), 661 (m), 641 (w, sh), 618 (m), 589 (w), 537 (m), 500 (w), 461 (w), 420 (w).
Synthesis of [SiNN‐Yb] (3): The complex [SiNSi‐Yb] (2) (0.200 g, 0.170 mmol) was dissolved in toluene (10 mL) and the resulting solution was heated at 80 °C for 48 h, which led to a color change of the solution from dark‐red to dark‐brown. All volatiles were removed under reduced pressure and the residue was extracted with n‐pentane (10 mL). Crystals suitable for X‐ray diffraction analysis were grown by slow evaporation of the n‐pentane solution at 20 °C. The mother liquor was removed by cannula transfer and the crystals were dried in vacuo for 20 min affording the title product as a yellow‐orange solid. Yield: 0.098 g (0.107 mmol), 63 %. Anal. Calcd for C36H72N7Si5Yb (916.50): C, 47.18; H, 7.92; N, 10.70. Found: C, 47.21; H, 7.05; N, 10.64. The thermolysis reaction of [SiNSi‐Yb] (2) in C6D6 at 80 °C was monitored by 29Si{1H} NMR and revealed the formation of a singlet at δ 75.8 ppm (see Figure S10). The latter was assigned to LPhSi‐SiLPh [24] by comparison to the signal of an authentic sample, thus confirming the formation of LPhSi‐SiLPh as a by‐product. 1H NMR (300.14 MHz, C6D6): δ [ppm]: 80.9 (br, 2 H, CH 2CH3), 35.2 (br, 3 H, CH2CH 3), 23.9 (br, 3 H, CH2CH 3), 22.2 (br, 2 H, CH 2CH3), 17.2 (d, 3 J HH=6.9 Hz, 1 H, CH Ph), 10.7 (t, 3 J HH=7.4 Hz, 1 H, CH Ph), 8.5 (br s, Δν1/2 ≈350 Hz, ca. 18 H), 8.3 (t, 3 J HH=7.4 Hz, ca. 1 H, CH Ph), 6.2 (br s, Δν1/2 ≈180 Hz, ca. 36 H, Si(CH 3)3), 5.9 (s, ca. 1 H, CH Ph), 5.3 (s, ca. 1 H, CH Ph), −0.7 (s, 1 H, CH pyr), −8.6 (s, 1 H, CH pyr), −12.6 (s, 1 H, CH pyr) (Assignment confirmed by 1H‐1H COSY experiment, see Figure S9). 13C{1H} NMR (75.48 MHz, C6D6): δ [ppm]: 195.7 (C quat), 147.5 (C quat), 140.3 (C quat), 137.4 (Ph), 132.7 (Ph), 132.4 (Ph), 130.7 (Ph), 128.8 (Ph), 113.3 (C pyr), 74.1 (C pyr), 70.7 (C quat), 63.0 (CH2CH3), 47.5 (C pyr), 41.8 (C(CH3)3), 40.8 (CH2 CH3), 7.8 (Si(CH3)3) (Assignment based on DEPT, 1H‐1H COSY and 1H‐13C HMBC experiments, 3 carbon signals could not be identified). 29Si{1H} NMR (59.62 MHz, C6D6): δ [ppm]: −109.4, −113.6 (no correlation signal could be detected in the 1H‐29Si HMBC experiments, probably due to the paramagnetic nature of the compound). IR (ATR): (cm−1)=3436 (w), 3061 (w), 2963 (vs), 2929 (s), 2900 (m), 2868 (m), 2163 (m), 2148 (m, sh), 1647 (s), 1613 (s, sh), 1598 (s, sh), 1574 (vs), 1498 (m, sh), 1485 (s, sh), 1476 (s), 1459 (s, sh), 1444 (vs), 1408 (m), 1386 (m, sh), 1357 (m), 1329 (w), 1286 (w), 1270 (w), 1241 (m), 1204 (s), 1155 (m), 1140 (m, sh), 1110 (w), 1097 (w), 1077 (m, sh), 1055 (m, sh), 1032 (s), 1020 (s, sh), 969 (m, sh), 928 (m), 884 (m), 869 (m), 830 (s, sh), 818 (s), 787 (w), 771 (m), 746 (m), 724 (m), 704 (s), 659 (m), 641 (w, sh), 619 (m), 605 (w, sh), 582 (w), 535 (w), 500 (w), 482 (w), 419 (w). EI‐MS: m/z (%): 916.441 [M]+ (3), 901.346 [M‐CH3]+ (2), with the corresponding isotopic patterns.
Synthesis of [SiNN‐Ca] (4): Method A: To a mixture of [SiNSi] (0.150 g, 0.220 mmol) and [Ca{N(SiMe3)2}2(THF)2] (0.111 g, 0.220 mmol) was condensed toluene at −78 °C. The resulting yellow solution was warmed up to room temperature, stirred for 1 h at room temperature and heated at 80 °C for 48 h, during which time the solution became orange‐brown. All the volatiles were removed under reduced pressure. The solid residue was extracted with n‐pentane (10 mL) and filtered. Only a few crystals of the title compound suitable for X‐ray diffraction studies could be obtained by slow evaporation of the n‐pentane solution at 20 °C. Method B: The deprotonation of N,N‐diethylpyridine‐2,6‐diamine [NNNH2] was performed according to the literature procedure. [34] [NNNH2] (0.500 g, 3.02 mmol) was dissolved in toluene (10 mL) and cooled to 0 °C. nBuLi (1.30 mL, 2.5 m in n‐hexane, 3.25 mmol) was slowly added and the reaction was stirred at room temperature for 2 h. The mixture was then cooled to −78 °C and a solution of LPhSiCl (0.891 g, 3.02 mmol) in toluene (10 mL) was added dropwise. The mixture was allowed to warm to room temperature and stirred overnight, after which the solution was filtered via cannula and all volatiles were removed under reduced pressure. The crude product was used without further purification for the next step. The crude product (0.168 g, 0.397 mmol) and [Ca{N(SiMe3)2}2(THF)2] (0.200 g, 0.397 mmol) were placed together into a Schlenk tube and toluene (20 mL) was condensed at −78 °C. The mixture was warmed to room temperature, stirred for 1 h and then heated at 80 °C for 48 h. All volatiles were removed under reduced pressure and the resulting solid was extracted with toluene (10 mL). Crystals suitable for X‐ray diffraction analysis were grown by slow evaporation of the toluene solution at 20 °C. Yield (based on crystals): 0.097 g (0.150 mmol), 38 %. Anal. Calcd for C30H54CaN6Si3 (0.25 C7H8) (646.17): C, 59.02; H, 8.74; N, 13.01. Found: C, 59.15; H, 8.19; N, 12.53. 1H NMR (400.30 MHz, [D8]THF, 298 K): δ [ppm]: 7.57–7.48 (m, 4 H, CH Ph), 7.39–7.37 (m, 1 H, CH Ph), 7.19–7.11 (m, residual CH toluene), 6.89 (dd, 3 J HH=8.4, 7.6 Hz, 1 H, p‐CH pyr), 5.49 (d, 3 J HH=8.4 Hz, 1 H, m‐CH pyr), 5.11 (d, 3 J HH=7.6 Hz, 1 H, m‐CH pyr), 3.15 (q, 3 J HH=6.9 Hz, 2 H, CH 2CH3), 3.08 (q, 3 J HH=6.9 Hz, 2 H, CH 2CH3), 2.30 (s, residual CH 3 toluene), 1.23 (t, 3 J HH=6.9 Hz, 3 H, CH2CH 3), 1.16 (s, 18 H, C(CH 3)3), 0.99 (t, 3 J HH=6.9 Hz, 3 H, CH2CH 3), 0.07 (s, 18 H, Si(CH 3)3). 1H NMR (400.30 MHz, C6D6, 343 K): δ [ppm]: 7.25–7.21 (m, 1 H, CH Ph), 7.12–6.98 (m, 5 H, CH Ph + p‐CH pyr + residual CH toluene), 6.12 (d, 3 J HH=8.1 Hz, 1 H, m‐CH pyr), 5.64 (d, 3 J HH=7.8 Hz, 1 H, m‐CH pyr), 3.67 (br m, 2 H, CH 2CH3), 3.11 (q, 3 J HH=6.9 Hz, 2 H, CH 2CH3), 2.13 (s, residual CH 3 toluene), 1.83 (br, 3 H, CH2CH 3), 1.27 (t, 3 J HH=6.7 Hz, 3 H, CH2CH 3), 1.09 (s, 18 H, C(CH 3)3), 0.37 (s, 18 H, Si(CH 3)3). 13C{1H} NMR (100.67 MHz, [D8]THF): δ [ppm]: 169.4 (o‐C pyr), 166.1 (NCN), 160.6 (o‐C pyr), 139.8 (p‐C pyr), 134.3 (C quat,Ph), 131.3 (C toluene), 131.2 (C toluene), 131.1 (CPh), 129.8 (CPh), 129.6 (CPh), 129.1 (C toluene), 129.0 (CPh), 129.0 (CPh), 127.0 (C toluene), 126.2 (C toluene), 95.6 (m‐C pyr), 86.4 (m‐C pyr), 53.8 (C(CH3)3), 42.1 (CH2CH3), 37.2 (CH2CH3), 31.7 (C(CH3)3), 21.7 (CH3 toluene), 16.5 (CH2 CH3), 16.3 (CH2 CH3), 7.2 (Si(CH3)3). 29Si{1H} NMR (79.52 MHz, [D8]THF): δ [ppm]: −9.0 (NSiN), −16.0 (SiMe3). IR (ATR): (cm−1)=3436 (w), 2963 (vs), 2929 (s, sh), 2900 (s), 2868 (s), 2837 (w, sh), 1647 (m), 1592 (vs, sh), 1577 (vs), 1558 (s), 1487 (s, sh), 1475 (s), 1457 (vs, sh), 1445 (vs), 1429 (s, sh), 1403 (m), 1387 (m), 1356 (m), 1333 (m), 1316 (m, sh), 1305 (m), 1252 (s), 1205 (w), 1189 (w, sh), 1174 (w, sh), 1155 (s), 1139 (w, sh), 1109 (w), 1072 (m), 1051 (m, sh), 1029 (s), 996 (m, sh), 969 (w, sh), 931 (m), 920 (m, sh), 879 (m), 845 (m, sh), 832 (s), 816 (s, sh), 777 (m), 757 (m, sh), 745 (m, sh), 723 (m), 705 (s), 683 (m), 661 (m), 617 (w), 587 (w), 514 (m), 482 (w), 465 (w), 429 (w). EI‐MS: m/z (%): 565.243 [M‐C4H9]+ (3), with the corresponding isotopic patterns.
Synthesis of [SiNSi‐Ca] (5): To a mixture of [SiNSi] (0.250 g, 0.367 mmol) and [Ca{N(SiMe3)2}2(THF)2] (0.184 g, 0.367 mmol) was condensed toluene (10 mL) at −78 °C. The mixture was allowed to warm to −30 °C, resulting in a yellow solution. After stirring at −30 °C for 1 h, the solution was placed in a freezer (−30 °C). Crystals suitable for X‐ray diffraction analysis were grown by slow evaporation of the toluene solution at −30 °C. The clear mother liquor was removed by cannula transfer and the crystals were dried in vacuo for 20 min affording the title product as a yellow solid. Yield (based on crystals): 0.169 g (0.162 mmol), 44 %. Anal. Calcd for C51H95CaN9Si6 (1042.97): C, 58.73; H, 9.18; N, 12.09. Found: C, 59.15; H, 8.19; N, 12.53. The compound is highly temperature sensitive (evolution above −30 °C). A 1H NMR experiment in tol‐d 8 at low temperature (−30 °C) was attempted but only revealed broad features. IR (ATR): (cm−1)=2991 (m, sh), 2966 (vs), 2931 (s, sh), 2900 (s), 2868 (m), 1647 (w), 1615 (m, sh), 1576 (vs), 1497 (w, sh), 1475 (m), 1459 (s, sh), 1440 (vs), 1410 (vs), 1392 (s, sh), 1363 (s), 1328 (m), 1307 (m), 1270 (m, sh), 1243 (vs), 1205 (s), 1178 (m, sh), 1153 (s), 1109 (w), 1098 (w), 1063 (s), 1036 (s), 1011 (s), 928 (m), 888 (m, sh), 873 (s), 820 (vs), 810 (s, sh), 783 (m), 768 (m), 748 (s), 724 (m), 706 (s), 660 (m), 641 (w, sh), 618 (m), 590 (m), 538 (w), 500 (w), 481 (w), 420 (w).
Reaction of [SiNSi‐Yb] (2) with THF: A J. Young NMR tube was charged with [SiNSi‐Yb] (2) (0.010 g, 0.009 mmol) and [D8]THF was condensed into this tube at −78 °C. The solution was warmed up to room temperature and an immediate color change from dark‐red to orange‐red was observed. Analysis of the 1H NMR spectrum clearly showed clean formation of the free [SiNSi] ligand and [Yb{N(SiMe3)2}2([D8]THF)n] (see Figure S16).
Reaction of [SiNSi‐Yb] (2) with NHCs (ITMe or IPr): Addition of ITMe (0.0042 g, 0.034 mmol) to [SiNSi‐Yb] (2) (0.020 g, 0.017 mmol) in C6D6 at room temperature resulted in a slight color change of the solution from dark‐red to dark‐purple. Analysis of the 1H and 13C{1H} NMR spectra revealed signals of free [SiNSi] ligand along with the characteristic signals of [(ITMe)2Yb{N(SiMe3)2}2] (see Figures S17‐S18). [18] In addition, the 29Si{1H} NMR spectrum confirmed de‐coordination of [SiNSi] as the signal at δ 18.5 ppm disappeared and signals corresponding to the free [SiNSi] ligand were observed at δ−14.0, −14.7 and −15.1 ppm (see Figure S19), in agreement with the reported signals. [10b]
Addition of IPr (0.010 g, 0.026 mmol) to [SiNSi‐Yb] (2) (0.010 g, 0.009 mmol) in C6D6 at room temperature did not lead to any obvious color change. Analysis of the 1H and 13C{1H} NMR spectra revealed that no reaction occurred between the two starting materials (see Figure S20). In conclusion, the [SiNSi] ligand is not displaced by the IPr ligand.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank Prof. Dr. D. Fenske for measuring the single crystals and the Karlsruhe Nano Micro Facility (KNMF) for measuring time. We also thank Ms. Helga Berberich for help with NMR measurements. Financial support by the DFG‐funded transregional collaborative research center SFB/TRR 88 “Cooperative Effects in Homo and Heterometallic Complexes (3MET)” project B3 is gratefully acknowledged. Open access funding enabled and organized by Projekt DEAL.
X. Sun, T. Simler, K. Reiter, F. Weigend, P. W. Roesky, Chem. Eur. J. 2020, 26, 14888.
References
- 1.
- 1a. Denk M., Lennon R., Hayashi R., West R., Belyakov A. V., Verne H. P., Haaland A., Wagner M., Metzler N., J. Am. Chem. Soc. 1994, 116, 2691; [Google Scholar]
- 1b. Hill N. J., West R., J. Organomet. Chem. 2004, 689, 4165. [Google Scholar]
- 2.
- 2a. Blom B., Gallego D., Driess M., Inorg. Chem. Front. 2014, 1, 134; [Google Scholar]
- 2b. Lipke M. C., Liberman-Martin A. L., Tilley T. D., Angew. Chem. Int. Ed. 2017, 56, 2260; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 2298; [Google Scholar]
- 2c. Qi X., Zheng T., Zhou J., Dong Y., Zuo X., Li X., Sun H., Fuhr O., Fenske D., Organometallics 2019, 38, 268; [Google Scholar]
- 2d. Zhou Y.-P., Driess M., Angew. Chem. Int. Ed. 2019, 58, 3715; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 3753. [Google Scholar]
- 3.
- 3a. Sen S. S., Khan S., Samuel P. P., Roesky H. W., Chem. Sci. 2012, 3, 659; [Google Scholar]
- 3b. Protchenko A. V., Vasko P., Do D. C. H., Hicks J., Fuentes M. Á., Jones C., Aldridge S., Angew. Chem. Int. Ed. 2019, 58, 1808; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 1822; [Google Scholar]
- 3c. Wang Y., Kostenko A., Hadlington T. J., Luecke M.-P., Yao S., Driess M., J. Am. Chem. Soc. 2019, 141, 626; [DOI] [PubMed] [Google Scholar]
- 3d. Li S., Wang Y., Yang W., Li K., Sun H., Li X., Fuhr O., Fenske D., Organometallics 2020, 39, 757; [Google Scholar]
- 3e. Yadav R., Simler T., Reichl S., Goswami B., Schoo C., Köppe R., Scheer M., Roesky P. W., J. Am. Chem. Soc. 2020, 142, 1190. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Denk M., Hayashi R. K., West R., J. Chem. Soc. Chem. Commun. 1994, 33; [Google Scholar]
- 4b. Gehrhus B., Hitchcock P. B., Lappert M. F., Maciejewski H., Organometallics 1998, 17, 5599; [Google Scholar]
- 4c. Schmedake T. A., Haaf M., Paradise B. J., Millevolte A. J., Powell D. R., West R., J. Organomet. Chem. 2001, 636, 17; [Google Scholar]
- 4d. Neumann E., Pfaltz A., Organometallics 2005, 24, 2008; [Google Scholar]
- 4e. Waterman R., Hayes P. G., Tilley T. D., Acc. Chem. Res. 2007, 40, 712; [DOI] [PubMed] [Google Scholar]
- 4f. Yang W., Fu H., Wang H., Chen M., Ding Y., Roesky H. W., Jana A., Inorg. Chem. 2009, 48, 5058; [DOI] [PubMed] [Google Scholar]
- 4g. Tavčar G., Sen S. S., Azhakar R., Thorn A., Roesky H. W., Inorg. Chem. 2010, 49, 10199; [DOI] [PubMed] [Google Scholar]
- 4h. Azhakar R., Sarish S. P., Roesky H. W., Hey J., Stalke D., Inorg. Chem. 2011, 50, 5039; [DOI] [PubMed] [Google Scholar]
- 4i. Azhakar R., Ghadwal R. S., Roesky H. W., Hey J., Stalke D., Chem. Asian J. 2012, 7, 528; [DOI] [PubMed] [Google Scholar]
- 4j. Azhakar R., Ghadwal R. S., Roesky H. W., Wolf H., Stalke D., J. Am. Chem. Soc. 2012, 134, 2423; [DOI] [PubMed] [Google Scholar]
- 4k. Brück A., Gallego D., Wang W., Irran E., Driess M., Hartwig J. F., Angew. Chem. Int. Ed. 2012, 51, 11478; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 11645; [Google Scholar]
- 4l. Blom B., Driess M., Gallego D., Inoue S., Chem. Eur. J. 2012, 18, 13355; [DOI] [PubMed] [Google Scholar]
- 4m. Wang W., Inoue S., Irran E., Driess M., Angew. Chem. Int. Ed. 2012, 51, 3691; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 3751; [Google Scholar]
- 4n. Schäfer S., Köppe R., Gamer M. T., Roesky P. W., Chem. Commun. 2014, 50, 11401; [DOI] [PubMed] [Google Scholar]
- 4o. Schäfer S., Köppe R., Roesky P. W., Chem. Eur. J. 2016, 22, 7127. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Cai X., Gehrhus B., Hitchcock P. B., Lappert M. F., Can. J. Chem. 2000, 78, 1484; [Google Scholar]
- 5b. Evans W. J., Perotti J. M., Ziller J. W., Moser D. F., West R., Organometallics 2003, 22, 1160; [Google Scholar]
- 5c. Zitz R., Arp H., Hlina J., Walewska M., Marschner C., Szilvási T., Blom B., Baumgartner J., Inorg. Chem. 2015, 54, 3306; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5d. Brackbill I. J., Douair I., Lussier D. J., Boreen M. A., Maron L., Arnold J., Chem. Eur. J. 2020, 26, 2360. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Pearson R. G., J. Am. Chem. Soc. 1963, 85, 3533; [Google Scholar]
- 6b. Pearson R. G., Inorg. Chim. Acta 1995, 240, 93. [Google Scholar]
- 7. Shannon R., Acta Crystallogr. Sect. A 1976, 32, 751. [Google Scholar]
- 8. Blom B., Klatt G., Gallego D., Tan G., Driess M., Dalton Trans. 2015, 44, 639. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. So C.-W., Roesky H. W., Magull J., Oswald R. B., Angew. Chem. Int. Ed. 2006, 45, 3948; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 4052; [Google Scholar]
- 9b. Sen S. S., Roesky H. W., Stern D., Henn J., Stalke D., J. Am. Chem. Soc. 2010, 132, 1123. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Ren H., Zhou Y.-P., Bai Y., Cui C., Driess M., Chem. Eur. J. 2017, 23, 5663; [DOI] [PubMed] [Google Scholar]
- 10b. Gallego D., Inoue S., Blom B., Driess M., Organometallics 2014, 33, 6885. [Google Scholar]
- 11.
- 11a. Gamer M. T., Roesky P. W., Konchenko S. N., Nava P., Ahlrichs R., Angew. Chem. Int. Ed. 2006, 45, 4447; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 4558; [Google Scholar]
- 11b. Arnold P. L., Liddle S. T., McMaster J., Jones C., Mills D. P., J. Am. Chem. Soc. 2007, 129, 5360; [DOI] [PubMed] [Google Scholar]
- 11c. Wiecko M., Roesky P. W., Organometallics 2007, 26, 4846; [Google Scholar]
- 11d. Jones C., Stasch A., Woodul W. D., Chem. Commun. 2009, 113; [DOI] [PubMed] [Google Scholar]
- 11e. Li S., Cheng J., Chen Y., Nishiura M., Hou Z., Angew. Chem. Int. Ed. 2011, 50, 6360; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 6484; [Google Scholar]
- 11f. Saleh L. M. A., Birjkumar K. H., Protchenko A. V., Schwarz A. D., Aldridge S., Jones C., Kaltsoyannis N., Mountford P., J. Am. Chem. Soc. 2011, 133, 3836; [DOI] [PubMed] [Google Scholar]
- 11g. Sanden T., Gamer M. T., Fagin A. A., Chudakova V. A., Konchenko S. N., Fedushkin I. L., Roesky P. W., Organometallics 2012, 31, 4331. [Google Scholar]
- 12. Simler T., Feuerstein T. J., Yadav R., Gamer M. T., Roesky P. W., Chem. Commun. 2019, 55, 222. [DOI] [PubMed] [Google Scholar]
- 13. Tilley T. D., Andersen R. A., Zalkin A., J. Am. Chem. Soc. 1982, 104, 3725. [Google Scholar]
- 14. Hitchcock P. B., Khvostov A. V., Lappert M. F., Protchenko A. V., J. Organomet. Chem. 2002, 647, 198. [Google Scholar]
- 15.
- 15a. Cordero B., Gómez V., Platero-Prats A. E., Revés M., Echeverría J., Cremades E., Barragán F., Alvarez S., Dalton Trans. 2008, 37, 2832; [DOI] [PubMed] [Google Scholar]
- 15b. Pyykkö P., Atsumi M., Chem. Eur. J. 2009, 15, 186; [DOI] [PubMed] [Google Scholar]
- 15c. Pyykkö P., J. Phys. Chem. A 2015, 119, 2326. [DOI] [PubMed] [Google Scholar]
- 16.
- 16a. Bochkarev L. N., Makarov V. M., Hrzhanovskaya Y. N., Zakharov L. N., Fukin G. K., Yanovsky A. I., Struchkov Y. T., J. Organomet. Chem. 1994, 467, C3; [Google Scholar]
- 16b. Zitz R., Hlina J., Gatterer K., Marschner C., Szilvási T., Baumgartner J., Inorg. Chem. 2015, 54, 7065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Niemeyer M., Inorg. Chem. 2006, 45, 9085. [DOI] [PubMed] [Google Scholar]
- 18. Yuan J., Hu H., Cui C., Chem. Eur. J. 2016, 22, 5778. [DOI] [PubMed] [Google Scholar]
- 19.
- 19a. Nelson D. J., Nolan S. P., Chem. Soc. Rev. 2013, 42, 6723; [DOI] [PubMed] [Google Scholar]
- 19b. Gómez-Suárez A., Nelson D. J., Nolan S. P., Chem. Commun. 2017, 53, 2650. [DOI] [PubMed] [Google Scholar]
- 20. Addison A. W., Rao T. N., Reedijk J., van Rijn J., Verschoor G. C., J. Chem. Soc. Dalton Trans. 1984, 1349. [Google Scholar]
- 21.
- 21a. Cabeza J. A., García-Álvarez P., González-Álvarez L., Chem. Commun. 2017, 53, 10275; [DOI] [PubMed] [Google Scholar]
- 21b. Mo Z., Kostenko A., Zhou Y.-P., Yao S., Driess M., Chem. Eur. J. 2018, 24, 14608. [DOI] [PubMed] [Google Scholar]
- 22. Deeken S., Motz G., Kempe R., Z. Anorg. Allg. Chem. 2007, 633, 320. [Google Scholar]
- 23.
- 23a. Qayyum S., Haberland K., Forsyth C. M., Junk P. C., Deacon G. B., Kempe R., Eur. J. Inorg. Chem. 2008, 557; [Google Scholar]
- 23b. Lyubov D. M., Cherkasov A. V., Fukin G. K., Ketkov S. Y., Shavyrin A. S., Trifonov A. A., Dalton Trans. 2014, 43, 14450; [DOI] [PubMed] [Google Scholar]
- 23c. Ku K. W., Au C. W., Chan H.-S., Lee H. K., Dalton Trans. 2013, 42, 2841. [DOI] [PubMed] [Google Scholar]
- 24. Sen S. S., Jana A., Roesky H. W., Schulzke C., Angew. Chem. Int. Ed. 2009, 48, 8536; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 8688. [Google Scholar]
- 25. Morss L. R., Chem. Rev. 1976, 76, 827. [Google Scholar]
- 26. Harder S., Angew. Chem. Int. Ed. 2004, 43, 2714; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2004, 116, 2768. [Google Scholar]
- 27. Sen S. S., Hey J., Herbst-Irmer R., Roesky H. W., Stalke D., J. Am. Chem. Soc. 2011, 133, 12311. [DOI] [PubMed] [Google Scholar]
- 28.
- 28a. Ortu F., Moxey G. J., Blake A. J., Lewis W., Kays D. L., Inorg. Chem. 2013, 52, 12429; [DOI] [PubMed] [Google Scholar]
- 28b. Loh C., Seupel S., Koch A., Görls H., Krieck S., Westerhausen M., Dalton Trans. 2014, 43, 14440; [DOI] [PubMed] [Google Scholar]
- 28c. Lapshin I. V., Yurova O. S., Basalov I. V., Rad'kov V. Y., Musina E. I., Cherkasov A. V., Fukin G. K., Karasik A. A., Trifonov A. A., Inorg. Chem. 2018, 57, 2942. [DOI] [PubMed] [Google Scholar]
- 29.
- 29a.TURBOMOLES V7.4 2020, a development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989–2007, TURBOMOLE GmbH, since 2007; available from http://www.turbomole.com;
- 29b. Balasubramani S. G., Chen G. P., Coriani S., Diedenhofen M., Frank M. S., Franzke Y. J., Furche F., Grotjahn R., Harding M. E., Hättig C., Hellweg A., Helmich-Paris B., Holzer C., Huniar U., Kaupp M., Marefat Khah A., Karbalaei Khani S., Müller T., Mack F., Nguyen B. D., Parker S. M., Perlt E., Rappoport D., Reiter K., Roy S., Rückert M., Schmitz G., Sierka M., Tapavicza E., Tew D. P., van Wüllen C., Voora V. K., Weigend F., Wodyński A., Yu J. M., J. Chem. Phys. 2020, 152, 184107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Perdew J. P., Ernzerhof M., Burke K., J. Chem. Phys. 1996, 105, 9982. [Google Scholar]
- 31. Weigend F., Ahlrichs R., Phys. Chem. Chem. Phys. 2005, 7, 3297. [DOI] [PubMed] [Google Scholar]
- 32. Caldeweyher E., Ehlert S., Hansen A., Neugebauer H., Spicher S., Bannwarth C., Grimme S., J. Chem. Phys. 2019, 150, 154122. [DOI] [PubMed] [Google Scholar]
- 33. McQuarrie D. A., Simon J. D., Molecular Thermodynamics, University Science Books, 1999. [Google Scholar]
- 34. Schröder-Holzhacker C., Stöger B., Pittenauer E., Allmaier G., Veiros L. F., Kirchner K., Monatsh. Chem. 2016, 147, 1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary