

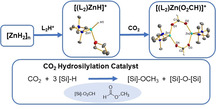
Abstract
Protonolysis of [ZnH2]n with the conjugated Brønsted acid of the bidentate diamine TMEDA (N,N,N′,N′‐tetramethylethane‐1,2‐diamine) and TEEDA (N,N,N′,N′‐tetraethylethane‐1,2‐diamine) gave the zinc hydride cation [(L2)ZnH]+, isolable either as the mononuclear THF adduct [(L2)ZnH(thf)]+[BArF 4]− (L2=TMEDA; BArF 4 −=[B(3,5‐(CF3)2‐C6H3)4]−) or as the dimer [{(L2)Zn)}2(μ‐H)2]2+[BArF 4]− 2 (L2=TEEDA). In contrast to [ZnH2]n, the cationic zinc hydrides are thermally stable and soluble in THF. [(L2)ZnH]+ was also shown to form di‐ and trinuclear adducts of the elusive neutral [(L2)ZnH2]. All hydride‐containing cations readily inserted CO2 to give the corresponding formate complexes. [(TMEDA)ZnH]+[BArF 4]− catalyzed the hydrosilylation of CO2 with tertiary hydrosilanes to give stepwise formoxy silane, methyl formate, and methoxy silane. The unexpected formation of methyl formate was shown to result from the zinc‐catalyzed transesterification of methoxy silane with formoxy silane, which was eventually converted into methoxy silane as well.
Keywords: carbon dioxide, chemoselective catalysis, hydrosilylation, Lewis acid, zinc hydride
Insoluble, thermally unstable zinc dihydride [ZnH2]n can be converted into robust zinc hydride cations [(L2)ZnH]+ which catalyze the homogeneous hydrosilylation of CO2 to give methoxy silane. Ligand L2 can be as simple as chelating TMEDA.
Introduction
The hydrosilylation of CO2 is catalyzed by a variety of transition metal‐ [1] and non‐metal‐based [2] catalysts to give formoxy silane, bis(silyl)acetal, methoxy silane, and methane. [3] The chemoselectivity appears to be determined by the Lewis acidity of the catalyst. A transparent structure‐selectivity relationship, however, remains unavailable, since both the nature of the hydrosilane and the activation mode of CO2 play critical roles during catalysis. By virtue of its pronounced Lewis acidity, molecular zinc compounds supported by chelating ligands have been successfully utilized as chemoselective catalysts for the hydrosilylation of CO2. [4] Most catalysts are neutral as the result of employing mono‐anionic LnX‐type ligands,[ 4a , 4b , 4c , 5 ] whereas cationic zinc hydrides remain relatively scarce. Assuming increased Lewis acidity due to the positive cationic charge, Rivard et al. showed improved activity of the zinc hydride cation precursor [(IPr)ZnH(THF)(OTf)] in ketone hydrosilylation when compared to its neutral analogue [(IPr)ZnH(μ‐H)]2 (IPr=1,3‐bis(2,6‐diisopropylphenyl)imidazol‐2‐ylidene) (Figure 1). [6] The presence of Lewis acids such as boranes was found to improve the reaction rate and to influence the degree of CO2 hydrosilylation, suggesting that zinc's inherent electrophilicity may not be sufficient on its own.[ 5 , 7 ] The optimum coordination number of a highly electrophilic zinc center can be conceived to be lower than four with the steric bulk of the ancillary ligand as small as possible.[ 4 , 5 , 7 , 12b ]
Figure 1.
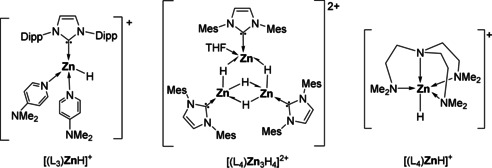
Cationic zinc hydrides reported in the literature.[ 6 , 7 , 8 ]
Here we report that protonolysis of zinc dihydride [9] with the conjugated Brønsted acid of the simple bidentate diamine TMEDA (N,N,N′,N′‐tetramethylethane‐1,2‐diamine) and TEEDA (N,N,N′,N′‐tetraethylethane‐1,2‐diamine), the zinc hydride cation [(L2)ZnH]+ and its derivatives become accessible. Using the non‐nucleophilic Kobayashi anion BArF 4 − (BArF 4 −=[B(3,5‐(CF3)2‐C6H3)4]−) was critical to obtain quasi tricoordinate zinc hydride cations, since other tetraarylborates were decomposed. [10] Hydrosilylation of CO2 was catalyzed by [(L2)ZnH]+ to give methoxy silane with formoxy silane and methyl formate as intermediates.
Results and Discussion
Synthesis and Structure of Zinc Hydride Cations
Zinc hydride cations 2 a and 2 b derived from [(L2)ZnH]+ were prepared by treating a suspension of [ZnH2]n [9] in THF at room temperature with the conjugated Brønsted acid of the bidentate diamine L2 1 a and 1 b (L2=TMEDA, TEEDA) (Scheme 1). Both complexes 2 a and 2 b are soluble in organic solvents such as THF or CH2Cl2, show no sign of decomposition even at 70 °C in solution, and were isolated as colorless crystals in practically quantitative yields. Notably, reacting [ZnH2]n with neat TMEDA or TEEDA did not afford any isolable compounds, [11] but rather resulted in the gradual decomposition of [ZnH2]n into metallic zinc and dihydrogen. The 1H NMR spectrum of 2 a in [D8]THF exhibits one set of two sharp singlets for the TMEDA ligand along with resonances of the borate anion in 1:1 ratio. Even after prolonged drying under vacuum THF could not be removed entirely. The zinc hydride resonance in 2 a was found as a singlet at δ=3.68 ppm in [D8]THF and as a broad singlet at δ=3.56 ppm in CD2Cl2 indicating fluxional THF coordination in solution. Generally, the resonance for terminal zinc hydrides is expected in the chemical shift region between 3 to 5 ppm,[ 6 , 8 ] whereas that for bridging zinc hydrides are usually shifted by 1 to 2 ppm toward higher field. [12]
Scheme 1.
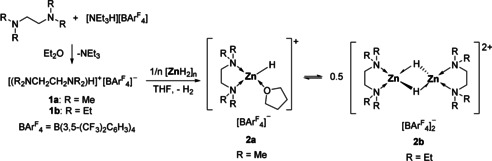
Synthesis of the zinc hydride cations 2 a and 2 b.
The 1H NMR spectra of 2 b in [D8]THF shows a set of signals for the TEEDA ligand in agreement with D 2h symmetry in addition to those for the borate anion. The CH2 resonances of the ligand backbone are observed as a singlet at δ=2.96 ppm, whereas the diastereotopic CH2 signals of the ethyl groups appear as two sets of multiplets of an A3BB′ spin system. The zinc hydride resonance is observed as a singlet at δ=3.73 ppm in [D8]THF and δ=4.28 ppm in CD2Cl2 respectively, in agreement with the presence of a dissociative equilibrium.
Single crystals of 2 a and 2 b suitable for X‐ray diffraction were obtained from CH2Cl2/n‐pentane solution at −30 °C (Figure 2). The molecular structure of 2 a confirms a monomeric, cationic zinc center tetrahedrally coordinated by a terminal hydrido ligand, one TMEDA and one THF ligand. The Zn−H bond length is 1.55(4) Å, comparable to 1.59(3) Å reported for [(Me6TREN)ZnH]+[BArF 4]− (Me6TREN=tris{2‐(dimethylamino)ethyl}amine). [7a] The solid state structure of 2 b reveals a dimeric structure. Both tetrahedrally coordinated zinc centers are bridged by two μ‐hydrido ligands and separated by 2.4397(4) Å. The Zn1‐H1 distance of 2.04(3) Å and the Zn1‐H1′ bond length of 2.06(4) Å are in the expected range. [12] Although the bias toward dimerization is pronounced when the sterically more encumbered TEEDA was used, we believe that a monomer‐dimer equilibrium exists in THF solution (Scheme 1).
Figure 2.
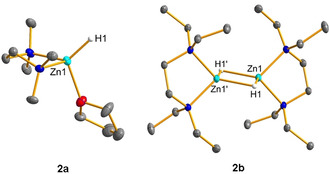
Cation portion in 2 a (left) and 2 b (right). Displacement parameters are set at 50 % probability. The borate anion and hydrogen atoms except of H1 in 2 a and H1 and H1′ in 2 b are omitted for clarity.
As coordinatively unsaturated zinc hydrides tend to aggregate to form clusters,[ 8 , 12 , 13 ] the Lewis acidic fragments [(L2)ZnH]+ 2 a and 2 b could be used to deaggregate polymeric [ZnH2]n. Treating a THF suspension of [ZnH2]n in the presence of one equivalent of TMEDA with 2 a at room temperature resulted in a clear solution within a few minutes. Depending on the stoichiometry, cationic zinc hydrides 3 and 4 were formed. They can also be synthesized directly from [ZnH2]n, using the appropriate amount of 1 and L2 (Scheme 2).
Scheme 2.
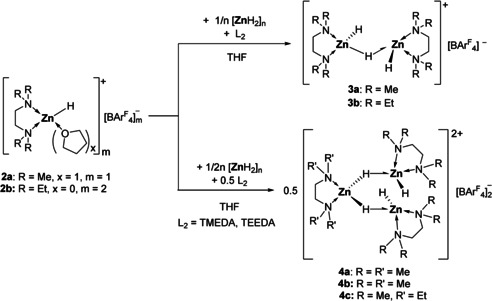
Formation of the cationic zinc hydrides 3 and 4.
Dinuclear 3 a is soluble in THF and CH2Cl2 and in contrast to 2 a and 2 b, sparingly soluble in benzene or toluene. Compound 3 a is stable at room temperature for at least two days but unlike 2 a and 2 b decomposes at elevated temperatures. Analogously, hydride cation 3 b was obtained from 2 b and TEEDA. The 1H NMR spectra in [D8]THF of 3 a and 3 b show the signals of the diamine and the borate anion in the expected region in a 2:1 ratio. At room temperature, no distinction is possible between the terminal and bridging hydrides: the hydride resonance is found as a singlet at δ=3.68 ppm. VT 1H NMR spectra of 3 a in CD2Cl2 (see SI) revealed splitting of the hydride resonance into two singlets in a 2:1 ratio and two sets for the TMEDA ligand at −50 °C. A formal combination of [ZnH]+ and [ZnH2]n agrees well with the structure obtained in the solid state. Single crystals of 3 a suitable for X‐ray diffraction were obtained from CH2Cl2/n‐hexane solution at −30 °C (Figure 3). In the dinuclear structure the two [(L2)ZnH]+ units are bridged by a single μ‐hydrido ligand. Both tetrahedral zinc centers are separated by 2.7860(7) Å. The terminal Zn−H bond lengths of Zn1‐H1 1.53(4) Å and Zn2‐H3 1.59(4) Å are in the same range as those determined in 2 a. The bridging hydride bonds are given as Zn1‐H2 1.71(4) Å and Zn2‐H2 1.62(4) Å, respectively. Conventionally, two metal centers bridged by three hydrides as in [(Me5TRENCH2)Lu(μ‐H)3Lu(Me6TREN)][BArF 4]2 [14] or [(triphos)Rh(μ‐H)3Rh(triphos)]BPh4 (triphos=1,1,1‐tris(diphenylphosphinomethyl)ethane) [15] are observed in a [M2H3] motif. A single bridging hydride in between two zinc centers remain hitherto unknown. Only few examples such as [(dippe)2Ni2H3][BPh4] (dippe=1,2‐bis(diisopropylphosphanyl)ethane) [16] and [{(tBu)2P(CH2)3P(tBu)2}2Pt2H3][BPh4] [17] were reported to feature this bridging mode.
Figure 3.
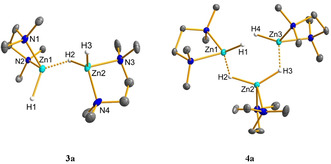
Molecular cations in 3 a (left) and 4 a (right). Displacement parameters are set at 50 % probability. The borate anion and hydrogen atoms except of H1, H2, H3 in 3 a and H1, H2, H3, H4 in 4 a are omitted for clarity.
It was possible to coordinate the zinc cation [(L2)ZnH]+ to both terminal hydrides of the putative neutral [(L2)ZnH2]. The reaction of 2 a with 0.5 equivalent of [ZnH2]n and TMEDA gave the cationic trinuclear zinc hydride 4 a in quantitative yield. Zinc hydride clusters 4 show good solubility in THF and CH2Cl2 as well as thermal stability up to 70 °C in solution. The 1H NMR spectrum displays signals for the ligand and the borate anion in a 3:2 ratio. The zinc hydride appears as a single resonance at δ=3.62 ppm. As observed for 3, it was not possible to distinguish between terminal and bridging hydrides at room temperature. Single crystals of 4 a suitable for X‐ray diffraction were obtained from a CH2Cl2/n‐pentane solution at −30 °C (Figure 3). In the solid state, the C 2‐symmetrical trinuclear zinc hydride dication consists of a [(L2)ZnH2] unit connected to two [(L2)ZnH]+ fragments by a μ‐H bridge (L2=TMEDA). The terminal Zn−H bond lengths of Zn1‐H1 1.60(3) and Zn3‐H4 1.59(3) Å are in the same range as the terminal zinc hydride bond found in 2 a, whereas Zn1‐H2 1.75(3) and Zn3‐H3 1.80(3) Å appear to be significantly longer than Zn2‐H2 1.61(3) and Zn2‐H3 1.64(3) Å. In contrast to the previously reported [Zn3H4]2+ core found in [(IMes)3Zn3H4(thf)][(BPh4)]2, which can be regarded as an adduct of neutral [(IMes)ZnH2]2 with dicationic [(IMes)Zn(thf)][BPh4]2,[ 7 , 8 ] complex 4 a can be considered as an adduct of [(TMEDA)ZnH2] coordinated to two [(TMEDA)ZnH]+ cations. The terminal zinc hydrides in 4 a can be attributed to a strong repulsion originated in both cationic zinc centers as found in [(IPr)ZnH(thf)(OTf)]. [6] When 2 a was reacted with [ZnH2]n and TEEDA, the mixed ligand trinuclear cation 4 c was isolated as the sole product. NMR spectroscopy as well as single crystal X‐ray diffraction (see SI) revealed an identical structural motif in both solution and solid state as compared to 4 a. Notably even at elevated temperatures (70 °C) was a ligand exchange not observed, indicating that the neutral [(TEEDA)ZnH2] in 4 originating from [ZnH2]n is kinetically inert.
To gain more insight into the electronic structure and nature of the zinc hydride bonds in 2–4, DFT and NBO calculations were performed (see SI). In 2 a, the Wiberg index for the Zn1‐H1 bond of 0.77 confirms a terminal hydride in the classical sense. For the optimized dimeric structure in 2 b much smaller Wiberg indices of 0.38 describe the bonding between Zn2 and the bridging hydrides (Zn2‐H1 and Zn2‐H2). Donation of these bonds to Zn1 (283.9 kcal mol−1 for Zn2‐H1→Zn1 and 266.5 kcal mol−1 for Zn2‐H2→Zn1) confirms that each Zn‐H‐Zn unit involves a 3c‐2e bond. Compound 3 a which is based on a [Zn2H3]+ fragment contains two terminal hydrides with Wiberg bond indices for Zn1‐H1 and Zn2‐H3 of 0.78, as expected. Considering the bridging hydride along Zn1‐H2 and Zn2‐H2, indices of 0.37 are obtained. A second order study demonstrates that the σ electron lone pair at H2 donates to the empty sp orbitals of both zinc centers, contributing 82 kcal mol−1 to Zn1 and 79 kcal mol−1 to Zn2. This is in line with a 3c‐2e bond along the Zn1‐H2‐Zn2 motif. The terminal hydride bonds Zn1‐H1 and Zn3‐H4 of the cation [Zn3H4]2+ in 4 a can again be regarded as classical hydride bonds with Wiberg indices of each 0.7. The bonding along the Zn2‐H3‐Zn3 and Zn2‐H2‐Zn1 motif differs from the situation found in 3 a. The Wiberg indices are given as 0.4 for the Zn2‐H2 and Zn2‐H3 bond and as 0.29 between Zn1‐H2 and Zn3‐H3, respectively. Second order analysis reveals a contribution of 76 kcal mol−1 of the σ electron lone pair at H2 to the empty sp orbital at Zn1, just as the s electron pair at H3 donates to the empty sp orbital at Zn3. This donation from the bridging hydrides to the empty sp orbitals of the cationic units weakens the Zn−H bonds on Zn2. Consequently, the cationic motif should result in significant Lewis acidity and 4 a can be considered as an adduct of neutral [ZnH2] stabilized by two [ZnH]+ cations.
Reaction of Zinc Hydride Cations with CO2
The cationic hydrides 2–4 readily reacted with CO2 (1 bar) in THF at room temperature to give formate complexes 5 a,b and 6 a,b in quantitative yields (Scheme 3). The 1H, 13C, 11B and 19F NMR resonances of the ligand and the anion in 5 a,b and 6 a,b are found in the expected regions. The formate proton in 5 a and 6 a was detected as a singlet at δ=8.23 and 8.26 ppm in the 1H NMR spectrum as well as by a single resonance at 170.06 and 170.07 ppm in the 13C NMR spectrum, respectively. Single crystal X‐ray diffraction of 5 a revealed a dimeric structure where two [(TMEDA)Zn(O2CH)]+ units are bridged by two formate ligands in a μ‐κ2‐O2CH fashion (Figure 4). The NMR spectroscopic data and structural parameters are comparable to those of the dimeric zinc formates [(MesBDI)Zn(O2CH)]2 [18] and [(DIPPBDI)Zn(O2CH)]2. [4b]
Scheme 3.
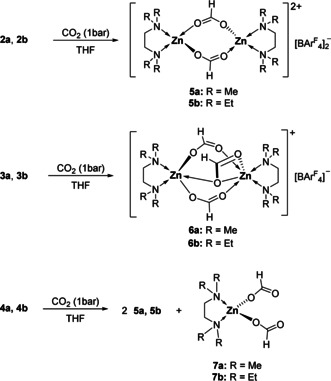
Reaction of zinc hydrides 2–4 with carbon dioxide.
Figure 4.
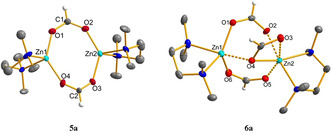
Molecular cations in 5 a (left) and 6 a (right). Displacement parameters are set at 50 % probability. The borate anion and hydrogen atoms except of the formate hydrogen are omitted for clarity.
Single crystals of 6 a suitable for X‐ray diffraction were obtained from a CH2Cl2/n‐pentane solution at −30 °C (Figure 4). The molecular structure of 6 a revealed a dimeric structure in which two [(TMEDA)Zn]+ ions are bridged by three formato ligands. In contrast to 5 a the Zn1 center is embedded in a square pyramidal coordination environment, whereas Zn2 shows a six‐coordinate zinc center with a distorted octahedral geometry. Zinc center Zn2 contains a μ‐κ2‐formate ligand. Six‐coordinate zinc centers are observed when small monodentate ligands are used or in the solid state structures of zinc‐containing MOFs.[ 13e , 19 ] Reaction of 4 with CO2 unexpectedly gave a mixture of doubly bridged 5 and neutral diformate 7. Compounds 7 a and 7 b were isolated from the reaction mixture and could also be synthesized independently from [ZnH2]n, diamine L2, and CO2 (see SI). They were characterized by NMR and IR spectroscopy. Single crystal X‐ray diffraction of 7 a confirmed the tetrahedral molecular structure with two κ1‐formate ligands. When the mixed ligand complex 4 c was exposed to CO2 atmosphere, a mixture of formates 5 a and 7 b was formed in a 2:1 ratio. Neither 5 b nor 7 a was obtained, suggesting that scrambling of the L2 ligands did not occur.
Hydrosilylation of CO2
The facile reaction of the cationic zinc hydrides toward CO2 prompted us to test their activity in the catalytic hydrosilylation of CO2. To convert CO2 into formoxy silane, silylacetals, and methoxy silane by zinc‐catalyzed hydrosilation, PhSiH3 or (EtO)3SiH have predominantly been used.[ 4b , 4d , 20 ] In addition to uncontrollable chemoselectivity, aryl and methoxy substituted silanes undergo scrambling of silicon substituents. [21] Therefore we applied tertiary alkyl‐substituted silanes such as nBuMe2SiH or Et3SiH which are more likely to be inert toward this exchange. [22] The CO2 hydrosilylation using these hydrosilanes was benchmarked by using 2 mol % of 2 a or 2 b in THF at 70 °C (Table 1).
Table 1.
Hydrosilane and catalyst screening for the hydrosilylation of CO2.[a]
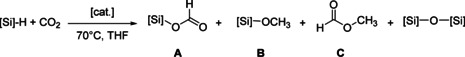
|
Entry[a] |
Hydrosilane |
Cat. [mol %] |
Conv. [%][b] |
Time [h] |
Selec. [%][b,e] A/B/C |
|---|---|---|---|---|---|
|
1 |
nBuMe2SiH |
2 a (2.0) |
>99 |
24 |
57/15/28 |
|
2 |
nBuMe2SiH |
2 b (2.0) |
>99 |
24 |
56/22/22 |
|
3 |
EtMe2SiH |
2 a (2.0) |
>99 |
16 |
63/11/26 |
|
4 |
Et3SiH |
2 a (2.0) |
66 |
48 |
93/3/4 |
|
5[c] |
nBuMe2SiH |
2 a (2.0) |
89 |
96 |
80/20/0 |
|
6 |
nBuMe2SiH |
[ZnH2]n (2.0) |
– |
24 |
– |
|
7[d] |
nBuMe2SiH |
[ZnH2]n (2.0) |
– |
24 |
– |
|
8 |
nBuMe2SiH |
[NEt3H][BArF 4] (10.0) |
– |
60 |
– |
|
9 |
nBuMe2SiH |
[Zn(OTf)2] (10.0) |
54 |
60 |
100/0/0 |
|
10 |
nBuMe2SiH |
BArF 18 (2.0) |
– |
24 |
– |
[a] n(hydrosilane)=0.18 mmol, 1 bar CO2, 0.5 mL of [D8]THF. [b] Conversion and selectivity determined by 1H NMR spectroscopy using 0.03 mM of hexamethylbenzene as internal standard. [c] Addition of 2 mol % of BPh3. [d] Addition of 1 equiv of TMEDA. [e] Disiloxane was formed in amounts corresponding to the overall stoichiometry (see Scheme 4).
When 2 a was used as catalyst, full conversion of nBuMe2SiH was achieved after 24 h. nBuMe2Si(O2CH) was identified as the major product in 57 % yield along with with 15 % of nBuMe2Si(OCH3) (Table 1, entry 1). Unexpectedly, apart from formoxy silane and methoxy silane methyl formate was detected in 28 % yield by 1H NMR spectroscopy (δ=3.66 (d,4 J H‐H=0.7 Hz), 8.02 (quart., 4 J H‐H=0.7 Hz) ppm).[ 23 , 24 ] When 2 b was used as catalyst, CO2 was hydrosilylated by nBuMe2SiH with comparable selectivity as using 2 a (Table 1, entry 2). Employing EtMe2SiH, full conversion of the hydrosilane was achieved after 16 h (Table 1, entry 3) with 63 % of formoxy silane, 11 % of methoxy silane, and 26 % of methyl formate. When sterically more demanding Et3SiH was used instead, the conversion rate was reduced (Table 1, entry 4), with chemoselectivity comparable to that when nBuMe2SiH or EtMe2SiH was used. Hydrosilanes commonly used in CO2 hydrosilylation such as PhSiH3 and (EtO)3SiH gave only poor or no conversion. Although boranes such as BPh3 can catalyze the reduction of CO2 to formoxy silane on its own, [2e] it has recently been shown that the activity of zinc hydrides can also be improved by the addition of boranes.[ 7a , 20 ] When a mixture containing 2 a (2 mol %) and BPh3 (2 mol %) was used as catalyst, activity and chemoselectivity were reduced giving formoxy silane as the major product only after 96 h (Table 1, entry 5). Methoxy silane was detected in 20 % yield, but in contrast to applying 2 a as the sole catalyst, methyl formate was formed only in trace amounts. Polymeric zinc dihydride showed no activity either with or without added TMEDA (Table 1, entry 6, 7). Methyl formate is not formed by Brønsted or Lewis acid catalysts, as employing [NEt3H][BArF 4] showed no conversion (Table 1, entry 8), while [Zn(OTf)2] gave exclusively formoxy silane (Table 1, entry 9). B(3,5‐(CF3)2‐C6H3)3 decomposed under the conditions of catalysis (Table 1, entry 10).
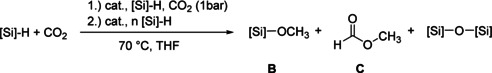
The chemoselective hydrosilylation of CO2 to give methoxy silane can be achieved in a two step one pot reaction at 70 °C (Table 2). Monitoring the reaction by NMR spectroscopy showed rapid formation of formoxy silane, whereas formation of methoxy silane and methyl formate was slow in the presence of CO2. When the remaining CO2 was removed and catalyst as well as hydrosilane were added, full consumption of the formoxy silane and increase in methyl formate concentration were observed. Using nBuMe2SiH as hydrosilane, a mixture containing 73 % of methoxy silane and 27 % of methyl formate was obtained after 16 h (Table 2, entry 1). When EtMe2SiH was applied instead, selectivity toward methoxy silane was slightly higher and the reaction time was reduced to 8 h (Table 2, entry 2). Only trace amounts of silylacetal are detected. When the reaction mixture was treated with an excess of hydrosilane after removal of CO2 and catalyst (2 equiv of hydrosilane, 4 mol % of catalyst), full conversion into methoxy silane and disiloxane was observed (Table 2, entry 3 and 4).
Table 2.
Catalytic hydrosilylation of CO2.
|
Entry[a] |
Hydrosilane |
Cat.[mol %] |
Conv. [%][b] |
Time [h] |
Selec. [%][b,e] B/C |
|---|---|---|---|---|---|
|
1[c] |
nBuMe2SiH |
2 a (4.0) |
>99 |
16 |
73/27 |
|
2[c] |
EtMe2SiH |
2 a (4.0) |
>99 |
8 |
62/38 |
|
3[d] |
nBuMe2SiH |
2 a (4.0) |
>99 |
24 |
>99/<1 |
|
4[d] |
EtMe2SiH |
2 a (4.0) |
>99 |
14 |
>99/<1 |
[a] n(hydrosilane)=0.18 mmol, 1 bar CO2, 0.5 mL of [D8]THF. [b] Conversion and selectivity determined by 1H NMR spectroscopy using 0.06 mM of hexamethylbenzene as internal standard. [c] Addition of 1 equiv, n(hydrosilane)=0.18 mmol and 4 mol % of catalyst in step 2. [d] Addition of 2 equiv, n(hydrosilane)=0.36 mmol and 4 mol % of catalyst in step 2. [e] Disiloxane was formed in amounts corresponding to the overall stoichiometry (see Scheme 4).
Consistent with the mechanism suggested in the literature, [3] fast insertion of CO2 into the zinc hydride bond and subsequent metathesis of zinc formate with hydrosilane forms formoxy silane. The latter is subsequently reduced to the silylacetal and eventually to methoxy silane and disiloxane. Notably, the formation of methyl formate in the initial phase of catalysis has not been reported. Methyl formate was found when formoxy silane underwent nucleophilic substitution with methanol using a ruthenium phosphine catalyst. [25] To gain more insight into the chemoselectivity, we probed the individual steps by stoichiometric reactions (Scheme 4). When the hydride cation 2 a was treated with nBuMe2Si(O2CH), an exchange reaction was observed giving the zinc formate cation 5 a and nBuMe2SiH. The nBuMe2SiH thus formed immediately reacted with 5 a to give methoxy silane, methyl formate, and disiloxane. In contrast, methoxy silane remained inert to such an exchange under the same conditions. When a mixture of isolated formoxy silane and methoxy silane was reacted in the presence of either 2 a or 5 a methyl formate and disiloxane were formed. In the absence of zinc catalysts formoxy silane and methoxy silane did not react even at elevated temperatures. We therefore suggest that methyl formate is produced by the nucleophilic attack of formoxy silane by methoxy silane, catalyzed by the Lewis acidic zinc cation (Scheme 4 c). When excess hydrosilane was present the formation of methoxy silane and disiloxane was observed exclusively (Table 2, entry 3 and 4), because methyl formate is known to form methoxy silane by hydrosilane in the presence of a suitable catalyst. [26] Therefore the cationic zinc hydride can act as a hydrosilylation catalyst to convert methyl formate into methoxy silane as soon as formoxy silane is fully consumed (Scheme 4 c). Formally methyl formate can be regarded as the equivalent of bis(silyl)acetal,[ 5 , 20 ] which was only detected in trace amounts under the catalytic conditions studied. We assume that the zinc hydride cation forms a zinc acetal [Zn]‐OCH2OCH3. This intermediate undergoes further attack by hydrosilane to give methoxy silane, disiloxane, and zinc hydride cation.
Scheme 4.
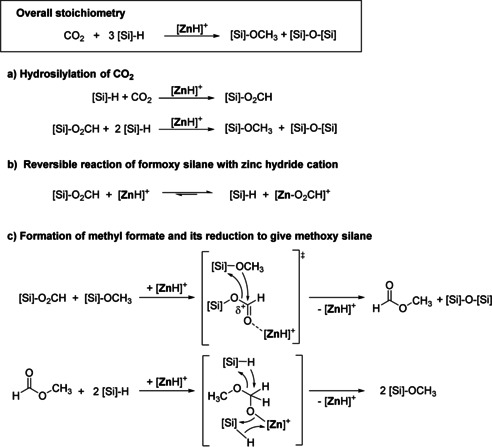
Catalytic hydrosilylation of CO2 by cationic zinc hydride [ZnH]+.
Conclusion
In conclusion, we have demonstrated that by using simple diamines TMEDA and TEEDA as supporting donor ligands, the quasi‐tricoordinate cationic zinc hydrides [(L2)ZnH]+ can be isolated as mono‐ or dinuclear complexes 2 and structurally characterized. Interestingly, [(L2)ZnH]+ forms adducts 3 and 4 with the hypothetical neutral molecular zinc dihydride [(L2)ZnH2]. Although the solid state structure of parent [ZnH2]n still remains unknown, the accessibility of [(L2)ZnH]+ facilitates the de‐aggregation of [ZnH2]n, resulting in molecular zinc hydrides of improved thermal stability and solubility. In agreement with the facile CO2 insertion into the Zn−H bond, the hydrosilylation of CO2 using hydosilanes to give methoxy silane are catalyzed by [ZnH]+ but not by [ZnH2]n itself. [13e] As a hitherto unknown intermediate of zinc‐catalyzed CO2 hydrosilylation, methyl formate has been identified. It results from the zinc‐catalyzed transesterification of kinetically preferred formoxy silane by methoxy silane and is eventually hydrosilylated to give methoxy silane as the thermodynamic product. As recently noted by Hazari et al. in a comprehensive study on the various effects influencing the chemoselectivity of group 10 metal‐catalyzed hydroboration of CO2, [27] the Lewis acidity of the metal center is critical but not the sole factor. With the access of [(L2)ZnH]+, a comparison of various homogeneous catalyst based on 3d metals [1] can be performed and the question addressed whether d‐electron configurations affect Lewis acidity. [4l]
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
The research was funded by the Deutsche Forschungsgemeinschaft. We thank Dr. G. Fink for NMR measurements and Profs. A. Venugopal and M. Ingleson for valuable discussions. Open access funding enabled and organized by Projekt DEAL.
F. Ritter, T. P. Spaniol, I. Douair, L. Maron, J. Okuda, Angew. Chem. Int. Ed. 2020, 59, 23335.
References
- 1.
- 1a. Koinuma H., Kawakami F., Kato H., Hirai H., J. Chem. Soc. Chem. Commun. 1981, 213–214; [Google Scholar]
- 1b. Deglmann P., Ember E., Hofmann P., Pitter S., Walter O., Chem. Eur. J. 2007, 13, 2864–2879; [DOI] [PubMed] [Google Scholar]
- 1c. Lalrempuia R., Iglesias M., Polo V., Sanz Miguel P. J., Fernández-Alvarez F. J., Pérez-Torrente J. J., Oro L. A., Angew. Chem. Int. Ed. 2012, 51, 12824–12827; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 12996–12999; [Google Scholar]
- 1d. Metsänen T. T., Oestreich M., Organometallics 2015, 34, 543–546; [Google Scholar]
- 1e. Scheuermann M. L., Semproni S. P., Pappas I., Chirik P. J., Inorg. Chem. 2014, 53, 9463–9465; [DOI] [PubMed] [Google Scholar]
- 1f. Li H., Gonçalves T. P., Zhao Q., Gong D., Lai Z., Wang Z., Zheng J., Huang K.-W., Chem. Commun. 2018, 54, 11395–11398; [DOI] [PubMed] [Google Scholar]
- 1g. Mazzotta M. G., Xiong M., Abu-Omar M. M., Organometallics 2017, 36, 1688–1691; [Google Scholar]
- 1h. Ríos P., Díez J., López-Serrano J., Rodríguez A., Conejero S., Chem. Eur. J. 2016, 22, 16791–16795; [DOI] [PubMed] [Google Scholar]
- 1i. Takaya J., Iwasawa N., J. Am. Chem. Soc. 2017, 139, 6074–6077; [DOI] [PubMed] [Google Scholar]
- 1j. Zhang L., Cheng J., Hou Z., Chem. Commun. 2013, 49, 4782–4784; [DOI] [PubMed] [Google Scholar]
- 1k.H. H. Cramer, B. Chatterjee, T. Weyhermüller, C. Werlé, W. Leitner, Angew. Chem. Int. Ed 2020, 59, in press; Angew. Chem 2020, 132, in press; [DOI] [PMC free article] [PubMed]
- 1l. Steinhoff P., Paul M., Schroers J. P., Tauchert M. E., Dalton Trans. 2019, 48, 1017–1022; [DOI] [PubMed] [Google Scholar]
- 1m. Bertini F., Glatz M., Stöger B., Peruzzini M., Veiros L. F., Kirchner K., Gonsalvi L., ACS Catal. 2019, 9, 632–639. [Google Scholar]
- 2.
- 2a. Berkefeld A., Piers W. E., Parvez M., J. Am. Chem. Soc. 2010, 132, 10660–10661; [DOI] [PubMed] [Google Scholar]
- 2b. Chen J., Falivene L., Caporaso L., Cavallo L., Chen E. Y. X., J. Am. Chem. Soc. 2016, 138, 5321–5333; [DOI] [PubMed] [Google Scholar]
- 2c. Courtemanche M.-A., Légaré M.-A., Rochette É., Fontaine F.-G., Chem. Commun. 2015, 51, 6858–6861; [DOI] [PubMed] [Google Scholar]
- 2d. Del Rio N., Lopez-Reyes M., Baceiredo A., Saffon-Merceron N., Lutters D., Müller T., Kato T., Angew. Chem. Int. Ed. 2017, 56, 1365–1370; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 1385–1390; [Google Scholar]
- 2e. Mukherjee D., Sauer D. F., Zanardi A., Okuda J., Chem. Eur. J. 2016, 22, 7730–7733; [DOI] [PubMed] [Google Scholar]
- 2f. Riduan S. N., Zhang Y., Ying J. Y., Angew. Chem. Int. Ed. 2009, 48, 3322–3325; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 3372–3375; [Google Scholar]
- 2g. Schäfer A., Saak W., Haase D., Müller T., Angew. Chem. Int. Ed. 2012, 51, 2981–2984; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 3035–3038; [Google Scholar]
- 2h. Motokura K., Nakagawa C., Pramudita R. A., Manaka Y., ACS Sustainable Chem. Eng. 2019, 7, 11056–11061; [Google Scholar]
- 2i. Huang W., Roisnel T., Dorcet V., Orione C., Kirillov E., Organometallics 2020, 39, 698–710. [Google Scholar]
- 3.
- 3a. Chen J., McGraw M., Chen E. Y.-X., ChemSusChem 2019, 12, 4543–4569; [DOI] [PubMed] [Google Scholar]
- 3b. Wang X., Xia C., Wu L., Green Chem. 2018, 20, 5415–5426; [Google Scholar]
- 3c. Zhang Y., Zhang T., Das S., Green Chem. 2020, 22, 1800–1820. [Google Scholar]
- 4.
- 4a. Ballmann G., Grams S., Elsen H., Harder S., Organometallics 2019, 38, 2824–2833; [Google Scholar]
- 4b. Feng G., Du C., Xiang L., del Rosal I., Li G., Leng X., Chen E. Y. X., Maron L., Chen Y., ACS Catal. 2018, 8, 4710–4718; [Google Scholar]
- 4c. Tüchler M., Gartner L., Fischer S., Boese A. D., Belaj F., Mösch-Zanetti N. C., Angew. Chem. Int. Ed. 2018, 57, 6906–6909; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 7022–7025; [Google Scholar]
- 4d. Sattler W., Parkin G., J. Am. Chem. Soc. 2012, 134, 17462–17465; [DOI] [PubMed] [Google Scholar]
- 4e. Deshmukh M. M., Sakaki S., Inorg. Chem. 2014, 53, 8485–8493; [DOI] [PubMed] [Google Scholar]
- 4f. Dawkins M. J. C., Middleton E., Kefalidis C. E., Dange D., Juckel M. M., Maron L., Jones C., Chem. Commun. 2016, 52, 10490–10492; for related hydrosilyation by zinc catalyst, see: [DOI] [PubMed] [Google Scholar]
- 4g. Nikonov G. I., ACS Catal. 2017, 7, 8454–8459; [Google Scholar]
- 4h. Boone C., Korobkov I., Nikonov G. I., ACS Catal. 2013, 3, 2336–2340; [Google Scholar]
- 4i. Alshakova I. D., Nikonov G. I., Synthesis 2019, 51, 3305–3312; [Google Scholar]
- 4j. Sahoo R. K., Mahato M., Jana A., Nembenna S., J. Org. Chem. 2020, 85, 11200–11210; [DOI] [PubMed] [Google Scholar]
- 4k. Brown N. J., Harris J. E., Yin X., Silverwood I., White A. J. P., Kazaria S. G., Hellgardt K., Shaffer M. S. P., Williams C. K., Organometallics 2014, 33, 1112–1119; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4l. Rauch M., Kar S., Kumar A., Avram L., Shimon L. J. W., Milstein D., J. Am. Chem. Soc. 2020, 142, 14513–14521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rauch M., Parkin G., J. Am. Chem. Soc. 2017, 139, 18162–18165. [DOI] [PubMed] [Google Scholar]
- 6. Lummis P. A., Momeni M. R., Lui M. W., McDonald R., Ferguson M. J., Miskolzie M., Brown A., Rivard E., Angew. Chem. Int. Ed. 2014, 53, 9347–9351; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 9501–9505. [Google Scholar]
- 7.
- 7a. Chambenahalli R., Andrews A. P., Ritter F., Okuda J., Venugopal A., Chem. Commun. 2019, 55, 2054–2057; [DOI] [PubMed] [Google Scholar]
- 7b. Specklin D., Hild F., Fliedel C., Gourlaouen C., Veiros L. F., Dagorne S., Chem. Eur. J. 2017, 23, 15908–15912; [DOI] [PubMed] [Google Scholar]
- 7c. Bruyere J.-C., Specklin D., Gourlaouen C., Lapenta R., Veiros L. F., Grassi A., Milione S., Ruhlmann L., Boudon C., Dagorne S., Chem. Eur. J. 2019, 25, 8061–8069; [DOI] [PubMed] [Google Scholar]
- 7d. Zhang Q., Fukaya N., Fujitani T., Choi J.-C., Bull. Chem. Soc. Jpn. 2019, 92, 1945–1949; [Google Scholar]
- 7e. Ballmann G., Martin J., Langer J., Färber C., Harder S., Z. Anorg. Allg. Chem. 2019, 6, 593–602. [Google Scholar]
- 8. Rit A., Zanardi A., Spaniol T. P., Maron L., Okuda J., Angew. Chem. Int. Ed. 2014, 53, 13273–13277; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 13489–13493. [Google Scholar]
- 9. Ashby E. C., Watkins J. J., Inorg. Chem. 1977, 16, 2070–2075. [Google Scholar]
- 10. Brookhart M., Grant B., Volpe A. F., Organometallics 1992, 11, 3920–3922. [Google Scholar]
- 11. Michalczyk M. J., Organometallics 1992, 11, 2307–2309. [Google Scholar]
- 12.
- 12a. Wiegand A.-K., Rit A., Okuda J., Coord. Chem. Rev. 2016, 314, 71–82; for the first example of a terminal zinc hydride, see: [Google Scholar]
- 12b. Spielmann J., Piesik D., Wittkamp B., Jansen G., Harder S., Chem. Commun. 2009, 3455–3456. [DOI] [PubMed] [Google Scholar]
- 13.
- 13a. Intemann J., Sirsch P., Harder S., Chem. Eur. J. 2014, 20, 11204–11213; [DOI] [PubMed] [Google Scholar]
- 13b. Neils T. L., Burlitch J. M., Inorg. Chem. 1989, 28, 1607–1609; [Google Scholar]
- 13c. Coles M. P., El-Hamruni S. M., Smith J. D., Hitchcock P. B., Angew. Chem. Int. Ed. 2008, 47, 10147–10150; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 10301–10304; [Google Scholar]
- 13d. Bell N. A., Coates G. E., J. Chem. Soc. A 1968, 823–826; [Google Scholar]
- 13e. Rit A., Spaniol T. P., Maron L., Okuda J., Angew. Chem. Int. Ed. 2013, 52, 4664–4667; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 4762–4765. [Google Scholar]
- 14. Venugopal A., Fegler W., Spaniol T. P., Maron L., Okuda J., J. Am. Chem. Soc. 2011, 133, 17574–17577. [DOI] [PubMed] [Google Scholar]
- 15. Bianchini C., Laschi F., Masi D., Mealli C., Meli A., Ottaviani F. M., Proserpio D. M., Sabat M., Zanello P., Inorg. Chem. 1989, 28, 2552–2560. [Google Scholar]
- 16. Tenorio M. J., Puerta M. C., Valerga P., J. Chem. Soc. Dalton Trans. 1996, 1305–1308. [Google Scholar]
- 17. Tulip T. H., Yamagata T., Yoshida T., Wilson R. D., Ibers J. A., Otsuka S., Inorg. Chem. 1979, 18, 2239–2250. [Google Scholar]
- 18. Schulz S., Eisenmann T., Schmidt S., Bläser D., Westphal U., Boese R., Chem. Commun. 2010, 46, 7226–7228. [DOI] [PubMed] [Google Scholar]
- 19.
- 19a. Karmakar A., Rubio G. M. D. M., da Silva M. F. C. G., Hazra S., Pombeiro A. J. L., Cryst. Growth Des. 2015, 15, 4185–4197; [Google Scholar]
- 19b. Bon V., Kavoosi N., Senkovska I., Muller P., Schaber J., Wallacher D., Tobbens D. M., Mueller U., Kaskel S., Dalton Trans. 2016, 45, 4407–4415. [DOI] [PubMed] [Google Scholar]
- 20. Rauch M., Strater Z., Parkin G., J. Am. Chem. Soc. 2019, 141, 17754–17762. [DOI] [PubMed] [Google Scholar]
- 21.
- 21a. Curtis M. D., Epstein P. S., Adv. Organomet. Chem. 1981, 19, 213–255; [Google Scholar]
- 21b. Hashimoto H., Tobita H., Ogino H., J. Organomet. Chem. 1995, 499, 205–211; [Google Scholar]
- 21c. Radu N. S., Hollander F. J., Tilley T. D., Rheingold A. L., Chem. Commun. 1996, 2459–2460; [Google Scholar]
- 21d. Park S., Kim B. G., Göttker-Schnetmann I., Brookhart M., ACS Catal. 2012, 2, 307–316; [Google Scholar]
- 21e. Corey J. Y., Chem. Rev. 2016, 116, 11291–11435; [DOI] [PubMed] [Google Scholar]
- 21f. Liu X., Xiang L., Louyriac E., Maron L., Leng X., Chen Y., J. Am. Chem. Soc. 2019, 141, 138–142. [DOI] [PubMed] [Google Scholar]
- 22. Schuhknecht D., Spaniol T. P., Maron L., Okuda J., Angew. Chem. Int. Ed. 2020, 59, 310–314; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 317–322. [Google Scholar]
- 23.Heterogeneous catalysts based on noble metals allow the formation of methyl formate directly from CO2 and H2 in the presence of additives such as triethylamine in a reaction medium such as methanol. Ruthenium is used in homogeneous catalysis. A zinc-catalyzed reaction is unknown, only a heterogeneous copper-zinc catalyst enables the formation of methyl formate from CO2.
- 24.
- 24a. Ziebart C., Federsel C., Anbarasan P., Jackstell R., Baumann W., Spannenberg A., Beller M., J. Am. Chem. Soc. 2012, 134, 20701–20704; [DOI] [PubMed] [Google Scholar]
- 24b. Jessop P. G., Hsiao Y., Ikariya T., Noyori R., J. Chem. Soc. Chem. Commun. 1995, 707–708; [Google Scholar]
- 24c. Kröcher O., Köppel R. A., Baiker A., Chem. Commun. 1997, 453–454; [Google Scholar]
- 24d. Corral-Pérez J. J., Bansode A., Praveen C. S., Kokalj A., Reymond H., Comas-Vives A., VandeVondele J., Copéret C., von Rohr P. R., Urakawa A., J. Chem. Soc. 2018, 140, 13884–13891; [DOI] [PubMed] [Google Scholar]
- 24e. Wu C., Zhang Z., Zhu Q., Han H., Yang Y., Han B., Green Chem. 2015, 17, 1467–1472; [Google Scholar]
- 24f. Filonenko G. A., Vrijburg W. L., Hensen E. J. M., Pidko E. A., J. Catal. 2016, 343, 97–105; [Google Scholar]
- 24g. Corral-Pérez J. J., Copéret C., Urakawa A., J. Catal. 2019, 380, 153–160; [Google Scholar]
- 24h. Huff C. A., Sanford M. S., J. Am. Chem. Soc. 2011, 133, 18122–18125; [DOI] [PubMed] [Google Scholar]
- 24i. Thenert K., Beydoun K., Wiesenthal J., Leitner W., Klankermayer J., Angew. Chem. Int. Ed. 2016, 55, 12266–12269; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 12454–12457; [Google Scholar]
- 24j. Westhues N., Belleflamme M., Klankermayer J., ChemCatChem 2019, 11, 5269–5274; [Google Scholar]
- 24k. Siebert M., Seibicke M., Siegle A. F., Krah S., Trapp O., J. Am. Chem. Soc. 2019, 141, 334–341; [DOI] [PubMed] [Google Scholar]
- 24l. Kerry Yu K. M., Tsang S. C., Catal. Lett. 2011, 141, 259–265. [Google Scholar]
- 25. Koo J., Kim S. H., Hong S. H., Chem. Commun. 2018, 54, 4995–4998. [DOI] [PubMed] [Google Scholar]
- 26.
- 26a. Mukhopadhyay T. K., Ghosh C., Flores M., Groy T. L., Trovitch R. J., Organometallics 2017, 36, 3477–3483; [Google Scholar]
- 26b. Mukhopadhyay T. K., Rock C. L., Hong M., Ashley D. C., Groy T. L., Baik M. H., Trovitch R. J., J. Am. Chem. Soc. 2017, 139, 4901–4915. [DOI] [PubMed] [Google Scholar]
- 27. Espinosa M. R., Charboneau D. J., Garcia de Oliveira A., Hazari N., ACS Catal. 2019, 9, 301–314. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary