

Abstract
Hereditary predisposition to cancer concerns between 5% and 10% of cancers. The main genes involved in the most frequent syndromes (hereditary breast and ovarian cancer syndrome, hereditary nonpolyposis colorectal cancer syndrome) were identified in the 1990s. Exploration of their functional pathways then identified novel genes for hereditary predisposition to cancer, and candidate genes whose involvement remains unclear. To determine the contribution of truncating variants in 11 candidate genes (BARD1, FAM175A, FANCM, MLH3, MRE11A, PMS1, RAD50, RAD51, RAD51B, RINT1, and XRCC2) to cancer predisposition in a population of interest, panel sequencing was performed in 849 patients with a suspected hereditary predisposition to cancer for whom a diagnostic panel of 38 genes identified no causal mutation. Sixteen truncating variants were found in FANCM (n = 7), RINT1 (n = 4), RAD50 (n = 2), BARD1, PMS1, and RAD51B. FANCM (adjusted P‐value: .03) and RINT1 (adjusted P‐value: .04) were significantly associated with hereditary breast and ovarian cancer. However, further studies are required to determinate the risk of cancer, including the segregation of the variants in the families of our cases. No mutation was identified in RAD51, MRE11A, FAM175A, XRCC2, or MLH3. The involvement of these genes in the hereditary predisposition to cancer cannot be ruled out, although if it exists it is rare or does not seem to involve truncating variants.
Keywords: breast and ovarian cancer syndrome, candidate genes, hereditary colorectal cancer, panel sequencing, predisposition to cancer
1. INTRODUCTION
Hereditary predisposition to cancer concerns between 5% and 10% of cancers. 1 The hereditary predisposition to breast and ovarian cancer (HBOC) is the most frequent of these. 2 The major genes involved in HBOC, BRCA1, and BRCA2, were identified in the 1990s. The exploration of candidate genes involved in the same biological pathways represented a logical approach and identified additional predisposition genes involved in this syndrome, including PALB2, RAD51C, and RAD51D. 3 However, the involvement of other homologous recombination genes, such as FANCM, RINT1, RAD50, RAD51, RAD51B, BARD1, MRE11A, FAM175A, or XRCC2, 4 in HBOC predisposition remains uncertain.
Another relatively frequent hereditary predisposition to cancer, hereditary nonpolyposis colorectal cancer (HNPCC) syndrome, predisposes mainly to colorectal and endometrial cancer and is caused by heterozygous pathogenic variants in the mismatch repair (MMR) genes MLH1, MSH2, MSH6, or PMS2. However, the involvement of other MMR genes, such as PMS1 or MLH3, is suspected but remains unclear. 5
Modern high‐throughput sequencing allows simultaneous analysis of multiple genes. With the patient's informed consent, candidate genes can thus be examined in parallel with the diagnostic gene panel routinely used for cancer predisposition. The research results are not reported to the patients because of their unknown clinical interest.
Our department explored 11 genes that are candidates for hereditary predisposition to cancer in 849 patients with personal and/or familial indications of hereditary risk and without any causal mutation identified by a diagnostic panel of 38 genes. Our goal was to explore the prevalence of pathogenic variants and their associated phenotypic presentation. The 11 genes included 9 genes involved in homologous recombination and suspected to be involved in HBOC (FANCM, RINT1, RAD50, RAD51, RAD51B, BARD1, MRE11A, FAM175A, and XRCC2) and two genes involved in MMR, and suspected to be implicated in HNPCC syndrome (PMS1, MLH3).
2. MATERIALS AND METHODS
2.1. Samples
Between 2016 and 2018, 849 patients from our Oncogenetics Department, who did not have a pathogenic or probably pathogenic variant identified be sequencing a 38‐gene diagnostic panel (Table 1) (including copy number variations) were analyzed for 11 research genes suspected to be involved in hereditary predisposition to cancer (Table 2). The patients presented HBOC and HNPCC as defined by National Comprehensive Cancer Network (NCCN) guidelines (Figure 1). 6 Cancer‐free patients were analyzed in the event of a family history with at least one first‐degree relative suggestive of one of these two syndromes but with no affected family member available for study. Each patient signed informed consent for genetic diagnosis of hereditary disease and for research. All genes were analyzed from peripheral blood on EDTA. DNA was extracted from peripheral blood using QIAamp DNA Blood maxikit (Qiagen).
TABLE 1.
Genes in the diagnosis panel
| Panel of diagnosis genes sequenced | |||
|---|---|---|---|
| APC | (LRG_130t1 & t2) | PALB2 | (LRG_308) |
| ATM | (LRG_135) | PMS2 | (LRG_161) |
| BAP1 | (LRG_529) | POLD1 | (LRG_785 t1) |
| BMPR1A | (LRG_298) | POLE | (LRG_789) |
| BRCA1 | (LRG_292) | PTEN | (LRG_311) |
| BRCA2 | (LRG_293) | RAD51C | (LRG_314) |
| BRIP1 | (LRG_300) | RAD51D | (LRG_516) |
| CDH1 | (LRG_301) | RET | (LRG_518 t1) |
| CDKN2A | (LRG_11t1 & t2) | SDHA | (NM_004168.3) |
| CHEK2 | (NM_007194.3) | SDHAF2 | (LRG_519) |
| EPCAM | (LRG_215) | SDHB | (LRG_316) |
| FLCN | (LRG_325) | SDHC | (LRG_317) |
| MAX | (LRG_530) | SDHD | (NM_003002.3) |
| MEN1 | (LRG_509 t2) | SMAD4 | (LRG_318) |
| MLH1 | (LRG_216) | STK11 | (LRG_319) |
| MSH2 | (LRG_218) | TMEM127 | (LRG_528) |
| MSH6 | (LRG_219) | TP53 | (LRG_321 t1) |
| MUTYH | (NM_001048171.1) | VHL | (LRG_322) |
| NBN | (LRG_158) | ||
| NF2 | (LRG_511 t1) | ||
Note: All patients had a 38 genes diagnosis panel analysis. All of them had no probably pathogenic or pathogenic variants as defined using the American College of Medical Genetic criteria.
TABLE 2.
Genes in the research panel of 11 genes
| Panel of research genes sequenced | |
|---|---|
| BARD1 | NM_000465.2 |
| FAM175A | NM_139076.2 |
| FANCM | NM_020937.2 |
| MLH3 | NM_001040108.1 |
| Mre11a | NM_05591.3 |
| PMS1 | NM_000534.4 |
| RAD50 | NM_005732.3 |
| RAD51 | NM_002875.4 |
| RAD51B | NM_133509.3 |
| RINT1 | NM_021930.4 |
| XRCC2 | NM_005431.1 |
Note: A panel of 11 genes was sequenced in 849 patients, including candidate genes to hereditary breast and ovarian cancer syndrome (BARD1, FAM175A, FANCM, Mre11a, RAD50, RAD51, RAD51B, RINT1 and XRCC2) and candidate genes to predisposition colorectal cancer (MLH1, PMS1).
FIGURE 1.
Indications of analyses and detection rates. HBOC, hereditary breast and ovarian cancer, HNPCC, hereditary nonpolyposis colorectal cancer
2.2. Panel sequencing
Sonic fragmentation of DNA was performed on a Bioruptor instrument (Diagenode). Kapa HTP library preparation and SeqCap EZ Choice probes and reagents (Roche) were used for library preparation and capture. Quality of fragmentation, library, and capture were controlled using a Bioanalyzer 2100 instrument (Agilent). Sequencing was performed using Miseq v2 kit (300 cycles) on Miseq Instrument (Illumina). All steps were performed following providers' guidelines.
2.3. Bioinformatic analysis
Demultiplexing was performed using bcl2fastq2 Conversion Software (Illumina). Alignment was performed on University of California Santa Cruz human genome reference build 19 using the Burrows‐Wheeler Aligner. Genome Analysis Toolkit (GATK) and PICARD tools were used for base quality score recalibration (BaseRecalibrator) and realignment (RealignerTargetCreator, IndelRealigner), as recommended by Eurogentest guidelines (Matthijs et al). Variant calling was performed using GATK HaplotypeCaller and annotated using EnsemblVariantEffectPredictor. Copy number variation analysis was performed using ExomeDef. Variants were filtered for quality score ≥ 30, depth ≥ 30x, and present in ≥ 20% of reads.
2.4. Variant interpretation
Variants interpretation was aided using ALAMUT (Interactive BioSoftware), which includes splice site analysis tools (SpliceSiteFinder, MaxEntScan), protein‐function prediction tools (SIFT, Polyphen 2.0), and links to different databases (ClinVar, LOVD). Only truncating variants were considered in this study: nonsense, frameshift, and canonical splice‐site variants that can be considered to be loss‐of‐function variants. The frequency of truncating variants in the general population was determined using gnomAD, which aggregates whole‐exome and whole‐genome sequencing data from approximately 140 000 individuals. Missense variants were not included due to unknown impact on function.
2.5. Statistical analyses
For each of the 11 genes, the frequency of variants observed in our dataset was compared to that of the gnomAD population in order to highlight differentially abundant variants. This comparison was made for our entire population, as well as for the HBOC and digestive cases separately. Due to low‐expected frequencies, the exact binomial test was preferred to an approximate test for proportions. A modified (for discrete test statistics) Benjamini‐Yekutieli multitesting correction (under P‐value dependence) was applied to the raw P‐values obtained from the whole study (n = 33). The tests were bilateral, and an adjusted P < .05 was considered statistically significant, indicating evidence against the hypothesis of equality of proportions. Statistical analysis was performed using R software, v. 3.6.1 (R Core Team, 2019, packages DiscreteFDR and MHTdiscrete).
3. RESULTS
HBOC accounted for 87.75% of cases (n = 745), while HNPCC accounted for 12.15% of cases (n = 104).
Sixteen truncating variants were identified, corresponding to a detection rate of 1.9%, all in a heterozygous state. The most frequently mutated genes were FANCM (n = 7), RINT1 (n = 4), and RAD50 (n = 2), followed by BARD1, PMS1, and RAD51B (n = 1). No truncating variants were found in FAM175A, MLH3, RAD51, MRE11A, or XRCC2. Truncating variants were found in 14 HBOC cases (87.5% of variants) and two digestive cases (12.5%) (Figure 2, Table 3).
FIGURE 2.
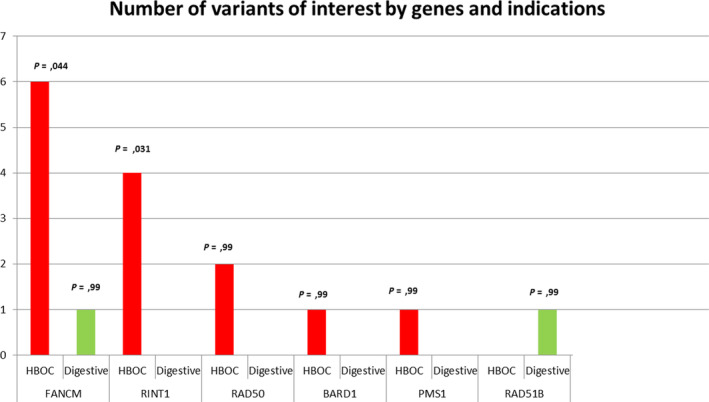
Number of variants of interest by genes and indications. Sixteen variants of interest were found in our study. FANCM and RINT1 were significantly associated with HBOC indication. No mutation was found in Mre11a, FAM175A, MLH3, RAD51, and XRCC2. HBOC, hereditary breast and ovarian cancer
TABLE 3.
Variants identified by gene and corresponding individual and familial phenotype
| Gene | Variant | Protein effect | Pathology of the proband | Familial history |
|---|---|---|---|---|
| FANCM | c.‐143_5340 + 20del | p.? | TNBC (81) | None |
| HGSOC (70) | ||||
| c.1972C > T | p.(Arg658*) | TNBC (59) | None | |
| c.2184del | p.(His729Ilefs*31) | HGSOC (55) | None | |
| c.2586_2589del | p.(Lys863Ilefs*12) | Medullary BC (40) | BC: Mother (87); Sister (57) | |
| c.5101C > T | p.(Gln1701*) | No cancer (Male) | HNPCC | |
| c.5791C > T | p.(Arg1931*) | Granulosa cell tumor of the ovary (52) |
Leukemia: Mother (45), Endometrial cancer: Sister (34); BC: Aunt (29) |
|
| c.5791C > T | p.(Arg1931*) | BC (45) | BC: Sister (42) | |
| RINT1 | c.515G > A | p.(Ser172Asn) | No cancer | BC: Mother (67); Sister (34) |
| c.1333 + 1G > A | p.? | BC (50) | BC: Mother (58), Aunt (73) | |
| c.2303dup | p.(Ile769Asnfs*10) | No cancer | HGSOC: Mother (50) BC: Grandmother and Aunt (50) | |
| c.2303dup | p.(Ile769Asnfs*10) | BC (43) | None | |
| RAD50 | c.130‐1G > T | p.? | HGSOC (55) | None |
| c.1048C > T | p.(Gln350*) | HGSOC (55) | None | |
| BARD1 | c.1935_1954dup20 | p.(Glu652Valfs*69) | HGSOC (78) | BC: Aunt (79) |
| RAD51B | c.139C > T | p.(Arg47*) | CRC (41); MSS | None |
| PMS1 | c.1705C > T | p.(Arg569*) | CRC (50); TNBC (57) | None |
Abbreviations: BC, breast cancer; CCR, colorectal cancer; HGSOC, high‐grade serous ovarian cancer; HNPCC, hereditary non polyposis colorectal cancer syndrome; MSS, microsatellite stable; TPN, triple negative.
3.1.1. FANCM
Seven truncating variants were identified in our cohort (0.82%, 95% CI 0.33%‐1.69%), which is significantly higher than the expected frequency in the general population (0.2%; adjusted P = .03), including six variants in HBOC cases (0.81%, 95% CI 0.29‐1.74; adjusted P = .04) and 1 incidental finding in an HNPCC case (0.96%, 95% CI 0.02%‐5.24%). The probands' phenotypes included breast cancer in four cases and ovarian cancer in two cases. One proband presented both breast and ovarian cancer. The median age of onset was 50.5 (STD: 19) for breast cancer and 55 (STD: 9.6) for ovarian cancer. A positive family history of cancer was found in 50% of cases, including only breast cancer, with a median age of onset of 50.5 (STD: 24.14). The variant c.2184del; p.(His729Ilefs*31) identified in a proband with ovarian cancer at 55 years of age, has not been previously described.
3.1.2. RINT1
Four previously undescribed truncating variants were identified in Rad50 interactor 1 (RINT1) in our cohort (0.47%, 95% CI 0.12%‐1.2%), which is significantly higher than the expected frequency in the general population (0.07; adjusted P = .04). All four variants were observed in HBOC families (0.54%, 95% CI 0.14%‐1.37%; adjusted P = .03).
One patient had breast cancer at 43 years of age but no family history of cancer, and one had breast cancer at 50 years of age with a family history of breast cancer. Two probands were unaffected men with a family history of HBOC analysed because all index cases in the family were deceased. Ovarian cancer in an unaffected male proband's mother occurred at 50 years of age; the other male proband's family included only breast cancer. The median age of onset of breast cancer in relatives was 54 (STD: 13.60). Interestingly, the variant c.2303dup; p.(Ile769Asnfs*10) was shared by two unrelated patients.
3.1.3. RAD50
Two truncating variants were found in RAD50 (0.24%, 95% CI 0.02%‐0.85%), both found in HBOC patients (0.40%, 95% CI 0.08%‐1.17%), who presented an isolated ovarian cancer, occurring in both cases at 55 years of age.
3.1.4. BARD1, RAD51B, PMS1
A previously undescribed truncating variant was found in BARD1 in a patient with ovarian cancer at 78 years of age, in RAD51B in a patient with an early onset colorectal cancer at 41 years old of age and PMS1 in a patient with rectal cancer at 50 years of age and triple‐negative breast cancer at 71 years of age.
4. DISCUSSION
The molecular exploration of 849 patients referred for oncogenetic indications and without any causal mutation identified using a diagnostic panel of 38 genes identified 16 loss‐of‐function mutations in seven research genes, including two genes significantly associated with HBOC patients (FANCM and RINT1).
FANCM is a regulator of genome maintenance, involved in DNA repair and genome duplication, acting as a tumor suppressor. 7 Bi‐allelic loss‐of‐function mutations are associated with Fanconi Anemia Like phenotype, or azoospermia and primary ovarian insufficiency. 8 , 9 Mono‐allelic mutations have been associated with breast cancer. 10 The two most frequent truncating variants in FANCM, c.5101C > T; p.(Gln1701*) and c.5791C > T; p.(Arg1931*), both observed in our study, increase breast cancer risk (OR: 2‐3.93). 11 , 12 A particular risk of early onset triple‐negative breast cancer has been reported for these variants (OR: 4.13), with a median age of diagnosis at 51 years of age. 13 This is confirmed in our study, with a median age at diagnosis comparable for probands (50.5 years of age), and including all familial breast cancers (49 years of age).
However, the triple‐negative sub‐type was not associated with the two variants previously described in our study. Moreover, our study describes for the first time an early onset medullary breast cancer at 40 years of age in a patient with a rare variant c.2586_2589del; p.(Lys863Ilefs*12) of FANCM. Medullary breast cancer is a rare form of cancer, representing less than 2% of breast carcinomas, and presenting triple‐negative features in more than 60% of cases and constitutional mutation of BRCA1 in more than 16% of cases. 14
FANCM has been also associated with ovarian cancer risk. 15 , 16 However, the penetrance of FANCM mutations remains unclear. In our study, the known variant c.5791C > T; p.(Arg1931*) is reported for the first time in a patient with an ovarian Granulosa cell tumor at 52 years of age, a rare cancer, with a strong familial history of breast cancer. Two patients with isolated high‐grade serous ovarian cancers carried previously unknown variants c.2184del; p.(His729Ilefs*31) and c.‐143_5340 + 20del; p.?. FANCM is significantly associated with HBOC patients in our study. The implication of FANCM in rare gynecological cancers, such as medullary breast cancer or ovarian Granulosa cell tumors, should be explored.
RINT1 also presents a significantly higher mutation rate in HBOC patients in our study, in comparison to the general population. RINT1 acts as a RAD50 interactor and is involved in cell cycle regulation, telomere elongation, centrosome integrity, and endoplasmic reticulum maintenance. 17 It has been associated with a low to moderate risk of breast cancer (OR = 2‐3.24), 17 , 18 as well as in Lynch syndrome, but not with ovarian cancer risk. The mechanism suspected in the predisposition to cancer is based on tumor‐suppressive or oncogene function. 19
The four variants found in our study have not been described before. All were identified in HBOC patients, including two breast cancer families and two breast and ovarian cancer families. It is important to note that two variants were present in two cancer‐free individuals belonging to HBOC families, in which the mutational status of the index cases was unknown. Thus, the involvement of RINT1 in the family phenotype can only be suspected.
Overall, FANCM and RINT1 seem to be associated with breast and ovarian cancer syndrome in our study. However, the determination of their impact in hereditary predisposition to cancer and their clinical interest require further investigation, including segregation analysis and more precise risk determination in mutation carriers.
Truncating variants have been identified in other genes, including RAD50, RAD51B, BARD1, PMS1, but these mutations were not significant in HBOC or HNPCC indications.
RAD50 forms a hetero‐trimer complex with Mre11a and NBS1, involving in DNA damage response and DNA double‐strand break repair. 20 Its biological function makes it a candidate for exploration in HBOC. However, large case‐control studies have not confirmed a risk in RAD50 mutation carriers. 21 , 22 In our study, truncating variants in RAD50 were identified in just two ovarian cancer cases that occurred at 55 years of age, and do not seem to be significantly associated with HBOC.
Only one variant was identified in RAD51B in our study, in a patient with colorectal cancer at 40 years of age. RAD51B is involved in homologous recombination repair. No truncating variants were found in HBOC patients, confirming their extreme rarity. 23 Although the breast cancer risk of truncating RAD51B mutations has not been clearly determined at this time, in both breast and ovarian cancer, their rarity induces a limited interest in screening them in HBOC patients. Missense variants in RAD51B have been associated with low breast cancer risk (OR 1.15 at 1.24) and were not explored here. 23
The detection rates of BARD1 and PMS1 were also low (0.09%). PMS1 forms a hetero‐trimer with MLH1 and PMS2, products of MMR genes involved in Lynch syndrome. 5 However, like MLH3 for which no mutation was found in our study, the biological function of PMS1 is unclear. One variant was identified in a patient with triple‐negative breast cancer at 79 years of age, and rectal cancer at 50 years of age. However, microsatellite instability data was not available from the rectal tumor, and the impact of this variant on the MMR system, and so in Lynch syndrome, remains unknown.
BARD1 interacts with BRCA1, known to be a major gene of predisposition to breast and ovarian cancer risk. Mutations in BARD1 have been associated with breast cancer risk (OR: 2.21 to 5), but not with ovarian cancer. 21 , 24 , 25 In our study, one variant has been found in BARD1 as an incidental finding in a late‐onset ovarian cancer patient.
No mutation has been identified in MRE11A, FAM175A, MLH3, RAD51, or XRCC2. Thus, these genes do not seem to be associated with a hereditary cancer predisposition in our study.
Only the truncating variants were included in our study, because their functional impact is probable, and does not require additional analyses. Thus, biological filters could have ruled out variants of interest other than truncating variants, in particular missense variants, which are more difficult to interpret. Their exploration would require functional analyses and/or case‐control studies of large cohorts.
FANCM and RINT1 have been reported to be associated with a risk of breast and/or ovarian cancer, and are significantly associated with HBOC patients in our study. A complementary segregation study would determinate more precisely the implication of these genes in this predisposition. The other genes analyzed are not significantly associated with a predisposition to cancer, or have no truncating variant in our study. Their involvement in the hereditary predisposition to cancer cannot be ruled out. However, if it exists, it is rare or does not seems to involve truncating variants.
ETHICAL STATEMENT
The study has been approved by local ethics committee; reference: 2020/CE54.
ACKNOWLEDGMENTS
The authors thank Centre Jean Perrin. The samples used in this study were conserved in the Biological Resource Center of Jean Perrin Comprehensive Cancer Center, identified under No. BB‐0033‐00075 (Clermont‐Ferrand, France).
Cavaillé M, Uhrhammer N, Privat M, et al. Analysis of 11 candidate genes in 849 adult patients with suspected hereditary cancer predisposition. Genes Chromosomes Cancer. 2021;60:73–78. 10.1002/gcc.22911
DATA AVAILABILITY STATEMENT
Data are available on request
REFERENCES
- 1. Wang Q. Cancer predisposition genes: molecular mechanisms and clinical impact on personalized cancer care: examples of Lynch and HBOC syndromes. Acta Pharmacol Sin. févr. 2016;37(2):143‐149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. NCCN Guidelines Insights: Genetic/Familial High‐Risk Assessment: Breast and Ovarian, Version 3.2019–January 18, 2019.
- 3. Chandler MR, Bilgili EP, Merner ND. A review of whole‐exome sequencing efforts toward hereditary breast cancer susceptibility gene discovery. Hum Mutat. 2016;37(9):835‐846. [DOI] [PubMed] [Google Scholar]
- 4. Shahi RB, De Brakeleer S, Caljon B, et al. Identification of candidate cancer predisposing variants by performing whole‐exome sequencing on index patients from BRCA1 and BRCA2‐negative breast cancer families. BMC Cancer. 4 avr. 2019;19(1):313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Smith CE, Mendillo ML, Bowen N, et al. Dominant mutations in S. cerevisiae PMS1 identify the Mlh1‐Pms1 endonuclease active site and an exonuclease 1‐independent mismatch repair pathway. PLoS Genet. 2013;9(10):e1003869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gupta S et al. NCCN guidelines insights: genetic/familial high‐risk assessment: colorectal, version 3.2017. J Natl Compr Canc Netw. 2017;15:1465‐1475. [DOI] [PubMed] [Google Scholar]
- 7. Lopes JL, Chaudhry S, Lopes GS, Levin NK, Tainsky MA. FANCM, RAD1, CHEK1 and TP53I3 act as BRCA‐like tumor suppressors and are mutated in hereditary ovarian cancer. Cancer Genet. 2019;235‐236:57‐64. [DOI] [PubMed] [Google Scholar]
- 8. Bogliolo M, Bluteau D, Lespinasse J, et al. Biallelic truncating FANCM mutations cause early‐onset cancer but not fanconi anemia. Genet Med. 2018;20(4):458‐463. [DOI] [PubMed] [Google Scholar]
- 9. Kasak L, Punab M, Nagirnaja L, et al. Bi‐allelic recessive loss‐of‐function variants in FANCM cause non‐obstructive Azoospermia. Am J Hum Genet. 2018;103(2):200‐212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Schubert S, van Luttikhuizen JL, Auber B, et al. The identification of pathogenic variants in BRCA1/2 negative, high risk, hereditary breast and/or ovarian cancer patients: high frequency of FANCM pathogenic variants. Int J Cancer. 2019;144(11):2683‐2694. [DOI] [PubMed] [Google Scholar]
- 11. Neidhardt G, Hauke J, Ramser J, et al. Association between loss‐of‐function mutations within the FANCM gene and early‐onset familial breast cancer. JAMA Oncol. 2017;3(9):1245‐1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Peterlongo P, Catucci I, Colombo M, et al. FANCM c.5791C>T nonsense mutation (rs144567652) induces exon skipping, affects DNA repair activity and is a familial breast cancer risk factor. Hum Mol Genet. 2015;24(18):5345‐5355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kiiski JI, Pelttari LM, Khan S, et al. Exome sequencing identifies FANCM as a susceptibility gene for triple‐negative breast cancer. Proc Natl Acad Sci USA. 2014;111(42):15172‐15177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dieci MV, Orvieto E, Dominici M, Conte P, Guarneri V. Rare breast cancer subtypes: histological, molecular, and clinical peculiarities. Oncologist. 2014;19:805‐813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nikolaidi A, Konstantopoulou I, Pistalmantzian N, Fostira F, Yannoukakos D, Athanasiadis I. A patient affected with serous ovarian/peritoneal carcinoma carrying the fancm mutation. Case Rep Oncol Med. 2019;2019:9357924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Figlioli G, Kvist A, Tham E, et al. The spectrum of FANCM protein truncating variants in european breast cancer cases. Cancers (Basel). 2020;12(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Li N, Thompson ER, Rowley SM, et al. Reevaluation of RINT1 as a breast cancer predisposition gene. Breast Cancer Res Treat. 2016;159(2):385‐392. [DOI] [PubMed] [Google Scholar]
- 18. Park DJ, Tao K, Le Calvez‐Kelm F, et al. Rare mutations in RINT1 predispose carriers to breast and Lynch syndrome‐spectrum cancers. Cancer Discov Juill. 2014;4(7):804‐815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Quayle SN, Chheda MG, Shukla SA, et al. Integrative functional genomics identifies RINT1 as a novel GBM oncogene. Neuro‐oncology. 2012;14(11):1325‐1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Situ Y, Chung L, Lee CS, Ho VMRN. (MRE11‐RAD50‐NBS1) complex in human cancer and prognostic implications in colorectal cancer. Int J Mol Sci. 2019;20(4):816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Couch FJ, Shimelis H, Hu C, et al. Associations between cancer predisposition testing panel genes and breast cancer. JAMA Oncol. 2017;3(9):1190‐1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lilyquist J, La Duca H, Polley E, et al. Frequency of mutations in a large series of clinically ascertained ovarian cancer cases tested on multi‐gene panels compared to reference controls. Gynecol Oncol. 2017;147(2):375‐380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pelttari LM, Khan S, Vuorela M, et al. RAD51B in familial breast cancer. PLoS ONE. 2016;11(5):e0153788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Weber‐Lassalle N, Borde J, Weber‐Lassalle K, et al. Germline loss‐of‐function variants in the BARD1 gene are associated with early‐onset familial breast cancer but not ovarian cancer. Breast Cancer Res. 2019;21(1):55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ramus SJ, Song H, Dicks E, et al. Germline mutations in the BRIP1, BARD1, PALB2, and NBN Genes in women with ovarian cancer. J Natl Cancer Inst. 2015;107(11):djv214. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data are available on request