

Abstract
The purpose of the present investigation is to analyze the in vivo genotoxicity dose–response data of ethylene oxide (EO) and the applicability of the derived point‐of‐departure (PoD) values when estimating permitted daily exposure (PDE) values. A total of 40 data sets were identified from the literature, and benchmark dose analyses were conducted using PROAST software to identify a PoD value. Studies employing the inhalation route of exposure and assessing gene or chromosomal mutations and chromosomal damage in various tissues were considered the most relevant for assessing risk from EO, since these effects are likely to contribute to adverse health consequences in exposed individuals. The PoD estimates were screened for precision and the values were divided by data‐derived adjustment factors. For gene mutations, the lowest PDE was 285 parts per trillion (ppt) based on the induction of lacI mutations in the testes of mice following 48 weeks of exposure to EO. The corresponding lowest PDE value for chromosomal mutations was 1,175 ppt for heritable translocations in mice following 8.5 weeks of EO exposure. The lowest PDE for chromosomal aberrations was 238 ppt in the mouse peripheral blood lymphocytes following 48 weeks of inhalation exposure. The diverse dose–response data for EO‐induced genotoxicity enabled the derivation of PoDs for various endpoints, tissues, and species and identified 238 ppt as the lowest PDE in this retrospective analysis.
Keywords: benchmark dose, chromosomal aberration, genotoxicity, mutation, permitted daily exposure
1. INTRODUCTION
Genotoxicity is routinely evaluated during safety assessments of synthetic and natural substances. Excitement over the promise of simple and inexpensive genotoxicity tests to predict cancer outcomes in rodents fueled the development of a multitude of short‐term tests starting in the 1970s. The primary focus of testing since that time has been to answer the qualitative question of whether a substance has genotoxic potential (Gollapudi et al., 2013; Gollapudi, 2017). Consequently, more attention has been paid in the field to maximize the dose levels evaluated in these assays in order to optimize the ability to detect an induced response. The analysis of dose–response and the identification of no‐effect‐levels attracted relatively little consideration. In recent years, however, there has been growing interest in using genotoxicity data to inform not only cancer mode of action but also to quantitatively assess risk from exposure to genotoxic agents (Pottenger et al., 2007; Pottenger and Gollapudi, 2009; 2010; Gollapudi et al., 2013; Johnson et al., 2014; MacGregor et al., 2015a, 2015b; Dearfield et al., 2017; Heflich et al., 2020; Klapacz and Gollapudi, 2020; White et al., 2020). The precedent came from Müller and Gocke (2009) who used mutagenicity data to derive permitted exposure levels of a human pharmaceutical contaminated with the mutagen ethyl methane sulfonate, which received acceptance from regulatory authorities (e.g., European Medicines Agency [EMEA], 2008). Recently, Luijten et al. (2020) applied quantitative dose–response analyses to the in vivo micronucleus (MN) test results to identify a point‐of‐departure (PoD) value to calculate permitted daily exposure (PDE) limits for benzene. As a result, the field of genetic toxicology is in the midst of a transformation, toward the vision of its founders who saw the potential of using genotoxicity data for risk assessments and for the development of practical exposure limits (International Commission for Protection Against Environmental Mutagens and Carcinogens [ICPEMC], 1980).
The applicability of the available genotoxicity data to risk assessment is an area of ongoing inquiry in the field. There are several substances whose published genotoxicity data are potentially useful for quantitative dose–response analyses to inform risk assessment. Ethylene oxide (EO; CAS Nr. 75‐21‐8) is one such chemical. It is a simple epoxide with a molecular formula C2H4O. It is extensively used in the chemical industry as an intermediate in the manufacture of other chemicals; it is also used as a sterilizing agent for medical equipment and as a fumigant for spices. There is strong evidence for tumorigenicity of EO following chronic inhalation exposures in animals but less than conclusive evidence in humans (U.S. Environmental Protection Agency [U.S. EPA], 2016; Texas Commission on Environmental Quality [TCEQ], 2020). In the absence of experimental evidence to the contrary, mutagenicity induced by EO (as reviewed below) was hypothesized as the default mode of action for its tumorigenicity (U.S. EPA, 2016; Jinot et al., 2018).
EO is produced metabolically in the liver from endogenously available ethylene (Ehrenberg et al., 1977; Törnqvist et al., 1989; Filser et al., 1992). EO in vivo, therefore, derives both from continuous endogenous production and from episodic exposures to exogenous sources including background EO in ambient air. Endogenous levels in human populations with negligible exogenous exposure to EO have been reported to range from 0.13 to 6.9 parts per billion, when expressed in terms of exogenous exposures of EO (Kirman and Hays, 2017). EO is a DNA‐reactive SN2 alkylating agent that reacts with macromolecules such as proteins and DNA (Li et al., 1992). Its DNA adduct profile, assessed from in vitro studies with naked DNA, reveals hydroxyethyl (HE) adducts formed primarily at the N7 position of guanine (N7‐HEG; 81%), the N3 position of adenine (N3‐HEA; 10%), and the N1 position of adenine (N1‐HEA; 7%) (Li et al., 1992). N6 position of adenine (N6‐HEA), N3 position of cytosine (N3‐HEC; which converts to uracil [N3‐HEU]), and N3 position of thymine (N3‐HET) adducts are also produced in small amounts (Li et al., 1992). Furthermore, earlier in vitro work reported formation of a small amount of O6 position of guanine (O6‐HEG; 0.4%) (Segerbäck, 1990). EO also has the potential to react with the DNA phosphate backbone (Ehrenberg and Hussain, 1981; Dellarco et al., 1990; Agurell et al., 1991).
The N7‐HEG adduct is by far the most abundant, but it is not considered mutagenic. N7‐HEG is chemically unstable and spontaneously depurinated, leaving behind abasic sites that are rapidly repaired (Boysen et al., 2009). Although N7‐HEG is not itself a pro‐mutagenic adduct (Philippin et al., 2014), its depurinations form abasic sites that could result in mutations if present during DNA replication. However, inhalation exposures of rats to 100 ppm (ppm) EO for 4 weeks (6 hr per day/5 days per week) did not reveal an increase in abasic sites over the exposure period (Rusyn et al., 2005). Although production by EO of its minor DNA adducts, some of which (e.g., O6‐HEG) may be more mutagenic, is proportional to its production of the major adduct (Li et al., 1992), this proportionality can vary across tissues and doses. Nonetheless, within a tissue, N7‐HEG is a useful surrogate for quantifying overall EO‐induced DNA damage.
Numerous studies have investigated the genotoxicity of EO. Although it is a relatively weak genotoxicant, the number of positive tests has fostered a contrary belief. The published genetic activity profile for EO shows that the average lowest‐effective‐exposure concentration required to give a positive result in the in vitro assays was between 1.0 and 100 μg/ml (Waters et al., 1999). EO blood levels of this magnitude require inhalation exposures greater than 150 ppm for 4 hr in mice (Brown et al., 1996). Illustrative of this, in pSP189 shuttle vector replicated in human Ad293 cells in vitro, exposure concentrations up to 2 mM induced only the non‐pro‐mutagenic N7‐HEG adducts with no increase in the frequency of mutations (Tompkins et al., 2009). Increased mutations were observed only when the concentrations were increased to 10–30 mM, concomitant with the appearance of pro‐mutagenic N1‐HEA, O6‐HEG, and N3‐HEU DNA adducts.
High acute doses of EO administered via intraperitoneal (IP) or intravenous (IV) injection or in drinking water have been shown to increase the frequencies of MN or chromosomal aberrations in mice and rats (Strekalova et al., 1971; Applegren et al., 1978; Conan et al., 1979; Jenssen and Ramel, 1980; Farooqi et al., 1993; Lorenti Garcia et al., 2001). However, these dosing methods are not relevant to human exposure scenarios. In inhalation studies, a route of exposure relevant to humans, no increases in chromosomal or MN aberrations were found in peripheral blood/splenic lymphocytes from rats exposed to EO at concentrations of 50–450 ppm for 1 or 3 days (6 hr/day) (Kligerman et al., 1983) or 50–200 ppm for 4 weeks (5 days/week, 6 hr/day) (van Sittert et al., 2000; Lorenti Garcia et al., 2001). Preston and Abernethy (1993) exposed rats for 1, 2, 3, or 4 weeks (6 hr/day, 5 days/week) to 150 ppm EO by inhalation and found no increase in the frequency of chromosomal aberrations in peripheral blood lymphocytes at any sampling time. In contrast, inhalation exposure of EO was reported to induce chromosomal aberrations in mice and increase frequency of MN or aberrations in both mice and rats. Ribeiro et al. (1987) reported that exposures of mice to 200–600 ppm EO by inhalation for 2 weeks induced chromosomal aberrations in bone marrow cells, whereas Vergnes and Pritts (1994) found significant increases in bone marrow MN in mice and rats following 4 weeks of inhalation exposure to 200 ppm EO (6 hr/day, 5 days/week). Donner et al. (2010) exposed mice to 0, 25, 50, 100, or 200 ppm EO by inhalation (6 hr/day, 5 days/week, up to 48 weeks) with sacrifice and found aberrations at intervals of 6, 12, 24, and 48 weeks. There were no statistically significant increases in aberration frequencies at any exposure concentration at the 6‐week sacrifice. Only the two highest concentrations showed significant increases in total aberration frequencies at 12 weeks. At 24 weeks, increases in aberrations were seen at the three highest doses while all exposure concentrations showed significant increases in aberrations at the 48‐week sacrifice.
EO has also been evaluated for the induction of mutations in endogenous and reporter genes such as the Hprt, K‐Ras, lacI, and cII in mice and rats (Sisk et al., 1997; Walker et al., 1997; 2000; Tates et al., 1999; van Sittert et al., 2000; Recio et al., 2004; Parsons et al., 2013; Manjanatha et al., 2017); both positive and negative results were reported depending on the target gene, exposure concentration, and duration of exposure. EO has also been investigated for the induction of germ cell mutations in mice (Generoso et al., 1980; 1986; 1990). Results from the above studies are summarized in conjunction with the benchmark dose (BMD) modeling results in Section 3.
The purpose of the present investigation is to analyze the publicly available in vivo genotoxicity dose–response data on EO from experimental mammalian models to identify a PoD and to explore the applicability of the PoD to calculate PDE levels to adequately protect exposed populations from adverse health outcomes resulting from mutagenic effects. in vitro genotoxicity data were not considered in this investigation since methodologies for in‐vitro‐to‐in‐vivo extrapolation of dose metrics and detoxification processes are not fully developed at this time. It is envisioned that the PDE value for genotoxicity is considered along with PDE values for other toxicity endpoints to determine the most sensitive endpoint to adequately protect humans against all adverse outcomes.
2. MATERIALS AND METHODS
The criteria for study inclusion and BMD analysis, as well as the selection of factors for extrapolation from animal to human, were determined before the BMD analysis was conducted. The methods are described in this section and discussed in greater detail in Section 4. The EPA's convention for terminology is used to report the results. BMD is used generically to refer to the BMD approach. In the specific cases of characterizing model results, BMD refers to central estimates and BMDL refers to the corresponding lower 95% confidence limit of the BMD.
2.1. Overall approach for inclusion criteria and consideration of study quality
Studies that reported in vivo positive genetic toxicity for EO were identified from a literature search and the U.S. EPA (2016) recent evaluation of EO inhalation carcinogenicity. Studies were screened for reporting statistically significant responses and employing at least three dose groups (including control dose) for dose–response analysis. The endpoints selected for evaluation included the induction of gene mutations, chromosomal mutations, and chromosomal aberrations (including MN). Studies conducted using cancer tissues, DNA‐adducts, and sister chromatid exchanges and those employing non‐standard parameters were excluded in these analyses. Data reported only in graphical format were also not included in the analysis. When a study reported multiple data sets, a number was assigned to each one (Table 1). One of the studies reporting chromosomal aberrations included gaps in the calculation of total aberrations (Ribeiro et al., 1987); however, total aberrations were recalculated by excluding the gaps for analysis. Ribeiro et al. (1987) also analyzed chromosomal aberrations in diakinesis/Metaphase 1 of mouse spermatocytes following EO exposure; these data, however, were not included in the analyses since not all types of events recorded as aberrations were considered to be treatment related (e.g., univalents) (Gollapudi et al., 1981).
TABLE 1.
List of ethylene oxide (EO) studies included for benchmark dose (BMD) analyses
| Study | Data set identifier | Species—strain | Route | Effect | Tissue | Endpoint | Exposure concentration | Frequency and duration |
|---|---|---|---|---|---|---|---|---|
| Applegren et al. (1978) | 1 | Mouse—NMRI | IV | Cytogenetic | BM | MN | 0, 50, 100, 150, 200, and 300 mg/kg | 1X/d, 2 days |
| 2 | Rat—Sprague Dawley | IV | Cytogenetic | BM | MN | 0, 100, and 150 mg/kg | 1X/d, 2 days | |
| Donner et al. (2010) | 1 | Mouse—B6C3F1 | IH | Cytogenetic | PBL | CA‐RT | 0, 25, 50, 100, and 200 ppm | 6 hr/day, 5 days/week; 12 weeks |
| 2 | Mouse—B6C3F1 | IH | Cytogenetic | PBL | CA‐RT | 0, 25, 50, 100, and 200 ppm | 6 hr/day, 5 days/week; 24 weeks | |
| 3 | Mouse—B6C3F1 | IH | Cytogenetic | PBL | CA‐RT | 0, 25, 50, 100, and 200 ppm | 6 hr/day, 5 day/week; 48 weeks | |
| 4 | Mouse—B6C3F1 | IH | Cytogenetic | PBL | CA‐TA | 0, 25, 50, 100, and 200 ppm | 6 hr/day, 5 day/week; 12 weeks | |
| 5 | Mouse—B6C3F1 | IH | Cytogenetic | PBL | CA‐TA | 0, 25, 50, 100, and 200 ppm | 6 hr/day, 5 day/week; 24 weeks | |
| 6 | Mouse—B6C3F1 | IH | Cytogenetic | PBL | CA‐TA | 0, 25, 50, 100, and 200 ppm | 6 hr/day, 5 days/week; 48 weeks | |
| 7 | Mouse—B6C3F1 | IH | Cytogenetic | S | CA‐RT | 0, 25, 50, 100, and 200 ppm | 6 hr/day, 5 days/week; 12 weeks | |
| 8 | Mouse—B6C3F1 | IH | Cytogenetic | S | CA‐RT | 0, 25, 50, 100, and 200 ppm | 6 hr/day, 5 days/week; 24 weeks | |
| 9 | Mouse—B6C3F1 | IH | Cytogenetic | S | CA‐RT | 0, 25, 50, 100, and 200 ppm | 6 hr/day, 5 days/week; 48 weeks | |
| Farooqi et al. ( 1993 ) | 1 | Mouse—Swiss albino | IP | Cytogenetic | BM | MN | 0, 0.68, 1.36, 2.72, and 3.40 mmol/kg | 1X |
| Lorenti Garcia et al. ( 2001 ) | 1 | Rat—Lewis | IP | Cytogenetic | SP | MN | 0, 50, and 100 mg/kg | 1X |
| 2 | Rat—Lewis | IP | Cytogenetic | BM | MN | 0, 50, and 100 mg/kg | 1X | |
| Generoso et al. ( 1980 ) | 1 | Mouse—T‐stock | IP | Mutation | GE | HTL | 0, 30, and 60 mg kg –1 day –1 | 5X/week; 5 weeks |
| Generoso et al. (1986) | 1 | Mouse—(C3H × 101)F1 | IH | Mutation | GE | DL | 0, 300, 400, and 500 ppm | 6 hr/day, 4 days (expr. time 6.5–7.5 days) |
| 2 | Mouse—(C3H × 101)F1 | IH | Mutation | GE | DL | 0, 300, 400, 500 ppm | 6 hr/day, 4 d (expr. time 4.5–5.5 days) | |
| Generoso et al. (1990) | 1 | Mouse—(C3H × 101)F1 | IH | Mutation | GE | DL | 0, 165, 204, 250, and 300 ppm | 8.5 weeks; 6 hr/day, 5 days/week for 6 weeks, then daily for 2.5 weeks |
| 2 | Mouse—(C3H × 101)F1 | IH | Mutation | GE | DL | 0, 165, 204, 250, and 300 ppm | 8.5 weeks; 6 hr/day, 5 days/week for 6 weeks, then daily for 2.5 weeks | |
| 3 | Mouse—(C3H × 101)F1 | IH | Mutation | GE | HTL | 0, 165, 204, 250, and 300 ppm | 8.5 weeks; 6 hr/day, 5 days/week for 6 weeks, then daily for 2.5 weeks | |
| 4 | Mouse—(C3H × 101)F1 | IH | Mutation | GE | HTL | 0, 165, 204, 250, and 300 ppm | 8.5 weeks; 6 hr/day, 5 days/week for 6 weeks, then daily for 2.5 weeks | |
| Jenssen and Ramel ( 1980 ) | 1 | Mouse—CBA | IP | Cytogenetic | BM | MN | 0, 50, 100, 125, 150, and 175 mg/kg | 1X |
| Lynch et al. (1984) | 1 | Monkey—Macaca fascicularis | IH | Cytogenetic | PBL | CA | 0, 50, and 100 ppm | 7 hr/day, 5 days/week; 2 years |
| Manjanatha et al. (2017) | 1 | Mouse—Big Blue B6C3F1 | IH | Mutation | LU | cII | 0, 10, 50, 100, and 200 ppm | 6 hr/day, 5 days/week; 4 weeks |
| 2 | Mouse—Big Blue B6C3F1 | IH | Mutation | LU | cII | 0, 100, and 200 ppm | 6 hr/day, 5 days/week; 8 weeks | |
| 3 | Mouse—Big Blue B6C3F1 | IH | Mutation | LU | cII | 0, 100, and 200 ppm | 6 hr/day, 5 days/week; 12 weeks | |
| Parsons et al. (2013) | 1 | Mouse—Big Blue B6C3F1 | IH | Mutation | LU | K‐ras | 0, 10, 50, 100, and 200 ppm | 6 hr/day, 5 days/week; 4 weeks |
| 2 | Mouse—Big Blue B6C3F1 | IH | Mutation | LU | K‐ras | 0, 10, 50, 100, and 200 ppm | 6 hr/day, 5 days/week; 4 weeks | |
| Recio et al. (2004) | 1 | Mouse—B6C3F1 lacI transg. | IH | Mutation | BM | lacI | 0, 25, 50, 100, and 200 ppm | 6 hr/day, 5 days/week; 48 weeks |
| 2 | Mouse—B6C3F1 lacI transg. | IH | Mutation | TE | lacI | 0, 25, 50, 100, and 200 ppm | 6 hr/day, 5 days/week; 48 weeks | |
| 3 | Mouse—B6C3F1 lacI transg. | IH | Mutation | TE | lacI | 0, 25, 50, 100, and 200 ppm | 6 hr/day, 5 days/week; 24 weeks | |
| Ribeiro et al. (1987) | 1 | Mouse—Swiss Webster | IH | Cytogenetic | BM | CA | 0, 200, 400, and 600 ppm | 6 hr, 1 day |
| 2 | Mouse—Swiss Webster | IH | Cytogenetic | BM | CA | 0, 200, and 400 ppm | 6 hr/day, 5 days/week; 2 weeks (total 12 days) | |
| Tates et al. ( 1999 ) | 1 | Rat—Lewis | IP | Mutation | SL | Hprt | 0, 20, 40, 80 mg/kg | 1X (expr. time 32 days) |
| 2 | Rat—Lewis | IP | Mutation | SL | Hprt | 0, 10, 20, 40, and 80 mg/kg | 1X (expr. time 35 days) | |
| 3 | Rat—Lewis | DW | Mutation | SL | Hprt | 0, 2, 5, and 10 mM | 30 days (expr. time 35 days) | |
| 4 | Rat—Lewis | DW | Mutation | SL | Hprt | 0, 2, 5, and 10 mM | 30 days (expr. time 41 days) | |
| 5 | Rat—Lewis | IH | Mutation | SL | Hprt | 0, 50, 100, and 200 ppm | 6 hr/day, 5 days/week; 4 weeks (expr. time 21 days) | |
| van Sittert et al. (2000) | 1 | Rat—Lewis | IH | Mutation | SL | Hprt | 0, 50, 100, and 200 ppm | 6 hr/days, 5 day/week; 4 weeks |
| Walker et al. (1997) | 1 | Mouse—B6C3F1 lacI transg. | IH | Mutation | SL | Hprt | 0, 50, 100, and 200 ppm | 6 hr/day, 5 days/week; 4 weeks |
Note: Inhalation studies are the key studies most relevant for derivation of an inhalation permitted daily exposure (PDE) value. Studies using different routes of exposure are italicized and presented for completeness.
Abbreviations: BM, bone marrow; CA, chromosomal aberration; CA‐RT, chromosomal aberration‐reciprocal translocation; CA‐TA, chromosomal aberration‐total aberration; cII, cII mutation; DL, dominant lethal mutation; DW, drinking water; GE, male germ cells; Hprt, hprt mutation; HTL, heritable translocation; IH, inhalation; IP, intraperitoneal; IV, intravenous; K‐ras, K‐ras mutation; lacI, lacI mutation; LU, lung cells; MN, micronucelus formation; PBL, peripheral blood lymphocytes; S, spermatogonia cells; SL, splenic lymphocytes; SP, splenocytes; TE, testes.
It was the intent of this manuscript to include as many studies as possible for BMD analysis that were reported in the U.S. EPA (2016) assessment as well as those published more recently. Therefore, BMD analysis was applied to all studies meeting the minimum requirements described above irrespective of exposure route or study quality. However, for the next step of deriving candidate PDE values for inhalation exposures, inhalation studies evaluating mutagenicity (i.e., chromosomal aberrations, chromosomal mutations, and gene mutations) and reporting the variability of the data (e.g., SD) were considered to be the most relevant experimental data (see Section 3 for further discussion). For purposes of comparison only (i.e., not considered candidate values) and consistent with our goal of transparent inclusiveness, PDE values were also calculated for inhalation studies that did not report variability by using a data‐derived factor to account for variability (described in detail in Section 2 on PDE derivation). In the final selection of the PDE value, focused attention was given to evaluating the relative quality of the inhalation studies that resulted in the lowest PDE values. At this final step, duration of exposure, group size, exposure levels tested, dose–response pattern of effect, and experimental design were evaluated, as discussed in detail in Section 3.
Several data sets reported mean response values but did not provide a measure of variance in terms of SD or SE that is necessary for BMD analysis. These studies were not considered to be of adequate quality for quantitative dose response analysis for risk assessment purposes. However, for purposes of comparison, these data sets were modeled in PROAST without variance, as the response of one animal for each dose group. Consequently, the BMDL is artificially elevated (less conservative) because of not having variance information. As described in greater detail in Section 4, the estimated BMDL50 values were divided by a data‐derived factor of five for these datasets.
An error in the published data was discovered during the evaluation of heritable translocation data from Generoso et al. (1990). Based on a review of Rhomberg et al. (1990) that summarized Generoso et al. (1990) data, it became apparent that the data reported in Generoso et al. (1990) in their Table 2 for (SEC × C57BL)F1 female stock is the same as combined data for both T‐stock and (SEC × C57BL)F1. For this reason, (SEC × C57BL)F1 data from Rhomberg et al. (1990) were modeled instead.
TABLE 2.
Results of benchmark dose (BMD) analysis grouped by endpoints
| Study | Data set | Species—strain | Route | Tissue | Endpoint | Data and modeling notes | Model | BMD a | BMDL a | BMDU a | BMDU: BMDL | CI b BMDL (ppm) | CI BMD (ppm) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Chromosomal aberrations | |||||||||||||
| Donner et al. (2010) | 1 | Mouse—B6C3F1 | IH | PBL | CA‐RT | 1 | Inv exponential | 28.42 | 20.5 | 44.8 | 2.2 | 3.7 | 5.1 |
| Donner et al. (2010) | 2 | Mouse—B6C3F1 | IH | PBL | CA‐RT | 1 | Hill | 61.44 | 16.7 | 84.8 | 5.1 | 3.0 | 11 |
| Donner et al. (2010) | 3 | Mouse—B6C3F1 | IH | PBL | CA‐RT | 1 | Exponential | 4.718 | 1.84 | 11.8 | 6.4 | 0.3 | 0.84 |
| Donner et al. (2010) | 4 | Mouse—B6C3F1 | IH | PBL | CA‐TA | 1, 2, 3 | Imprecise BMD result | NA | NA | NA | NA | NA | NA |
| Donner et al. (2010) | 5 | Mouse—B6C3F1 | IH | PBL | CA‐TA | 1 | Exponential | 0.6354 | 0.0326 | 4.38 | 134 | 0.0058 | 0.11 |
| Donner et al. (2010) | 6 | Mouse—B6C3F1 | IH | PBL | CA‐TA | 1 | Inv exponential | 5.078 | 0.422 | 13 | 31 | 0.075 | 0.91 |
| Donner et al. (2010) | 7 | Mouse—B6C3F1 | IH | S | CA‐RT | 1, 2, 3 | Imprecise BMD result | NA | NA | NA | NA | NA | NA |
| Donner et al. (2010) | 8 | Mouse—B6C3F1 | IH | S | CA‐RT | 1, 4 | No dose–response | NA | NA | NA | NA | NA | NA |
| Donner et al. (2010) | 9 | Mouse—B6C3F1 | IH | S | CA‐RT | 1, 4 | No dose–response | NA | NA | NA | NA | NA | NA |
| Lynch et al. (1984) | 1 | Monkey—Macaca fascicularis | IH | PBL | CA | 5, 6 | Exponential | 6.364 | 1.38 | 14.2 | 10 | 0.3 | 1.3 |
| Ribeiro et al. (1987) | 1 | Mouse—Swiss Webster | IH | BM | CA | 7, 8 | Exponential | 118 | 118 | 118 | 1 | 4.2 | 21.1 |
| Ribeiro et al. (1987) | 2 | Mouse—Swiss Webster | IH | BM | CA | 3, 7, 8 | Imprecise BMD result | NA | NA | NA | NA | NA | NA |
| Chromosomal mutations | |||||||||||||
| Generoso et al. ( 1980 ) | 1 | Mouse—T‐stock | IP | GE | HTL | 1, 7, 9 | Exponential | 0.0183 | 0.0181 | 0.0185 | 1 | 0.001 | 0.061 |
| Generoso et al. (1986) | 1 | Mouse—(C3H × 101)F1 | IH | GE | DL | 7, 10 | Inv exponential | 463.8 | 449 | 477 | 1.1 | 16 | 83 |
| Generoso et al. (1986) | 2 | Mouse—(C3H × 101)F1 | IH | GE | DL | 7, 10 | Exponential | 479.4 | 467 | 493 | 1.1 | 17 | 86 |
| Generoso et al. (1990) | 1 | Mouse—(C3H × 101)F1 | IH | GE | DL | 7 | Lognormal | 288.1 | 283 | 292 | 1 | 10 | 51 |
| Generoso et al. (1990) | 2 | Mouse—(C3H × 101)F1 | IH | GE | DL | 7 | Lognormal | 303.7 | 297 | 312 | 1.1 | 11 | 54 |
| Generoso et al. (1990) | 3 | Mouse—(C3H × 101)F1 | IH | GE | HTL | 2, 5, 9 | Inv exponential | 61.34 | 40.5 | 79.3 | 2 | 7.2 | 11 |
| Generoso et al. (1990) | 4 | Mouse—(C3H × 101)F1 | IH | GE | HTL | 2, 5, 9, 11 | Exponential | 21.32 | 20.8 | 21.4 | 1 | 3.7 | 3.8 |
| Gene mutations | |||||||||||||
| Manjanatha et al. (2017) | 1 | Mouse—big blue B6C3F1 | IH | LU | cII | 4, 5 | No dose–response | NA | NA | NA | NA | NA | NA |
| Manjanatha et al. (2017) | 2 | Mouse—big blue B6C3F1 | IH | LU | cII | 5 | Exponential and hill | 113.5 | 45.1 | 178 | 3.9 | 8.1 | 20 |
| Manjanatha et al. (2017) | 3 | Mouse—big blue B6C3F1 | IH | LU | cII | 5 | Inv exponential | 198.4 | 141 | 324 | 2.3 | 25 | 35 |
| Parsons et al. (2013) | 1 | Mouse—big blue B6C3F1 | IH | LU | K‐ras | 7, 12, 13, 14 | Exponential | 11.87 | 8.12 | 25.3 | 3.1 | 0.29 | 2.1 |
| Parsons et al. (2013) | 2 | Mouse—big blue B6C3F1 | IH | LU | K‐ras | 7, 12, 13, 14 | Exponential | 6.64 | 5.3 | 7.82 | 1.5 | 0.19 | 1.2 |
| Recio et al. (2004) | 1 | Mouse—B6C3F1 lacI transg. | IH | BM | lacI | 5 | Exponential | 83.84 | 31 | 147 | 4.7 | 5.5 | 15 |
| Recio et al. (2004) | 2 | Mouse—B6C3F1 lacI transg | IH | TE | lacI | 5 12, 13 | Hill | 2.54 | 0.504 | 40.3 | 80 | 0.09 | 0.45 |
| Recio et al. (2004) | 3 | Mouse—B6C3F1 lacI transg | IH | TE | lacI | 5, 4 | No dose–response | NA | NA | NA | NA | NA | NA |
| van Sittert et al. (2000) | 1 | Rat—Lewis | IH | SL | Hprt | 5, 12, 15 | Inv exponential | 110 | 49.4 | 293 | 5.9 | 9 | 20 |
| Tates et al. ( 1999 ) | 1 | Rat—Lewis | IP | SL | Hprt | 5, 16 | Exponential | 46.89 | 14.5 | 86.6 | 6 | 68 | 219 |
| Tates et al. ( 1999 ) | 2 | Rat—Lewis | IP | SL | Hprt | 4, 5, 16 | No dose–response | NA | NA | NA | NA | NA | NA |
| Tates et al. ( 1999 ) | 3 | Rat—Lewis | DW | SL | Hprt | 4, 5, 16 | No dose–response | NA | NA | NA | NA | NA | NA |
| Tates et al. ( 1999 ) | 4 | Rat—Lewis | DW | SL | Hprt | 4, 5, 16 | No dose–response | NA | NA | NA | NA | NA | NA |
| Tates et al. (1999) | 5 | Rat—Lewis | IH | SL | Hprt | 4, 5, 16 | No dose–response | NA | NA | NA | NA | NA | NA |
| Walker et al. (1997) | 1 | Mouse—B6C3F1 lacI transg. | IH | SL | Hprt | 5 | Inv exponential | 35.65 | 21.3 | 54.6 | 2.6 | 4 | 6.4 |
|
Micronucleus formation | |||||||||||||
| Applegren et al. (1978) | 1 | Mouse—NMRI | IV | BM | MN | 7 | Inv exponential | 0.08822 | 0.0547 | 0.123 | 2.2 | 51 | 412 |
| Applegren et al. (1978) | 2 | Rat—Sprague Dawley | IV | BM | MN | 4, 7 | No dose–response | NA | NA | NA | NA | NA | NA |
| Farooqi et al. ( 1993 ) | 1 | Mouse—Swiss albino | IP | BM | MN | 7 | Inv exponential | 0.02352 | 0.00724 | 0.05550 | 7.7 | 0.3 | 4.8 |
| Jenssen and Ramel ( 1980 ) | 1 | Mouse—CBA | IP | BM | MN | 5, 12 | Exponential | 115.6 | 92.3 | 148 | 1.6 | 431 | 539 |
| Lorenti Garcia et al. ( 2001 ) | 1 | Rat—Lewis | IP | SP | MN | 5 | Exponential | 23.14 | 13.8 | 33.6 | 2.4 | 64 | 108 |
| Lorenti Garcia et al. ( 2001 ) | 2 | Rat—Lewis | IP | BM | MN | 5 | Exponential and hill | 57.19 | 24.7 | 85.5 | 3.5 | 115 | 267 |
Note: Only inhalation studies were used for derivation of an inhalation permitted daily exposure (PDE) value. Studies using different routes of exposure (italicized) are presented for completeness. Data and modeling notes: (1) Reported data include zero response and/or zero variability. Surrogate values applied as described in the text. (2) PROAST noted potential data issue (AIC difference of full and best‐fit model >2). (3) Model results rejected due to imprecise BMDL and BMDU values (e.g., BMDU:BMDL>10,000, BMDL = 0) and potential data issue flag from PROAST. (4) Lack of dose–response trend was found by PROAST or authors of this paper (e.g., flat responses across administered doses). (5) Complete summary statistics or individual data were reported. (6) Reported summary statistic data were modeled to avoid zero values in the reported individual data. (7) Report lacked response variability. Modeled with reported mean response only. Modeled BMDL is divided by a factor of 5 (see the text). (8) Total aberration excluding gaps was modeled to avoid double‐counting. (9) The data set reported pups as experimental units and, as a result, do not take into account of litter effect from male sires. (10) All three concurrent control values were included in the modeling. Evaluated both Day 4.5–5.5 and 6.5–7.5 periods because authors indicate 4.5–7.5 is significant. Number of living per female is modeled since calculation of percent dominant lethal would result in control group with 0%. (11) Data shown in Generoso et al. (1990) for HTL for female SEC C57BL F1 stock were actually both female stocks combined. The companion paper by Rhomberg et al. (1990) reported Generoso et al. (1990) data for SEC C57BL F1 stock alone, and they were used for modeling. (12) Some models were biologically implausible (e.g., sharp S‐curve) and excluded. Best‐fit (lowest AIC) model selected from remaining models. (13) Highest dose group excluded in order for PROAST to successfully model dose–response. (14) Response values <1 × 10−5 mutations were replaced with 1 × 10−5 mutations, the reported experimental limit of detection. (15) Some models yielded imprecise BMD results (e.g., BMDL > BMDU) and excluded. Best‐fit (lowest AIC) model selected from remaining models. (16) Multiple expression time results were reported; expression time with significant response or most significant response is modeled. Assume control group results are concurrent with modeled expression time.
Abbreviations: AIC,Akaike information criterion; BM, bone marrow; CA, chromosomal aberration; CA‐RT, chromosomal aberration‐reciprocal translocation; CA‐TA, chromosomal aberration‐total aberration; CI, continuous inhalation; cII, cII mutation; DL, dominant lethal mutation; DW, drinking water; GE, male germ cells; Hprt, hprt mutation; HTL, heritable translocation; IH, inhalation; IP, intraperitoneal; IV, intravenous; K‐ras, K‐ras mutation; lacI, lacI mutation; LU, lung cells; MN, micronucelus formation; PBL, peripheral blood lymphocytes; S, spermatogonia cells; SL, splenic lymphocytes; SP, splenocytes; TE, testes.
BMD50 central estimate (BMD), BMD50 lower 95% confidence limit (BMDL), and BMD50 upper 95% confidence limit (BMDU) are estimated in the original units as shown in Table 1.
Modeled BMD and BMDL are adjusted to 24 hr/day and 7 days/week continuous exposure. IP and IV are converted to inhalation exposures where applicable (see the text).
2.2. BMD analysis
The BMD analysis using PROAST v. 67.0 (Dutch National Institute for Public Health and the Environment [RIVM], 2019) was conducted on the data sets to identify PoD values for EO‐induced genotoxicity. PROAST was chosen instead of the U.S. EPA BMD software to be consistent with previous BMD analyses of the genetic toxicology data (e.g., Gollapudi et al. 2013; Johnson et al. 2014). The critical effect size (CES; or benchmark response, BMR) of 50% was used as described in greater detail in Section 4. The data sets in their original exposure units were modeled in PROAST for continuous response data.
Some data sets included zero values in their response metrics for one or more dose levels, and those values are not amenable to modeling in PROAST, where responses are modeled in logarithmic scale. A conventional approach of using surrogate values to address those instances was used by replacing the zero value in the numerator with an incidence rate of 1 for the affected metric. For reported zero SD or SE values at a given dose, they were also replaced with surrogate variance values by assuming that the coefficient of variation of response at that dose is similar to that of the next highest dose having nonzero variance value. The surrogate variance value is calculated as the reported mean response at the given dose multiplied by the coefficient of variation of the next nonzero variance dose.
Results of dominant lethal mutation studies reported zero or negative response values since the response metrics of percent dominant lethality is defined as the difference of fetal deaths between treatment and the control group (1—average living treated/average living controls) (Generoso et al., 1986). By definition, the percent dominant lethals is always zero for the control group, and the result can be negative for the treated group if average living is greater than in controls. Since PROAST does not allow entry of zero or negative response values, the results are expressed as one minus the percent dominant lethals for the modeling analysis.
The data sets were not further transformed for the BMD analysis in PROAST. PROAST evaluated the data sets using four models (exponential, hill, inverse exponential, and lognormal) and provided model estimates for BMD (BMD central estimate), 90% confidence interval around the central estimate as BMDL (lower 95% confidence limit), BMDU (upper 95% confidence limit), and model average results. The best fit model is selected based on having the lowest Akaike information criterion (AIC) value and visual examination of model fit. In instances when visual examination of the fitted model showed a biologically unreasonable fit (e.g., nonmonotonic, very steep S‐curve), the result of that model was excluded from consideration. A model result is also excluded from consideration where the BMDL values appear unreliable, such as when the BMDU/BMDL ratio is >100, the BMDL value is estimated as greater than the BMDU, BMDL is zero, or BMDU is infinity.
2.3. Dose conversion to continuous inhalation concentrations
The dose metric used in all the BMD analyses presented here is the experimental exposure concentration, rather than the continuous exposure concentration that is relevant to human health risk assessment. Because inhalation studies on EO reviewed were conducted over 6 or 7 hr/day and 5 days/week (whole‐body exposure), all the estimated BMD50 and BMDL50 values were then converted to continuous inhalation (CI) concentration (CIBMD and CIBMDL50) over 24 hr/day and 7 days/week. For inhalation, the approach used in U.S. EPA (2016) for EO was followed. The BMD values in the original units were multiplied by hours of test exposure per 24 hr and by 5 days test exposure per 7 days to obtain continuous daily exposure values.
Although EO exposures by routes other than inhalation were not used to determine PDEs, quantitative analyses of dose responses in these studies were made for comparative purposes. For IP or IV injections only, the equivalent human CI exposures are obtained following the approach used in U.S. EPA (1996); the BMD values in the original units were multiplied by 70 kg20 m−3 day−1, with an additional factor of 5 days test exposure/7 days for repeat dose exposures, and the result was divided by 0.75 to account for the fraction of EO in air being retained during inhalation in humans (Brugnone et al., 1986).
2.4. Derivation of PDE levels via inhalation
For the derivation of candidate PDE for inhalation exposure, the inhalation studies reporting some measure of variability (e.g., SD or SE) evaluating mutagenicity (i.e., chromosomal aberration, chromosomal mutations, and gene mutations) were considered the most relevant experimental data for human risk assessment. However, PDEs were also derived for studies that did not meet these criteria, with additional adjustments as described below and in greater detail in Section 4.
where PDE = permitted daily exposure; CIBMDL50 = CI 50% response BMD lower 95% confidence limit; F1 = a data‐derived interspecies factor of 3.16 was applied instead of the standard 10, consistent with U.S. EPA (2016) use of 1 for the toxicokinetic interspecies subfactor; F2 = a factor of 10 to account for inter‐individual variability; F3 = A variable factor to account for studies of short‐term exposure; F3 = 1 for studies that last at least one‐half lifetime (1 year for rodents or rabbits; 7 years for cats, dogs, and monkeys); F3 = 1 for reproductive studies in which the whole period of organogenesis is covered; F3 = 2 for a 6‐month study in rodents or a 3.5‐year study in nonrodents; F3 = 5 for a 3‐month study in rodents or a 2‐year study in nonrodents; F3 = 10 for studies of a shorter duration; and F4 = a factor of 10 was used for severity of the effect induced by genotoxicity/mutagenicity.
In the final step of selecting the PDE from among the lowest candidate PDEs, the following factors were considered: duration of treatment, group size (minimum of 5), BMDU:BMDL ratio, number and range of exposure levels tested, dose–response pattern, and experimental design.
3. RESULTS
3.1. Data used for BMD analysis
Forty data sets were identified for dose–response analyses (Table 1). Except for one study in monkeys, all other studies were conducted in either rats or mice. All these studies were conducted by treating males except for Farooqi et al. (1993) who used female mice in their study and Applegren et al. (1978) who did not specify the sex of mice and rats used in their study. The routes of exposure included IP or IV injection, drinking water, and inhalation. The primary route of human exposure to EO is via inhalation, and as such, laboratory studies using inhalation exposure are considered the most relevant for human risk assessment. Data from studies using IP, IV, or drinking water administration were nevertheless included in the dose–response analysis for comparative purposes only (Table 2). The duration of treatments ranged from a single administration to exposures up to 2 years. Data derived from studies conducted with longer treatment durations are considered the most appropriate for chronic risk assessment as compared to acute or shorter‐term exposures. Both somatic and germ cells were investigated for genotoxicity across these studies.
The 40 data sets cover a wide range of genotoxicity endpoints, including MN formation, chromosomal aberrations, reciprocal translocations, gene mutations in the endogenous (Hprt locus) and transgenic reporter (lacI and cII) genes, dominant lethal mutations, and heritable translocations. Distinguishing between “mutagenicity” and “genotoxicity” is critical when using these data for risk assessment purposes. The term “genotoxicity” describes a continuum of events that may or may not lead to mutations. Mutagenicity, on the other hand, refers to changes in the DNA nucleotide sequence of the genome, transmissible from one cell to the daughter cells or from one generation to the other. Mutations differ from all other endpoints in the genotoxicity continuum in that they are the apical effects and as such are not repairable by the normal cellular defense mechanisms. Although not all mutations have a phenotypic consequence, the specific gene mutations evaluated in EO‐exposed rats and mice (i.e., Hprt, lacI, and cII) are detected by selection‐based methodologies, and as such, they are not silent mutations.
For human health risk assessment purposes, dose–response data for gene mutations and chromosomal effects that satisfied the quality criteria listed in Section 2 were considered the most appropriate endpoints. Chromosomal aberrations (including MN formation) were also considered to be relevant for risk assessment since their incidence has been shown to correlate to cancer in human biomonitoring studies (Norppa et al., 2006; Bolognesi et al., 2015). The mutational endpoints and chromosomal aberrations analyzed in the experimental studies reviewed in here are good predictors of the type of genetic damage that can lead to adverse health outcomes.
3.2. BMD analyses
Results from BMD50 analyses are presented in Table 2. Of the 40 data sets evaluated, 28 data sets successfully estimated BMD values. Nearly all data sets have low (<10) BMDU/BMDL ratios, suggesting a high level of precision of the BMD50 estimates. Higher BMDU/BMDL ratios of 31 and 134 (Donner et al., 2010) (chromosomal aberration in peripheral blood lymphocytes), and 47 and 80 (Recio et al., 2004) (lacI mutations in testes) indicate that the BMD estimates from these data are not as precise as the other data sets. The results with unreasonably large uncertainty, defined here as having a BMDU/BMDL ratio >100 (i.e., ratio of 134 from Donner et al. (2010)) were excluded from PDE derivation. For several data sets that did not report SD or SE, adjusted BMDL estimates were also listed in the table. Figure 1 shows sample PROAST modeling outputs using Donner et al. (2010) data set 6, Generoso et al. (1990) data set 4, and Recio et al. (2004) data set 2.
FIGURE 1.
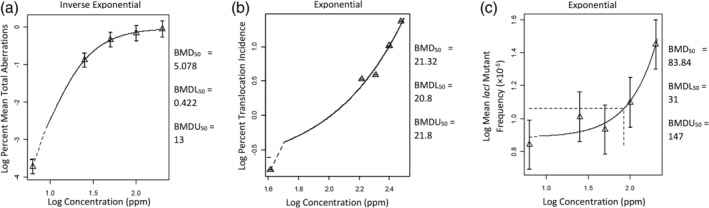
Examples of benchmark dose (BMD) analysis results adapted from PROAST outputs (see Appendix S1 for actual model output that provide greater details). Shown in the figure are the best‐fit models for the lowest permitted daily exposure (PDE) derivation for (a) chromosomal aberration (Donner et al. (2010) data set 6), (b) chromosomal mutation (Generoso et al. (1990) data set 4), and (c) gene mutations (Recio et al. (2004) data set 1). Name of the fitted model is shown above each figure. Triangle symbols and error bars are the geometic mean and confidence interval of the reported data. Solid curves are the modeled relationships between exposure and response. The controls are plotted at an arbitrary (but low) location on the log‐dose scale because log of zero is undefined; for that reason the model curve at the lower end is represented as a dashed line. Horizontal and vertical dashed lines indicate exposure associated with 50% response, but they may be difficult to see in (a) and (b) because they are very close to the axes. BMD50 is the 50% response benchmark dose central estimate, BMDL50 is the lower 95% confidence limit, and BMDU50 is the upper 95% confidence limit
3.3. Identification of PoD for the derivation of candidate PDE
As stated earlier, inhalation studies are considered the most relevant for human risk assessment on EO, and they are the basis for candidate PDE derivation. There are six data sets for gene mutations (cII, lacI, and Hprt) with model fit and acceptable BMDU/BMDL ratios (Table 3) and six acceptable data sets each for chromosomal mutations (dominant lethals and heritable translocations; Table 4) and chromosomal aberrations (Table 5). Candidate PDE values were not derived from studies that reported MN formation since the routes of exposure were IP or IV. The following sections provide detailed descriptions of weight of evidence, including consideration of study quality and dose–response pattern for the lowest candidate PDE values.
TABLE 3.
Point‐of‐departure (PoD) estimates for inhalation gene mutation studies and the corresponding permitted daily exposure (PDE) values
| Study | Data set | Species and strain | Group size | Tissue | Endpoint | Exposure concentration | Frequency and duration | CI a BMDL (ppm) | CI BMD a (ppm) | Total UF b | PDE (ppt) from CI BMDL | PDE (ppt) from CI BMD |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Manjanatha et al. (2017) | 2 | Mouse, big blue B6C3F1 | 10 | LU | cII | 0, 100, and 200 ppm | 6 hr/day, 5 days/week; 8 weeks | 8.1 | 20 | 3,162 c | 2,547 | 6,409 |
| Manjanatha et al. (2017) | 3 | Mouse, big blue B6C3F1 | 10 | LU | cII | 0, 100, 200 ppm | 6 hr/day, 5 days/week; 12 weeks | 25 | 35 | 1,581 d | 15,924 | 22,407 |
| Parsons et al. ( 2013 ) | 1 | Mouse—big blue B6C3F1 | 10 | LU | K‐ras | 0, 10, 50, 100, and 200 ppm | 6 hr/day, 5 days/week; 4 weeks | 0.29 | 2.1 | 3,162 c | 92 | 670 |
| Parsons et al. ( 2013 ) | 2 | Mouse—big blue B6C3F1 | 10 | LU | K‐ras | 0, 10, 50, 100, and 200 ppm | 6 hr/day, 5 days/week; 4 weeks | 0.19 | 1.2 | 3,162 c | 60 | 375 |
| Recio et al. (2004) | 1 | Mouse, B6C3F1 | 6 | BM | lacI | 0, 25, 50, 100, and 200 ppm | 6 hr/days, 5 days/week; 48 weeks | 5.5 | 15 | 316 e | 17,505 | 47,344 |
| Recio et al. (2004) | 2 | Mouse, B6C3F1 | 6 | TE | lacI | 0, 25, 50, 100, and 200 ppm | 6 hr/day, 5 days/week; 48 weeks | 0.09 | 0.45 | 316 e | 285 | 1,434 |
| van Sittert et al. (2000) | 1 | Rat—Lewis | 6–8 | SL | Hprt | 0, 50, 100, and 200 ppm | 6 hr/day, 5 days/week; 4 weeks | 9 | 20 | 3,162 c | 2,790 | 6,212 |
| Walker et al. (1997) | 1 | Mouse, B6C3F1 | 7–9 | SL | Hprt | 0, 50, 100, and 200 ppm | 6 hr/day, 5 days/week; 4 weeks | 4 | 6.4 | 3,162 c | 1,203 | 2,013 |
Note: Inhalation studies that reported variability of the data (SD or SEM) were used for PDE consideration are bolded. For reference (italicized), the PDE for inhalation studies that did not report variability were derived by dividing the BMDL by 5 (see the text).
Abbreviations: BM, bone marrow; CI, continuous inhalation; cII, cII mutation; Hprt, hprt mutation; lacI, lacI mutation; K‐ras, K‐ras mutation; LU, lung cells; SL, splenic lymphocytes; SP, splenocytes; TE, testes.
Modeled BMD and BMDL are adjusted to 24 hr/day and 7 days/week continuous exposure (see the text).
Total UF—total uncertainty factor based on F1 for interspecies toxicokinetic, F2 for inter‐individual variability, F3 for study duration, and F4 for effect severity (see the text).
Total UF based on F1 (TD only) 3.16, F2 10, F3 10, and F4 10.
Total UF based on F1 (TD only) 3.16, F2 10, F3 5, and F4 10.
Total UF based on F1 (TD only) 3.16, F2 10, F3 1, and F4 10.
TABLE 4.
Point‐of‐departure (PoD) estimates for inhalation heritable translocation and dominant lethal mutation studies, and the corresponding permitted daily exposure (PDE) values
| Study | Data set | Species and strain | Group size | Tissue | Endpoint | Exposure concentration | Frequency and duration | CI a BMDL (ppm) | CI BMD (ppm) | Total UF b | PDE (ppt) from CI BMDL | PDE (ppt) from CI BMD |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Generoso et al. ( 1986 ) | 1 | Mouse, (C3H × 101) F1 | 24 | GE | DL | 0, 300, 400, 500 ppm | 6 hr/day, 4 days; expression time 6.5–7.5 days | 16 | 83 | 3,162 c | 5,071 | 26,190 |
| Generoso et al. (1986) | 2 | Mouse, (C3H × 101) F1 | 24 | GE | DL | 0, 300, 400, 500 ppm | 6 hr/day, 4 days; expression time 4.5–5.5 days | 17 | 86 | 3,162 c | 5,274 | 27,071 |
| Generoso et al. ( 1990 ) | 1 | Mouse, (C3H × 101) F1 | 24 | GE | DL | 0, 165, 204, 250, and 300 ppm | 6 hr/day, 5 days/week for 8.5 weeks; daily for 2.5 weeks | 10 | 51 | 3,162 c | 3,196 | 16,269 |
| Generoso et al. ( 1990 ) | 2 | Mouse, (C3H × 101) F1 | 24 | GE | DL | 0, 165, 204, 250, and 300 ppm | 6 hr/day, 5 days/week for 8.5 weeks; daily for 2.5 weeks | 11 | 54 | 3,162 c | 3,354 | 17,150 |
| Generoso et al. (1990) | 3 | Mouse, (C3H × 101) F1 | 24 d | GE | HTL | 0, 165, 204, 250, and 300 ppm | 6 hr/day, 5 days/week for 8.5 weeks; daily for 2.5 weeks | 7.2 | 11 | 3,162 c | 2,287 | 3,464 |
| Generoso et al. (1990) | 4 | Mouse, (C3H × 101) F1 | 24 d | GE | HTL | 0, 165, 204, 250, and 300 ppm | 6 hr/day, 5 days/week for 8.5 weeks: daily for 2.5 weeks | 3.7 | 3.8 | 3,162 c | 1,175 | 1,204 |
Note: Inhalation studies that reported variability of the data (SD or SEM) were used for PDE consideration are bolded. For reference (italicized), the PDE for inhalation studies that did not report variability were derived by dividing the BMDL by 5 (see the text).
Abbreviations: CI, continuous inhalation; GE, Male germ cell; DL, dominant lethal mutation; HTL, heritable translocation.
Modeled BMD and BMDL are adjusted to 24 hr/day and 7 days/week continuous exposure (see the text).
Total UF—total uncertainty factor based on F1 for interspecies toxicokinetic, F2 for inter‐individual variability, F3 for study duration, and F4 for effect severity (see the text).
Total UF based on F1 (TD only) 3.16, F2 10, F3 10, and F4 10.
Number of male sires per dose group exposed to ethylene oxide. Reported results for heritable translocation were based on the total number of progenies per dose group.
TABLE 5.
Point‐of‐departure (PoD) estimates for inhalation chromosomal aberration studies and the corresponding permitted daily exposure (PDE) values
| Study | Data set | Species and strain | Group size | Tissue | Endpoint | Exposure concentration | Frequency and duration | CI BMDL a (ppm) | CI BMD (ppm) | Total UF b | PDE (ppt) from CI BMDL | PDE (ppt) from CI BMD |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Donner et al. (2010) | 1 | Mouse, B6C3F1 | 4–5 | PBL | CA‐RT | 0, 25, 50, 100, and 200 ppm | 6 hr/day; 5 days/week; 12 weeks | 3.7 | 5.1 | 1,581 c | 2,315 | 3,210 |
| Donner et al. (2010) | 2 | Mouse, B6C3F1 | 4–7 | PBL | CA‐RT | 0, 25, 50, 100, and 200 ppm | 6 hr/day; 5 days/week; 24 weeks | 3.0 | 11 | 632 d | 4,715 | 17,347 |
| Donner et al. (2010) | 3 | Mouse, B6C3F1 | 6–8 | PBL | CA‐RT | 0, 25, 50, 100, and 200 ppm | 6 hr/day; 5 days/week; 48 weeks | 0.3 | 0.84 | 316 e | 1,039 | 2,604 |
| Donner et al. (2010) | 6 | Mouse, B6C3F1 | 6–8 | PBL | CA‐TA | 0, 25, 50, 100, and 200 ppm | 6 hr/day; 5 days/week; 48 weeks | 0.075 | 0.91 | 316 e | 238 | 2,868 |
| Lynch et al. (1984) | 1 | Monkey, Macaca facicularis | 11–12 | PBL | CA | 0, 50, and 100 ppm | 7 hr/day; 5 days/week; 2 years | 0.3 | 1.3 | 316 e | 909 | 4,193 |
| Ribeiro et al. ( 1987 ) | 1 | Mouse, Swiss Webster | 10 | BM | CA | 0, 200, 400, and 600 ppm | 6 hr; 1 day | 4.2 | 4.2 | 3,162 f | 1,333 | 6,669 |
Note: Inhalation studies that reported variability of the data (SD or SEM) were used for PDE consideration are bolded. For reference (italicized), the PDE for inhalation studies that did not report variability were derived by dividing the BMDL by 5 (see the text).
Abbreviations: BM, bone marrow; CA, chromosomal aberration; CA‐RT, chromosomal aberrations‐reciprocal translocation; CA‐TA, chromosomal aberrations‐total aberration; CI, continuous inhalation; PBL, peripheral blood lymphocytes.
Modeled BMD and BMDL are adjusted to 24 hr/day and 7 days/week continuous exposure (see the text).
Total UF—total uncertainty factor based on F1 for interspecies toxicokinetic, F2 for inter‐individual variability, F3 for study duration, and F4 for effect severity (see the text).
Total UF based on F1 (TD only) 3.16, F2 10, F3 5, and F4 10.
Total UF based on F1 (TD only) 3.16, F2 10, F3 2, and F4 10.
Total UF based on F1 (TD only) 3.16, F2 10, F3 1, and F4 10.
Total UF based on F1 (TD only) 3.16, F2 10, F3 10, and F4 10.
3.3.1. Mutations
The three categories of genotoxicity endpoints analyzed here are gene mutations, chromosome mutations, and chromosome aberrations. As indicated above, mutations are nonrepairable changes in DNA sequences that are heritable and change genetic information content. Gene and chromosome mutations completely fulfill this definition, the latter referring to chromosome level changes measured in germ cells. Chromosome aberrations, however, are somewhat different in that, even though being non‐repairable events that change genetic content, the most common kind of aberrations scored are of the chromatid type and therefore, strictly speaking, not heritable because they are cell lethal. However, some chromosome aberrations are heritable, fulfilling all criteria for the strict definition of mutations. For example, the Donner et al. (2010) study included here measured reciprocal translocations, in addition to total chromosomal aberrations. Reciprocal translocations are heritable. Since chromosome aberrations capture mechanisms that underlie all mutations, they are included in the analyses presented here.
3.3.2. Gene mutations
For gene mutations, the lowest CIBMDL value was 0.09 ppm based on the induction of gene mutations in testes (seminiferous tubules) of mice following 48 weeks of inhalation exposure to EO (Table 3; (Recio et al., 2004). The study design was considered robust for deriving a PDE because of the chronic duration of exposure, because of the inclusion of 5 exposure levels including control, and because the exposure levels ranged from 25 to 200 ppm, a range below that causing metabolic saturation in mice. However, the BMDL value is a conservative PoD based on the dose–response pattern. The target gene analyzed in this study was a bacterial gene (lacI) incorporated into the genome of a mouse. The PDE value based on this CIBMDL was 285 parts per trillion (ppt). The data used for the BMD analysis was extracted from Table 4 of Recio et al. The lacI mutant frequencies (× 10−5; mean ± SD) were 3.6 ± 3.2, 6.2 ± 2.2, 7.7 ± 3.4, 8.5 ± 6.5, and 4.2 ± 2.7 at exposure concentrations of 0, 25, 50, 100, and 200 ppm EO, respectively. The mutant frequency values for 25, 50, and 100, but not for 200 ppm, were statistically different from 0 ppm. PROAST initially did not find a significant dose–response with the inclusion of the 200‐ppm dose. The BMD analysis for this data set was successful after excluding the 200‐ppm dose. A closer examination of the data reveals that the response observed was marginally dose related, and the precision of the BMD estimates are at the borderline of acceptability with a BMDU to BMDL ratio of 80. Furthermore, the spectrum of lacI mutations in EO‐exposed mice in the above study was not significantly different from the lacI background mutations. It is also noteworthy that, in the same study, the mutational spectrum for EO‐exposed bone marrow cells differed significantly from the background with a significant increase in the expected AT to TA transversions—a molecular signature of EO‐induced mutations. Additionally, there is a high level of variation at each dose, along with overlapping levels of variation between each dose and the vehicle control. Due to this high variation, it is inconclusive whether there is a dose response or not. Thus, although a PDE value was calculated from this data set, it needs to be pointed out that the value derived from this data set should be viewed with a greater degree of caution given (a) the lack of molecular signature, (b) the high BMDU to BMDL ratio, and (c) the wide variation in the data.
Measurements by Parsons et al. (2013) of K‐ras Codon 12 GGT → GAT mutations in lung cells of male mice exposed to 10, 50, 100, or 200 ppm EO by inhalation for 4 weeks resulted in the next lowest CIBMDL value of 0.19 ppm. Measurements for Codon 12 GGT → GTT mutations resulted in a CIBMDL value of 0.29 ppm. This data set did not meet criteria for derivation of a PDE because no response variance was reported.
A CIBMDL value of 4 ppm was calculated for Hprt mutations in splenic lymphocytes of mice exposed to EO by inhalation for 4 weeks (Walker et al., 1997). The BMD analysis used the following mean (±SE) of observed mutant frequencies from Walker et al. (1997): 2.2 (0.3), 3.8 (0.5), 6.8 (0.9), and 14.1 (1.1) at exposure concentrations of 0, 50, 100, and 200 ppm, respectively. The BMD estimate for this data set has an acceptable degree of precision as assessed from the BMDU:BMDL ratio of 2.6. The target gene in this study (Hprt) is an endogenous gene that responds to small basepair insertions, deletions, and substitutions in DNA, as well as large (mega basepair) deletions (Stout and Caskey, 1985; Lippert et al., 1995). The PDE calculated using this data set was 1,203 ppt.
van Sittert et al. (2000) also investigated the induction of Hprt mutations in splenic lymphocytes of rats after 4 weeks of inhalation exposure to EO. Although exposure levels and duration are the same as the ones observed by Walker et al. (1997), the increases in mutant frequencies observed by van Sittert et al. (2000) were only marginal, with mean (SD) values of 4.41 (1.45), 3.21 (1.06), 5.49 (2.10), and 6.41 (2.33) at 0, 50, 100, and 200 ppm, respectively. Only the mutant frequency observed at the 200‐ppm level was significantly different from the control value. The CIBMDL value for this data was 9 ppm with a corresponding PDE value of 2,790 ppt. Thus, the PDE value based on the rat data is 2.3‐fold higher than the corresponding value resulting from the analysis of data obtained in the mouse using a similar experimental design.
Recio et al. (2004) observed significant increase in lacI mutant frequency in the bone marrow of transgenic mice after 48 weeks of exposure to 100 and 200 ppm (but not to 25 or 50 ppm) EO. No increases in mutant frequencies were noted at any of the above exposure levels following 12 or 24 weeks of exposure. Manjanatha et al. (2017), on the other hand, observed significant (<2.5‐fold) increases in cII mutant frequency in the lungs of transgenic mice at 8 and 12 weeks of exposure, but not at 4 weeks. The CIBMDL values and the corresponding PDE estimates are also listed in Table 3 for comparative purpose.
3.3.3. Chromosomal mutations
Dominant lethal mutations are due to chromosomal aberrations in the germ cells leading to the death of embryos following fertilization, while heritable translocations lead to semi‐sterility in the progeny of exposed individuals that carry such events (Table 4). Both these assays are conducted in the germ cells of males. The effect of EO on these endpoints was investigated by Generoso and colleagues via inhalation exposure in two strains of mice (Generoso et al., 1986; 1990). BMD analysis was conducted for the reported data sets; however, the authors recognized that the responses were reported in terms of pups (N > 100 and up to 2,068 per dose group), while the experimental units should have been based on the exposed male sires (N = 24 per dose group) to account for litter effects. Quantitative BMD analysis based on pups instead of male sires is not appropriate for risk assessment purposes because it inflates the sample size. The lowest CIBMDL value was 3.7 ppm for heritable translocations in (C3H × 101)F1 mouse strain mated with (SEC × C57Bl)F1 females, which is followed by the value 7.2 ppm for the same endpoint in the mating of (C3H × 101)F1 treated males with T‐stock female mice. The corresponding PDE values were 1,175 and 2,287 ppt for the two female stocks, respectively. For dominant lethal mutations, the CIBMDL values were in the range of 10–17 ppm depending upon the strain and exposure levels tested with the corresponding PDE values ranging from 3,196 to 5,274 ppt.
3.3.4. Chromosomal aberrations
The endpoints in this category include reciprocal translocations and total chromosome aberrations evaluated in metaphase cells following exposure to EO (Table 5).
The lowest CIBMDL for chromosomal aberrations with an acceptable BMDU to BMDL ratio (i.e., <100) was 0.075 ppm for the peripheral blood lymphocytes of B6C3F1 mice exposed by inhalation to EO for 48 weeks (Donner et al., 2010). The study design is considered robust for deriving a PDE value because of the chronic exposure duration, the inclusion of five exposure levels including controls, and the adequate group size of 6–8. The PDE value after applying the adjustment factors to this CIBMDL estimate is 238 ppt. The dose‐related increase in aberrations was relatively weak, with the highest concentration (200 ppm) inducing less than 1% aberrations. In this study, there was no effect on aberration incidence at 6 weeks of exposure to 25, 50, 100, and 200 ppm EO, and the increases observed at 12 and 24 weeks were also weak (<1%). The overall weak response is likely related to the limited number of chromosomes interrogated for aberrations (4 pairs of painted chromosomes out of 20 pairs). For reciprocal translocations reported by Donner et al. (2010), the PDE values ranged from 1,039 to 4,715 ppt. Unfortunately, the data on reciprocal translocations in the primary spermatocytes of mice reported by Donner et al. (2010) did not fit any of the dose–response models in PROAST, and as such, no BMD values could be calculated from these data.
In the study by Lynch et al. (1984), chromosomal aberrations in peripheral blood lymphocytes of monkeys were evaluated following exposure to EO (50 or 100 ppm) for 2 years. This study could be considered the most relevant for human exposures based on the relevant species, the longest chronic exposure duration, and ample group size. A small but statistically significant increase in aberrant cells was noticed in this study at both concentrations: 0.6% in the 0‐ppm group versus 2.0 and 3.7% at 50 and 100 ppm EO exposure groups, respectively. The CIBMDL value for this study was 0.3 ppm, and the corresponding PDE estimate is 909 ppt.
Ribeiro et al. (1987) investigated chromosomal aberrations in the bone marrow of mice after a single 6‐hr exposure to EO at concentrations of 200, 400, and 600 ppm. The incidence of cells with aberrations (after excluding gaps from the reported values) was 0.4, 1.0, 3.0, and 3.4% at 0, 200, 400, and 600 ppm, respectively. The CIBMDL for this data set was 4.2 ppm with an estimated PDE of 1,333 ppt.
4. DISCUSSION
4.1. In vivo genotoxicity of EO
EO induced gene mutations, chromosomal mutations, and chromosomal aberrations in somatic and germ cells of experimental animals following inhalation exposure—the route of exposure most relevant to human risk assessment. The positive response in these studies appears to be dependent upon the concentration and duration of exposure. Some of the studies employed exposure concentrations ≥300 ppm. In mice, but not in rats, the detoxification of EO is saturated at an exposure concentration of 330 ppm (Brown et al., 1996). Since these high concentrations have no environmental relevance and exceeded the kinetically derived maximum dose (KMD), the information derived from such studies should be viewed with caution for human risk assessment, especially if the adverse effects were observed only at doses exceeding the KMD (Bus, 2017). EO is also a relatively weak genotoxicant, both in the magnitude of the observed response and the doses and exposure durations needed to elicit a significant response (Recio et al., 2004; Donner et al., 2010).
4.2. Selection of BMR and extrapolation/uncertainty factors
As described in Section 2, an a priori approach for BMD analysis was developed. The rationale behind several of the decisions made for this EO case study merits further discussion.
The BMD analysis for EO is not intended to identify a threshold dose level but to identify a PoD dose that induces a predetermined change in the response rate of the endpoint of interest, which can be used for risk assessment purposes. Recent analyses have shown that a CES or BMR in the range of 50% is more appropriate for in vivo mutagenicity data (Zeller et al., 2017) than the CES of 5% (Hardy et al., 2017) or 10% (Johnson et al., 2014; MacGregor et al., 2015a, 2015b). A CES of 50%, or BMR50, was selected for identifying the PoD using the approaches from Slob (2017) and Zeller et al. (2017). These two different approaches are based on determining a low but measurable increase above the background that can supersede the no‐observed‐effect level (NOEL) as the most suitable PoD metric for calculating human exposure limits. Both independent approaches have been used by the Genetic Toxicology Technical Committee of the Health and Environmental Sciences Institute (Washington, DC) to define endpoint‐specific CES of 50% for the in vivo MN assay and the transgenic rodent (TGR) mutation assays. These endpoint‐specific CES ensure that the PoD is precisely derived and biologically relevant for that endpoint. Note that for these continuous endpoints, a 50% CES or BMR50 is comparable to a 1.5‐fold increase above the background. An interesting comparison is that many toxicological endpoints, including TGR mutant frequency, have the pairwise testing significance level set at a twofold increase. Therefore, the NOEL can be anything below a twofold increase. An additional “severity” factor of 10 was applied to all BMDL estimates to account for the potential severity of the effect induced by genotoxicity/mutagenicity. Further discussion among the scientific community is needed regarding the appropriateness of this factor for all genotoxicity or mutagenicity endpoints. Studies that did not provide some measure of variability (e.g., SD or SE) were considered inadequate for PDE derivation but were included for comparison purposes. The BMDL estimate for studies that do not report some measure of variability is artificially elevated (less conservative). The BMDL was divided by an adjustment factor of five based on the ratio of BMD and BMDL values from inhalation studies that provided variance data, which showed average BMD:BMDL ratios as high as 4.9. The BMD:BMDL ratios averaged 4.9 for chromosomal aberrations (5 studies), 1.3 for chromosomal mutations (2 studies), 2.7 for gene mutations (7 studies), and 3.3 overall (14 studies across all endpoints). Thus, an adjustment factor of five was selected to be conservative (protective).
Candidate PDE values were calculated for inhalation studies using general guidance from international regulatory agencies. Consistent with U.S. EPA (2014) guidance on data‐derived extrapolation factors, quantitative data were used to replace default uncertainty factors for interspecies and intraspecies extrapolation. Data are available for the toxicokinetic component of the F1 factor to extrapolate from animal to human. Specifically, no interspecies scaling for rodent‐to‐human exposure concentration is needed based on U.S. EPA (2016) comparisons of measured EO air: blood partition coefficients in humans, mice, and rats, which showed partition coefficients in rodents are higher than in humans (i.e., assuming equivalent exposure concentration is conservative). This approach is also supported by validated the physiologically based pharmacokinetic (PBPK) model of Fennell and Brown (2001) that showed blood EO area‐under‐the‐curves with respect to air concentration are similar for rodents and humans at up to 200 ppm. Similarly, no monkey‐to‐human scaling for the Lynch et al. (1984) data set is needed since monkey pharmacokinetics are presumed to be more like humans than rodents.
Although studies not using the inhalation route of exposure did not meet our a priori study criteria for inclusion, a PDE was derived for these studies for comparison purposes. A data‐derived adjustment factor of 0.75 was applied to account for the fact that 100% of the IP and IV doses are delivered, whereas only 75% of EO in air is retained by humans exposed via inhalation (Brugnone et al., 1986).
While the adjustment factors described above were considered appropriate for the EO analyses presented here, we realize that they are subject to further debate among the scientific community. This issue was discussed at length recently by White et al. (2020). One of the most contentious adjustment factors is the variable factor used for the duration of the study (see F3 in Section 2). Typically, the dosing regimen in genetic toxicity studies is acute or subacute (28 days) and often use doses orders of magnitude higher than anticipated human exposure. Such a dose regimen is consistent with the recommendations of various regulatory agencies and international guidelines for testing. The logical question then is whether there is a need for an uncertainty factor of 10 for a genetic toxicity study conducted for less than 3 months in rodents. We elected to apply the standard uncertainty factors for the analyses presented here since this issue is still unresolved and arguments can be presented for and against the use of this factor. For example, both Donner et al. (2010) and Recio et al. (2004) have clearly shown an exposure duration dependent increase in chromosomal aberrations and gene mutations in mice, respectively, with Recio et al. observing increases in mutations in the bone marrow only at 48 weeks but not at 12 or 24 weeks of exposure to EO.
4.3. Genotoxicity of EO as an endpoint for risk assessment
The recognition and the emphasis in recent years that genotoxicity data could and should be used on its own merit for human risk assessment (Gollapudi et al., 2013; Johnson et al., 2014; Heflich et al., 2020; Klapacz and Gollapudi, 2020) provided the impetus to examine the available EO dose–response data for the identification of a PoD for the derivation of a PDE. The in vivo genotoxicity of EO has been investigated via various routes of exposure. Data from other routes of exposure were also analyzed and presented in Table 2, for comparative purposes only.
Genotoxicity of EO has been examined in multiple species: mice, rats, and monkeys. This poses the question of which species is the most relevant to human risk assessment. Non‐human primates are assumed to be more relevant to humans than rats and mice in terms of physiology and breathing patterns. Based on the PBPK model of Fennell and Brown (2001), rats, mice and humans are similar in terms of internal dosimetry of EO at exposures less than 200 ppm. At exposure levels >200 ppm, rats are more similar to humans than mice in terms of internal dosimetry of EO, whereas mice diverge from rats and humans. For the purpose of EO mutagenicity risk assessment, data from all three species (mice, rats, and monkeys) were considered equally informative since genotoxicity of EO was evident in these species at exposure concentrations around and below 200 ppm. Similarly, no distinction was made on the strain of mouse used in these investigations due to a paucity of data on strain specific EO metabolism and dosimetry. Furthermore, in the analysis reported here, no remarkable differences in BMD estimates were observed in most cases when different strains (albeit limited in number) were investigated for the same endpoint (see Table 5).
As discussed previously, data on gene and chromosomal mutations along with chromosome aberrations were considered the most relevant for risk assessment. Although multiple tissues/cell types (bone marrow, peripheral blood lymphocytes, splenic lymphocytes, and germ cells) were investigated for EO‐induced genotoxicity, the effects observed in all these tissues were considered equally informative since the objective is to identify a dose level of EO that provides an acceptable level of protection to the genome of an individual. Such a dose level is also believed to protect the individual from all the adverse effects mediated through mutagenicity such as heritable diseases, cancer, and diseases associated with somatic mosaicism (Heflich et al., 2020).
To identify the lowest PDE values for different endpoints, only those values derived from data sets reporting a variability metric in Tables 3, 4, 5 were considered. For chromosomal mutations, the CIBMDL for heritable translocations in (C3H × 101)F1 mice from 8.5 weeks of EO exposures reported by Generoso et al. (1990) resulted in the lowest PDE value of 1,175 ppt. However, this candidate PDE value is not appropriate to use for risk assessment purposes because the experimental unit of measurement should have been based on the exposed male sires and not the individual pups. For gene mutations, the lowest PDE value of 285 ppt is derived from the CIBMDL for lacI mutations in male germ cells following 48‐week exposure reported by Recio et al. (2004). As discussed before, there are limitations regarding the data set used to derive this value, including the absence of a dose–response and lack of molecular signature consistent with EO‐induced mutagenesis. Furthermore, the analysis for lacI mutants in the bone marrows of the same mice as those used for interrogating germ cell mutations (following 48 weeks of exposure to EO) gave a clear dose response accompanied by a mutational spectrum consistent with EO‐induced mutagenicity. The PDE value derived from the bone marrow data was 17,505 ppt. Such a huge difference in PDE between bone marrow and testes (17,505 ppt versus 285 ppt for testes) is mechanistically unexplainable since germ cells are considered to be relatively less sensitive to EO‐induced mutagenicity due to the prolonged G1 phase of the spermatogonial cell cycle, providing greater opportunity for the repair of induced genetic damage (Donner et al., 2010). Despite the limitations of the germ cell data from Recio et al. (2004), the corresponding PDE value (i.e., 285 ppt) was still considered a worst‐case scenario for this case study. Although it would be ideal to verify the germ cell findings of Recio et al. in an independent study, such a replicate study is unrealistic given the complexity and expense of this chronic study. The PDE values (based on CIBMDL) for other studies listed in Tables 3 and 4 for gene and chromosomal mutations ranged from 2,287 to 15,924 ppt. For chromosomal aberrations, the lowest PDE value of 238 ppt was estimated from the data reported in Donner et al. (2010) for peripheral blood lymphocytes of mice following 48 weeks of exposure to EO (Table 5).
Collectively, the lowest PDE estimates for chromosomal mutations, gene mutations, and chromosomal aberrations were 1,175, 285, and 238 ppt, respectively. While the estimates for gene and chromosomal mutations were derived from male germ cells, the PDE value for chromosomal aberrations was derived from somatic cells. The lowest of the three values (238 ppt derived for chromosomal aberration in peripheral blood lymphocytes from Donner et al. (2010)) could thus represent the permissible daily exposure to EO based on the quantitative analysis of the genetic toxicology data on EO for risk assessment purposes.
The PDE of 238 ppt proposed in this publication is more than three orders of magnitude higher than the 0.1 ppt established by the U.S. EPA (2016) and similar to the 240 ppt estimated from TCEQ (2020) risk values for 1‐in‐106 (1/M) extra risk. Both agencies based their risk assessments on the same cohort of sterilizer workers (Steenland et al., 2003; 2004). The major reason for the >2,000‐fold difference in the 1/M extra risk level is the selection of two quite different statistical models used for low‐dose extrapolation. The U.S. EPA Integrated Risk Information System (2016) applied a supralinear two‐piece linear spline model based on statistical significance and a major emphasis on visual fit of different models with categorical grouped data points. These grouped data were few in number (5 or 10) and not representative of the much larger (53 and 233) number of cases that were actually modeled. The supralinear model implies a very steep increase in extra risk at lower exposures compared to higher exposure levels. In contrast, TCEQ (2020) selected the standard Cox proportional hazards (Cox PH) model based on the biological cancer mode of action as the primary basis for informing the low‐dose extrapolation, statistical significance, and consideration of which model accurately predicts the observed data overall and in the lower exposure regions. The Cox PH model, also selected by the European Commission Scientific Committee on Occupational Exposure Limits for EO (SCOEL, 2012), is linear over exposures of interest (TCEQ, 2020). For additional perspective, the TCEQ cancer risk value for 1/M extra risk of 240 ppt and the proposed PDE value of 238 are below mean endogenous equivalent EO exposures of 1,900 ± 1,300 ppt measured in humans (Kirman and Hays, 2017).
4.4. Concluding remarks
The diverse dose–response data for EO‐induced genotoxicity available from the literature enabled the identification of PoDs for various endpoints, tissues, and species in this retrospective analysis. Generating such diverse data prospectively from studies that are compliant with the Organization for Economic Co‐operation and Development test guidelines, while desirable, is prohibitively expensive and time consuming.
Several challenges in the BMD analysis were encountered during this study. Some of the challenges included data quality, experimental design, and PROAST model evaluation. While some challenges were addressed with ad hoc solutions, as documented in Section 2, others involved inherent data quality issue that could not be resolved completely. In general, given the maturity of the genotoxicity literature on EO, the 40 data sets reviewed herein were not designed with the objective of conducting quantitative BMD analysis for PDE derivation. In some cases, the studies were published more than two decades ago and do not meet current standards for data quality (e.g., variance not reported). Our novel approach of carrying out BMD analysis on each dose response, even those without optimum study design, provided an additional and very useful method of assessment. Precision of the BMD through the BMD CI range and certain data sets showing no dose response at the doses tested enabled enhanced discussion around those data sets and provided increased focus on and confidence in the studies used for PDE assessment.
To improve the quality of genotoxicity studies for quantitative analysis, we identified several important issues. First, studies should have at least three exposure levels in addition to the control. As Slob et al. (2005) explained, BMD analysis benefits from a design with more dose groups. More dose groups (e.g., 3 or 4 treated exposure levels), each with an adequate number of animals (e.g., ≥5), can better characterize dose–response relationships and identify the BMD without losing statistical precision. Ideally, these dose levels should be relevant to human exposure levels avoiding excessive toxicity (e.g., consideration of clinical observations, histopathology, and metabolic saturation). Second, the traditional approach in genotoxicity studies is to report data with litter as the experimental unit for dominant lethal studies but with individual pups as the experimental unit for heritable translocations. In the case of the heritable translocation test, the male sires are the experimental unit (i.e., they were the ones that were exposed). If their sperm is affected, then the male sire and not the individual pups in the litter spawned by them should be considered the experimental unit for quantitative analysis (Piegorsch and Haseman, 1991). This is consistent with guidance developed by U.S. EPA (1991) for developmental toxicity risk assessment. Third, several data sets included zero response such that surrogate values had to be developed with assumptions the authors consider reasonable so they could be included in this analysis, but which nevertheless, are assumptions. Fourth, BMD results should be examined carefully against the data to determine whether the results are biologically plausible and consistent with the authors' conclusions. Due to either inherent data quality, dose spacing, and/or modeling algorithm limitations, some results could be numerically illogical (e.g., lower confidence limit higher than upper confidence limit) or, more critically, not biologically reasonable, as exemplified by the huge difference in CIBMDL for lacI mutations between bone marrow (5.5 ppm) and testes (0.09 ppm). There were also instances where goodness‐of‐fit statistics conveyed good model fit to the data, yet visual examination of the modeled dose–response curve found its shape biologically unreasonable (e.g., sharp‐S). Additional quality considerations that could aid in selecting studies for PDE derivation include characterization of test material, evidence of monitoring chamber concentrations during exposure, and detection of an effect in the positive control to demonstrate technical competence in identifying an induced effect.
As quantitative approaches for the application of genotoxicity data advance, an issue that merits further discussion is whether to derive acceptable exposure levels separately for somatic and germ cells. Some scientists might consider the PDE value for somatic cells more relevant for protecting against carcinogenic effects in the exposed population while the PDE for germ cells might be more relevant for protecting future generations. Obviously, the lower of the two values would afford protection against both adverse outcomes. However, data from germ cells are seldom available for a vast majority of substances, precluding the derivation of PDE for germ cells. While the PDE values derived for somatic and germ cell endpoints are approximately the same for EO, further analyses with a broader set of substances are necessary to determine whether the value derived from the somatic cells is predictive of the PDE for germ cells.
As the field of genetic toxicology moves from qualitative hazard identification to quantitative risk assessment, it is recommended that genetic toxicology testing be designed such that high‐quality dose–response data are available for quantitative analysis and the identification of PoD values with high levels of precision. The species of choice and route of exposure for testing should be based on considerations relevant to humans. In the specific case of EO, there are two chronic inhalation mutagenicity studies of adequate quality for BMD analysis that provide converging evidence supporting the lowest PDE of 238 ppt.
CONFLICT OF INTEREST
Funding for this work was provided by the American Chemistry Council, Ethylene Oxide Task Force, and LyondellBasell. Decisions concerning analysis and conclusion were made solely by the authors. The opinions of the authors do not necessarily reflect the opinions of the sponsors.
AUTHOR CONTRIBUTIONS
B. Bhaskar Gollapudi, Abby A. Li, and Richard J. Albertini contributed equally to the conceptualization of the project. All authors contributed equally to data analysis, interpretation, and drafting of the manuscript.
Supporting information
Appendix S1: BMD50 analysis
ACKNOWLEDGMENTS
The authors thank Drs. Bas Bokkers and Wout Slob of the Dutch National Institute for Public Health and the Environment (RIVM) for their patient and enlightening responses to our many PROAST‐related queries.
Gollapudi BB, Su S, Li AA, Johnson GE, Reiss R, Albertini RJ. Genotoxicity as a toxicologically relevant endpoint to inform risk assessment: A case study with ethylene oxide. Environ Mol Mutagen. 2020;61:852–871. 10.1002/em.22408
Accepted by: P. White
Funding information American Chemistry Council; Ethylene Oxide Task Force; LyondellBasell
REFERENCES
- Agurell, E. , Cederberg, H. , Ehrenberg, L. , Lindahl‐Kiessling, K. , Rannug, U. and Törnqvist, M. (1991) Genotoxic effects of ethylene oxide and propylene oxide: a comparative study. Mutation Research, 250(1–2), 229–237. [DOI] [PubMed] [Google Scholar]
- Applegren, L.E. , Eneroth, G. , Grant, C. , Lanström, L.E. and Tenghagen, K. (1978) Testing of ethylene oxide for mutagenicity using the micronucleus test in mice and rats. Acta Pharmacologica et Toxicologica, 43, 69–67. [DOI] [PubMed] [Google Scholar]
- Bolognesi, C. , Bonassi, S. , Knasmueller, S. , Fenech, M. , Bruzzone, M. , Lando, C. and Ceppi, M. (2015) Clinical application of micronucleus test in exfoliated buccal cells: a systematic review and metanalysis. Mutation Research, Reviews in Mutation Research, 766, 20–31. [DOI] [PubMed] [Google Scholar]
- Boysen, G. , Pachkowski, B.F. , Nakamura, J. and Swenberg, J.A. (2009) The formation and biological significance of N7‐guanine adducts. Mutation Research, 678(2), 76–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, C.D. , Wong, B.A. and Fennell, T.R. (1996) In vivo and in vitro kinetics of ethylene oxide metabolism in rats and mice. Toxicology and Applied Pharmacology, 136(1), 8–19. [DOI] [PubMed] [Google Scholar]
- Brugnone, F. , Perbellini, L. , Faccini, G.B. , Pasini, F. , Bartolucci, G.B. and DeRosa, E. (1986) Ethylene oxide exposure: biological monitoring by analysis of alveolar air and blood. International Archives of Occupational and Environmental Health, 58, 105–112. [DOI] [PubMed] [Google Scholar]
- Bus, J.S. (2017) “The dose makes the poison”: key implications for mode of action (mechanistic) research in a 21st century toxicology paradigm. Current Opinions in Toxicology, 3, 87–91. [Google Scholar]
- Conan, R.A. , Waggy, G.T. , Spiegel, M.H. and Berglund, R.L. (1979) Study of the mutagenic action of ethylene oxide, ethylene glycol, and 2‐chloroethanol residues in plastic material sterilized by ethylene oxide. Annales des Falsifications et De L'Expertise Chimique et Toxicologique, 72, 141–151. [Google Scholar]
- Dearfield, K.L. , Gollapudi, B.B. , Bemis, J.C. , Benz, R.D. , Douglas, G.R. , Elespuru, R.K. , Johnson, G.E. , Kirkland, D.J. , LeBaron, M.J. , Li, A.P. , Marchetti, F. , Pottenger, L.H. , Rorije, E. , Tanir, J.Y. , Thybaud, V. , van Benthem, J. , Yauk, C.L. , Zeiger, E. and Luijten, M. (2017) Next generation testing strategy for assessment of genomic damage: a conceptual framework and considerations. Environmental and Molecular Mutagenesis, 58(5), 264–283. [DOI] [PubMed] [Google Scholar]
- Dellarco, V.L. , Generoso, W.M. , Sega, G.A. , Fowle, J.R. and Jacobson‐Kram, D. (1990) Review of the mutagenicity of ethylene oxide (Review). Environmental and Molecular Mutagenesis, 16, 85–103. [DOI] [PubMed] [Google Scholar]
- Donner, E.M. , Wong, B.A. , James, R.A. and Preston, R.J. (2010) Reciprocal translocations in somatic and germ cells of mice chronically exposed by inhalation to ethylene oxide: implications for risk assessment. Mutagenesis, 25(1), 49–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutch National Institute for Public Health and the Environment (RIVM) 2019. PROAST. Version 67.0 (menu‐driven). Available at: https://www.rivm.nl/en/proast [Accessed 29th October 2019].
- Ehrenberg, L. and Hussain, S. (1981) Genetic toxicity of some important epoxides (Review). DNA Repair, 86, 1–113. [DOI] [PubMed] [Google Scholar]
- Ehrenberg, L. , Osterman‐Golkar, S. , Segerbäck, D. , Svensson, K. and Calleman, C.J. (1977) Evaluation of genetic risks of alkylating agents. III. Alkylation of haemoglobin after metabolic conversion of ethene to ethene oxide in vivo. Mutation Research, 45(2), 175–184. [DOI] [PubMed] [Google Scholar]
- European Commission Scientific Committee on Occupational Exposure Limits (SCOEL) . (2012). Recommendation from the Scientific Committee on Occupational Exposure Limits for ethylene oxide. Available at: https://ec.europa.eu/social/BlobServlet?docId=7879. [Accessed 16th April 2020]. [Google Scholar]
- European Medicines Agency (EMEA) . (2008). Studies assessed by the European Medicines Agency indicate no increased risk of developing cancer for patients who have taken Viracept contaminated with ethyl mesilate. Press release. Available at: https://www.ema.europa.eu/en/news/studies-assessed-european-medicines-agency-indicate-no-increased-risk-developing-cancer-patients-who. [Accessed 16th April 2020].
- Farooqi, Z. , Törnqvist, M. , Ehrenberg, L. and Natarajan, A.T. (1993) Genotoxic effects of ethylene oxide and propylene oxide in mouse bone marrow cells. Mutation Research, 299, 223–228. [DOI] [PubMed] [Google Scholar]
- Fennell, T.R. and Brown, C.D. (2001) A physiologically based pharmacokinetic model for ethylene oxide in mouse, rat, and human. Toxicology and Applied Pharmacology, 173, 161–175. [DOI] [PubMed] [Google Scholar]
- Filser, J.G. , Denk, B. , Törnqvist, M. , Kessler, W. and Ehrenberg, L. (1992) Pharmacokinetics of ethylene in man; body burden with ethylene oxide and hydroxyethylation of hemoglobin due to endogenous and environmental ethylene. Archives of Toxicology, 66, 157–163. [DOI] [PubMed] [Google Scholar]
- Generoso, W.M. , Cain, K.T. , Cornett, C.V. , Cacheiro, N.L. and Hughes, L.A. (1990) Concentration–response curves for ethylene‐oxide‐induced heritable translocations and dominant lethal mutations. Environmental and Molecular Mutagenesis, 16(2), 126–131. [DOI] [PubMed] [Google Scholar]
- Generoso, W.M. , Cain, K.T. , Hughes, L.A. , Sega, G.A. , Braden, P.W. , Gosslee, D.G. and Shelby, M.D. (1986) Ethylene oxide dose and dose‐rate effects in the mouse dominant‐lethal test. Environmental Mutagenesis, 8(1), 1–7. [DOI] [PubMed] [Google Scholar]
- Generoso, W.M. , Cain, K.T. , Krishna, M. , Sheu, C.W. and Gryder, R.M. (1980) Heritable translocation and dominant‐lethal mutation induction with ethylene oxide in mice. Mutation Research, 73(1), 133–142. [DOI] [PubMed] [Google Scholar]
- Gollapudi, B.B. (2017) An ongoing journey toward a risk‐based testing in genetic toxicology. Current Opinion in Toxicology, 3, 71–74. [Google Scholar]
- Gollapudi, B.B. , Johnson, G.E. , Hernandez, L.G. , Pottenger, L.H. , Dearfield, K.L. , Jeffrey, A.M. , Julien, E. , Kim, J.H. , Lovell, D.P. , Macgregor, J.T. , Moore, M.M. , van Benthem, J. , White, P.A. , Zeiger, E. and Thybaud, V. (2013) Quantitative approaches for assessing dose–response relationships in genetic toxicology studies. Environmental and Molecular Mutagenesis, 54(1), 8–18. [DOI] [PubMed] [Google Scholar]
- Gollapudi, B.B. , Kamra, O.P. and Blecher, S.R. (1981) A search for a genetic basis for gonosomal univalency in mice. Cytogenetics and Cell Genetics, 29(4), 241–249. [DOI] [PubMed] [Google Scholar]
- Hardy, A. , Benford, D. , Halldorsson, T. , Jeger, M.J. , Knutsen, K.H. , More, S. , Mortensen, A. , Naegeli, H. , Noteborn, H. , Ockleford, C. , Ricci, A. , Rychen, G. , Silano, V. , Solecki, R. , Turck, D. , Aerts, M. , Bodin, L. , Davis, A. , Edler, L. , Gundert‐Remy, U. , Sand, S. , Slob, W. , Bottex, B. , Abrahantes, J.C. , Marques, D.C. , Kass, G. and Schlatter, J.R. (2017) Update: use of the benchmark dose approach in risk assessment. EFSA Journal, 15(1), 4658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heflich, R.H. , Johnson, G.E. , Zeller, A. , Marchetti, F. , Douglas, G.R. , Witt, K.L. , Gollapudi, B.B. and White, P.A. (2020) Mutation as a toxicological endpoint for regulatory decision‐making. Environmental and Molecular Mutagenesis, 61(1), 34–41. [DOI] [PubMed] [Google Scholar]
- International Commission for Protection Against Environmental Mutagens and Carcinogens (ICPEMC) . (1980) ICPEMC news no. 2. Environmental Health Perspectives, 34, 223–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenssen, D. and Ramel, C. (1980) The micronucleus test is part of a short‐term mutagenicity test program for the prediction of carcinogenicity evaluated by 143 agents test. Mutation Research, 75, 191–202. [DOI] [PubMed] [Google Scholar]
- Jinot, J. , Fritz, J.M. , Vulimiri, S.V. and Keshava, N. (2018) Carcinogenicity of ethylene oxide: key findings and scientific issues. Toxicology Mechanisms and Methods, 28(5), 386–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson, G.E. , Soeteman‐Hernández, L.G. , Gollapudi, B.B. , Bodger, O.G. , Dearfield, K.L. , Heflich, R.H. , Hixon, J.G. , Lovell, D.P. , MacGregor, J.T. , Pottenger, L.H. , Thompson, C.M. , Abraham, L. , Thybaud, V. , Tanir, J.Y. , Zeiger, E. , van Benthem, J. and White, P.A. (2014) Derivation of point of departure (PoD) estimates in genetic toxicology studies and their potential applications in risk assessment. Environmental and Molecular Mutagenesis, 55(8), 609–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirman, C.R. and Hays, S.M. (2017) Derivation of endogenous equivalent values to support risk assessment and risk management decisions for an endogenous carcinogen: ethylene oxide. Regulatory Toxicology and Pharmacology, 91, 165–172. [DOI] [PubMed] [Google Scholar]
- Klapacz, J. and Gollapudi, B.B. (2020) Considerations for the use of mutation as a regulatory endpoint in risk assessment. Environmental and Molecular Mutagenesis, 61(1), 84–93. [DOI] [PubMed] [Google Scholar]
- Kligerman, A.D. , Erexson, G.L. , Phelps, M.E. and Wilmer, J.L. (1983) Sister‐chromatid exchange induction in peripheral blood lymphocytes of rats exposed to ethylene oxide by inhalation. Mutation Research, 120, 37–44. [DOI] [PubMed] [Google Scholar]
- Li, F. , Segal, A. and Solomon, J.J. (1992) In vitro reaction of ethylene oxide with DNA and characterization of DNA adducts. Chemico‐Biological Interactions, 83(1), 35–54. [DOI] [PubMed] [Google Scholar]
- Lippert, M.J. , Nicklas, J.A. , Hunter, T.C. and Albertini, R.J. (1995) Pulsed field analysis of hprt T‐cell large deletions: telomeric region breakpoint spectrum. Mutation Research, 326(1), 51–64. [DOI] [PubMed] [Google Scholar]
- Lorenti Garcia, C. , Darroudi, F. , Tates, A.D. and Natarajan, A.T. (2001) Induction and persistence of micronuclei, sister‐chromatid exchanges and chromosomal aberrations in splenocytes and bone‐marrow cells of rats exposed to ethylene oxide. Mutation Research, 492, 59–67. [DOI] [PubMed] [Google Scholar]
- Luijten, M. , Ball, N.S. , Dearfield, K.L. , Gollapudi, B.B. , Johnson, G.E. , Madia, F. , Peel, L. , Pfuhler, S. , Settivari, R.S. , Ter Burg, W. , White, P.A. and van Benthem, J. (2020) Utility of a next generation framework for assessment of genomic damage: a case study using the industrial chemical benzene. Environmental and Molecular Mutagenesis, 61(1), 94–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch, D.W. , Lewis, T.R. , Moorman, W.J. , Burg, J.R. , Gulati, D.K. , Kaur, P. and Sabharwal, P.S. (1984) Sister‐chromatid exchanges and chromosome aberrations in lymphocytes from monkeys exposed to ethylene oxide and propylene oxide by inhalation. Toxicology and Applied Pharmacology, 76(1), 85–95. [DOI] [PubMed] [Google Scholar]
- MacGregor, J.T. , Frötschl, R. , White, P.A. , Crump, K.S. , Eastmond, D.A. , Fukushima, S. , Guérard, M. , Hayashi, M. , Soeteman‐Hernández, L.G. , Johnson, G.E. , Kasamatsu, T. , Levy, D.D. , Morita, T. , Müller, L. , Schoeny, R. , Schuler, M.J. and Thybaud, V. (2015b) IWGT report on quantitative approaches to genotoxicity risk assessment II. Use of point‐of‐departure (PoD) metrics in defining acceptable exposure limits and assessing human risk. Mutation Research, Genetic Toxicology and Environmental Mutagenesis, 783, 66–78. [DOI] [PubMed] [Google Scholar]
- MacGregor, J.T. , Frötschl, R. , White, P.A. , Crump, K.S. , Eastmond, D.A. , Fukushima, S. , Guérard, M. , Hayashi, M. , Soeteman‐Hernández, L.G. , Kasamatsu, T. , Levy, D.D. , Morita, T. , Müller, L. , Schoeny, R. , Schuler, M.J. , Thybaud, V. and Johnson, G.E. (2015a) IWGT report on quantitative approaches to genotoxicity risk assessment I. methods and metrics for defining exposure‐response relationships and points of departure (PoDs). Mutation Research, Genetic Toxicology and Environmental Mutagenesis, 783, 55–65. [DOI] [PubMed] [Google Scholar]
- Manjanatha, M.G. , Shelton, S.D. , Chen, Y. , Parsons, B.L. , Myers, M.B. , McKim, K.L. , Gollapudi, B.B. , Moore, N.P. , Haber, L.T. , Allen, B. and Moore, M.M. (2017) Dose and temporal evaluation of ethylene oxide‐induced mutagenicity in the lungs of male big blue mice following inhalation exposure to carcinogenic concentrations. Environmental and Molecular Mutagenesis, 58(3), 122–134. [DOI] [PubMed] [Google Scholar]
- Müller, L. and Gocke, E. (2009) Considerations regarding a permitted daily exposure calculation for ethyl methanesulfonate. Toxicology Letters, 190(3), 330–332. [DOI] [PubMed] [Google Scholar]
- Norppa, H. , Bonassi, S. , Hansteen, I.L. , Hagmar, L. , Strömberg, U. , Rössner, P. , Boffetta, P. , Lindholm, C. , Gundy, S. , Lazutka, J. , Cebulska‐Wasilewska, A. , Fabiánová, E. , Srám, R.J. , Knudsen, L.E. , Barale, R. and Fucic, A. (2006) Chromosomal aberrations and SCEs as biomarkers of cancer risk. Mutation Research, 600(1–2), 37–45. [DOI] [PubMed] [Google Scholar]
- Parsons, B.L. , Manjanatha, M.G. , Myers, M.B. , McKim, K.L. , Shelton, S.D. , Wang, Y. , Gollapudi, B.B. , Moore, N.P. , Haber, L.T. and Moore, M.M. (2013) Temporal changes in K‐ras mutant fraction in lung tissue of big blue B6C3F1 mice exposed to ethylene oxide. Toxicological Sciences, 136(1), 26–38. [DOI] [PubMed] [Google Scholar]
- Philippin, G. , Cadet, J. , Gasparutto, D. , Mazon, G. and Fuchs, R.P. (2014) Ethylene oxide and propylene oxide derived N7‐alkylguanine adducts are bypassed accurately in vivo. DNA Repair (Amst), 22, 133–136. [DOI] [PubMed] [Google Scholar]
- Piegorsch, W.W. and Haseman, J.K. (1991) Statistical methods for analyzing developmental toxicity data. Teratogenesis, Carcinogenesis, and Mutagenesis, 11, 115–133. [DOI] [PubMed] [Google Scholar]
- Pottenger, L.H. , Bus, J.S. and Gollapudi, B.B. (2007) Genetic toxicity assessment: employing the best science for human safety evaluation part VI: when salt and sugar and vegetables are positive, how can genotoxicity data serve to inform risk assessment? Toxicological Sciences, 98(2), 327–331. [DOI] [PubMed] [Google Scholar]
- Pottenger, L.H. and Gollapudi, B.B. (2009) A case for a new paradigm in genetic toxicology testing. Mutation Research, 678(2), 148–151. [DOI] [PubMed] [Google Scholar]
- Pottenger, L.H. and Gollapudi, B.B. (2010) Genotoxicity testing: moving beyond qualitative “screen and bin” approach towards characterization of dose–response and thresholds. Environmental and Molecular Mutagenesis, 51(8–9), 792–799. [DOI] [PubMed] [Google Scholar]
- Preston, R.J. and Abernethy, D.J. (1993) Studies on the induction of chromosomal aberration and sister chromatid exchange in rats exposed to styrene by inhalation. IARC Scientific Publications, 127, 225–233. [PubMed] [Google Scholar]
- Recio, L. , Donner, M. , Abernethy, D. , Pluta, L. , Steen, A.M. , Wong, B.A. , James, A. and Preston, R.J. (2004) In vivo mutagenicity and mutation spectrum in the bone marrow and testes of B6C3F1 lacI transgenic mice following inhalation exposure to ethylene oxide. Mutagenesis, 19, 215–222. [DOI] [PubMed] [Google Scholar]
- Rhomberg, L. , Dellarco, V.L. , Siegel‐Scott, C. , Dearfield, L.K. and Jacobson‐Kram, D. (1990) Quantitative estimation of the genetic risk associated with the induction of heritable translocations at low‐dose exposure: ethylene oxide as an example. Environmental and Molecular Mutagenesis, 16, 104–125. [DOI] [PubMed] [Google Scholar]
- Ribeiro, L.R. , Tabello‐Gay, M.N. , Salvadori, D.M. , Pereira, C.A. and Becak, W. (1987) Cytogenetic effects of inhaled ethylene oxide in somatic and germ cells of mice. Archives of Toxicology, 59, 332–335. [DOI] [PubMed] [Google Scholar]
- Rusyn, I. , Asakura, S. , Li, Y. , Kosyk, O. , Koc, H. , Nakamura, J. , Upton, P.B. and Swenberg, J.A. (2005) Effects of ethylene oxide and ethylene inhalation on DNA adducts, apurinic/apyrimidinic sites and expression of base excision DNA repair genes in rat brain, spleen, and liver. DNA Repair, 4, 1099–1110. [DOI] [PubMed] [Google Scholar]
- Segerbäck, D. (1990) Reaction products in hemoglobin and DNA after in vitro treatment with ethylene oxide and N‐(2‐hydroxyethyl)‐N‐nitrosourea. Carcinogenesis, 11(2), 307–312. [DOI] [PubMed] [Google Scholar]
- Sisk, S.C. , Pluta, L.J. , Meyer, K.G. , Wong, B.C. and Recio, L. (1997) Assessment of the in vivo mutagenicity of ethylene oxide in the tissues of B6C3F1 lacI transgenic mice following inhalation exposure. Mutation Research, 391, 153–164. [DOI] [PubMed] [Google Scholar]
- Slob, W. (2017) A general theory of effect size, and its consequences for defining the benchmark response (BMR) for continuous endpoints. Critical Reviews in Toxicology, 47(4), 342–351. [DOI] [PubMed] [Google Scholar]
- Slob, W. , Moerbeek, M. , Rauniomaa, E. and Piersma, A.H. (2005) A statistical evaluation of toxicity study designs for the estimation of the benchmark dose in continuous endpoints. Toxicological Sciences, 84, 167–185. [DOI] [PubMed] [Google Scholar]
- Steenland, K. , Stayner, L. and Deddens, J. (2004) Mortality analyses in a cohort of 18 235 ethylene oxide exposed workers: follow up extended from 1987 to 1998. Occupational and Environmental Medicine, 61, 2–7. [PMC free article] [PubMed] [Google Scholar]
- Steenland, K. , Whelan, E. , Deddens, J. , Stayner, L. and Ward, E. (2003) Ethylene oxide and breast cancer incidence in a cohort study of 7576 women (United States). Cancer Causes & Control, 14, 531–539. [DOI] [PubMed] [Google Scholar]
- Stout, J.T. and Caskey, C.T. (1985) HPRT: gene structure, expression, and mutation. Annual Review of Genetics, 19, 127–148. [DOI] [PubMed] [Google Scholar]
- Strekalova, E.Y. , Chirkova, E.M. and Golubovich, E. (1971) Mutagenic action of ethylene oxide on sex and somatic cells in male white rats. Toksikol Nov Prom Khim Veshchestv, 14, 11–16. [Google Scholar]
- Tates, A.D. , van Dam, F.J. , Natarajan, A.T. , van Teylingen, C.M.M. , de Zwart, F.A. , Zwinderman, A.H. , van Sittert, N.J. , Nilsen, A. , Nilsern, O.G. , Zahlsen, K. , Magnusson, A.L. and Törnqvist, M. (1999) Measurement of HPRT mutations in splenic lymphocytes and haemoglobin adducts in erythrocytes of Lewis rats exposed to ethylene oxide. Mutation Research, 431, 397–415. [DOI] [PubMed] [Google Scholar]
- Texas Commission on Environmental Quality (TCEQ) . (2020). Ethylene Oxide Carcinogenic Dose‐Response Assessment. CAS Registry Number: 75‐21‐8. Development Support Document. Final, May 15, 2020. Houston, Texas: Texas Commission on Environmental Quality. [Google Scholar]
- Tompkins, E.M. , McLuckie, K.I. , Jones, D.J. , Farmer, P.B. and Brown, K. (2009) Mutagenicity of DNA adducts derived from ethylene oxide exposure in the pSP189 shuttle vector replicated in human Ad293 cells. Mutation Research, 678(2), 129–137. [DOI] [PubMed] [Google Scholar]
- Törnqvist, M.A. , Almberg, J.G. , Bergmark, E.N. , Nilsson, S. and Osterman‐Golkar, S.M. (1989) Ethylene oxide doses in ethene‐exposed fruit store workers. Scandinavian Journal of Work, Environment & Health, 15, 436–438. [DOI] [PubMed] [Google Scholar]
- U.S. Environmental Protection Agency (U.S. EPA) . (1991) Guidelines for Developmental Toxicity Risk Assessment. EPA/600/FR‐91/001. Risk Assessment Forum. Washington, DC: U.S. Environmental Protection Agency. [Google Scholar]
- U.S. Environmental Protection Agency (U.S. EPA) . (1996) Soil Screening Guidance. Technical Background Document. Appendix B: Route‐to‐Route Extrapolation of Inhalation Benchmarks. EPA/540/R‐96/018. Office of Emergency and Remedial Response. Washington, DC: U.S. Environmental Protection Agency. [Google Scholar]
- U.S. Environmental Protection Agency (U.S. EPA) . (2016). Evaluation of inhalation carcinogenicity of ethylene oxide. EPA/635/R‐16/350Fa. National Center for Environmental Assessment. Office of Research and Development. Washington, DC: U.S. Environmental Protection Agency. [Google Scholar]
- U.S. Environmental Protection Agency (US EPA) . (2014) Guidance for Applying Quantitative Data to Develop Data‐Derived Extrapolation Factors for Interspecies and Intraspecies Extrapolation. EPA/R‐14/002F. Washington, DC: Office of the Science Advisor. Risk Assessment Forum. U.S. Environmental Protection Agency. [Google Scholar]
- van Sittert, N.J. , Boogaard, P.J. , Natarajan, A.T. , Tates, A.D. , Ehrenberg, L.G. and Törnqvist, M. (2000) Formation of DNA adducts and induction of mutagenic effects in rats following 4 weeks inhalation exposure to ethylene oxide as a basis for cancer risk assessment. Mutation Research, 447, 27–48. [DOI] [PubMed] [Google Scholar]
- Vergnes, J.S. and Pritts, I.M. (1994) Effects of ethylene on micronucleus formation in the bone marrow of rats and mice following four week of inhalation exposure. Mutation Research, 324, 87–91. [DOI] [PubMed] [Google Scholar]
- Walker, V.E. , Sisk, S.C. , Upton, P.B. , Wong, B.A. and Recio, L. (1997) In vivo mutagenicity of ethylene oxide at the hprt locus in T‐lymphocytes of B6C3F1 lacI transgenic mice following inhalation exposure. Mutation Research, 392, 211–222. [DOI] [PubMed] [Google Scholar]
- Walker, V.E. , Wu, K.Y. , Upton, P.B. , Ranasinghe, A. , Scheller, N. , Cho, M.H. , Vergnes, J.S. , Skopek, T.R. and Swenberg, J.A. (2000) Biomarkers of exposure and effect as indicators of potential carcinogenic risk arising from in vivo metabolism of ethylene to ethylene oxide. Carcinogenesis, 21(9), 1661–1669. [DOI] [PubMed] [Google Scholar]
- Waters, M.D. , Stack, H.F. and Jackson, M.A. (1999) Genetic toxicology data in the evaluation of potential human environmental carcinogens. Mutation Research, 437, 21–49. [DOI] [PubMed] [Google Scholar]
- White, P.A. , Long, A.S. and Johnson, G.E. (2020) Quantitative interpretation of genetic toxicity dose‐response data for risk assessment and regulatory decision‐making: current status and emerging priorities. Environmental and Molecular Mutagenesis, 61(1), 66–83. [DOI] [PubMed] [Google Scholar]
- Zeller, A. , Duran‐Pacheco, G. and Guerard, M. (2017) An appraisal of critical effect sizes for the benchmark dose approach to assess dose–response relationships in genetic toxicology. Archives of Toxicology, 91(12), 3799–3807. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1: BMD50 analysis