

Abstract
Objective
Microglial phagocytosis of apoptotic cells is an essential component of the brain regenerative response during neurodegeneration. Whereas it is very efficient in physiological conditions, it is impaired in mouse and human mesial temporal lobe epilepsy, and now we extend our studies to a model of progressive myoclonus epilepsy type 1 in mice lacking cystatin B (CSTB).
Methods
We used confocal imaging and stereology‐based quantification of apoptosis and phagocytosis of the hippocampus of Cstb knockout (KO) mice, an in vitro model of phagocytosis and siRNAs to acutely reduce Cstb expression, and a virtual three‐dimensional (3D) model to analyze the physical relationship between apoptosis, phagocytosis, and active hippocampal neurons.
Results
Microglial phagocytosis was impaired in the hippocampus of Cstb KO mice at 1 month of age, when seizures arise and hippocampal atrophy begins. This impairment was not related to the lack of Cstb in microglia alone, as shown by in vitro experiments with microglial Cstb depletion. The phagocytosis impairment was also unrelated to seizures, as it was also present in Cstb KO mice at postnatal day 14, before seizures begin. Importantly, phagocytosis impairment was restricted to the granule cell layer and spared the subgranular zone, where there are no active neurons. Furthermore, apoptotic cells (both phagocytosed and not phagocytosed) in Cstb‐deficient mice were at close proximity to active cFos+ neurons, and a virtual 3D model demonstrated that the physical relationship between apoptotic cells and cFos+ neurons was specific for Cstb KO mice.
Significance
These results suggest a complex crosstalk between apoptosis, phagocytosis, and neuronal activity, hinting that local neuronal activity could be related to phagocytosis dysfunction in Cstb KO mice. Overall, these data suggest that phagocytosis impairment is an early feature of hippocampal damage in epilepsy and opens novel therapeutic approaches for epileptic patients based on targeting microglial phagocytosis.
Keywords: apoptosis, epilepsy, hippocampus, microglia, phagocytosis, seizures
Key Points.
-
•
Microglial phagocytosis of apoptotic cells is impaired in the dentate gyrus of young Cstb KO mice (postnatal days 14 and 30)
-
•
Cstb deficiency in cultured microglia does not impair phagocytosis
-
•
Phagocytosis deficiency is restricted to the granule cell layer, whereas the subgranular zone is spared
-
•
A virtual 3D model shows that apoptotic cells are closer than expected (3 μm) to active cFos+ neurons
-
•
Microglial phagocytosis impairment in Cstb KO mice is unrelated to seizures but may be related to local neuronal activity
1. INTRODUCTION
Progressive myoclonus epilepsy 1 (EPM1) or Unverricht‐Lundborg disease is a genetic neurodegenerative disease characterized by disabling myoclonus, epileptic seizures, and ataxia. The disease is caused by biallelic loss‐of‐function mutations in the cystatin B gene (CSTB) encoding the cystatin B protein, 1 , 2 , 3 a protease inhibitor that regulates and limits the activity of nuclear and cytoplasmic cysteine proteases known as cathepsins. 4 , 5 Cstb knockout (KO) mice recapitulate key clinical features of EPM1, 5 , 6 as they develop seizures at 1 month and progressive ataxia at around 6 months of age, 7 , 8 , 9 , 10 consistent with findings in humans. In addition, Cstb KO mice present early alterations in the inflammatory response of microglia, the brain immune cells. 11 , 12 , 13 In addition to controlling the release of inflammatory mediators, microglia are also the brain professional macrophages. 14 We have recently reported that in both mouse and human mesial temporal lobe epilepsy (MTLE), microglial phagocytosis of apoptotic cells is impaired, 15 leading us to question whether the phagocytosis impairment would also occur in Cstb KO mice.
Phagocytosis is a highly regulated response that prevents the spillover of cytotoxic content that results from the cell death, is immunomodulatory, and actively participates in maintaining tissue homeostasis. 14 , 16 , 17 In physiological conditions, microglia are very efficient phagocytes, as their processes constantly scan the brain parenchyma and express a plethora of receptors for “find‐me” and “eat‐me” signals produced by apoptotic cells. 18 , 19 , 20 , 21 Microglia are similarly efficient facing increased apoptotic cell numbers generated during excitotoxicity and inflammation, as they use several strategies to cope with increased apoptosis: (1) recruit more phagocytic cells, (2) increase the number of apoptotic cells phagocytosed per microglial cell, and/or (3) increase the microglial numbers. 15 , 22 As a consequence, microglial phagocytosis is proportional to the number of apoptotic cells, that is, microglial phagocytosis is tightly coupled to apoptosis. 15 However, during MTLE the hyperactivity of the neuronal network and the massive release of the “find‐me” signal adenosine triphosphate (ATP) during seizures interferes with the recognition by microglia, resulting in delayed dead cell clearance and inflammation. 15
To test whether microglial phagocytosis was affected in other forms of epilepsy, such as EPM1, we analyzed microglial phagocytosis efficiency in Cstb KO mice. Most of the CSTB deficiency‐related pathological changes have been reported in the cerebellum and the cortex, 7 , 9 , 10 , 11 but here we focused on the dentate gyrus (DG) of the hippocampus, in whose subgranular zone (SGZ) there is ongoing production of newborn neurons through adulthood. 23 Most of these newborn neurons naturally undergo apoptosis and are immediately phagocytosed by microglia, allowing us to establish the baseline phagocytosis efficiency. 22 , 24 We here show DG atrophy and microglial phagocytosis impairment at the age when seizures arise, at postnatal day 30 (P30). We then studied whether this effect was related to a cell‐autonomous effect on microglia, using an in vitro model of phagocytosis and siRNA‐mediated depletion; or was related to altered neuronal function, by studying presymptomatic mice at P14, before seizures arise. We found that in the neurogenic region, the SGZ, phagocytosis was spared; in contrast, phagocytosis was specifically impaired in the granule neuron cell layer. We finally used direct analysis and mathematical modeling to determine that apoptotic cells were in close proximity to cFos+, activated neurons, in the absence of seizures. Our data suggest a complex environmental signaling in Cstb KO mice that results in granule cell layer–specific microglial phagocytosis impairment and point toward microglia as a potential target of novel treatments aimed at reducing neuronal damage in epilepsy patients.
2. MATERIALS AND METHODS
Full methods can be found in Appendix S1.
3. RESULTS
3.1. Microglial phagocytosis is uncoupled to apoptosis in Cstb KO mice at P30
To determine the apoptosis/phagocytosis dynamics in Cstb KO mice, we focused on the hippocampal DG, as the ongoing neurogenesis and the natural apoptosis of newborn cells allow us to compare phagocytosis efficiency between control and KO mice. 22 We focused on P30 mice, an age when cerebellar apoptosis and cortical atrophy occur and seizures begin. 7 , 9 We analyzed the granule cell population by immunostaining the DG of both wild‐type (WT) and Cstb KO mice with the nuclear marker 4,6‐diamidino‐2‐phenylindole (DAPI) to assess apoptosis by karyorrhexis and/or pyknosis, and with Iba1 to visualize microglia (Figure 1A). We found a significant increase in the total number of apoptotic cells (Figure 1B) that was translated into a decrease in the total granule cell population in Cstb KO mice compared to WT mice (Figure 1C). However, the total granule cell density was similar in both genotypes, with a trend toward a reduction in the volume of the septal hippocampus (P = .052; Figure 1D,E). This increased apoptosis and trend toward decreased hippocampal volume in Cstb KO mice at P30 are consistent with the cerebellar and cortical atrophy previously described. 9 , 10 , 11
FIGURE 1.
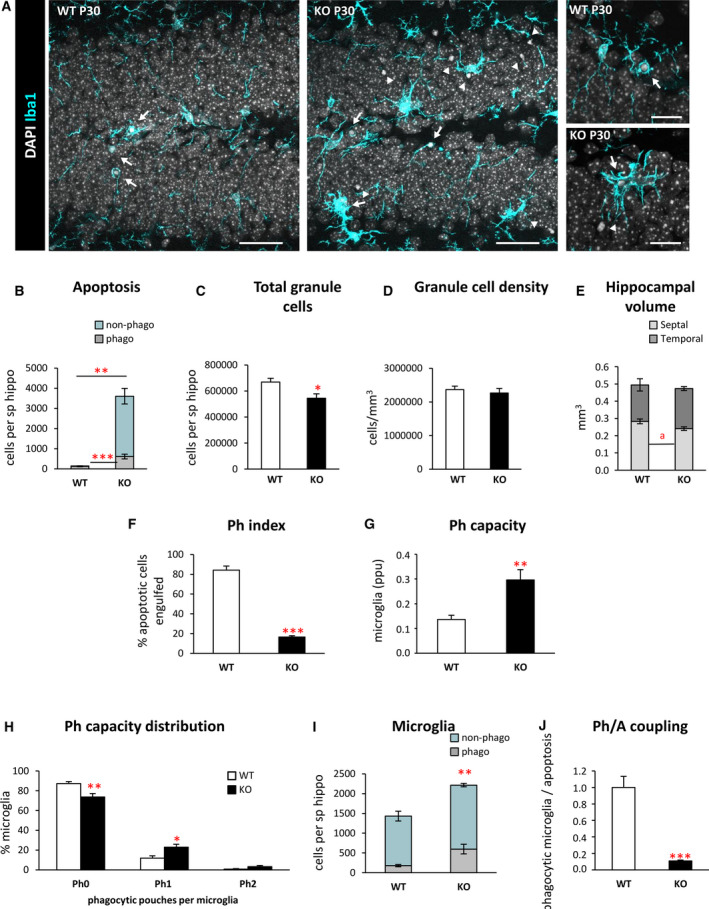
Microglial phagocytosis is impaired in the dentate gyrus (DG) of postnatal day 30 (P30) Cstb knockout (KO) mice. A, Representative confocal images of the DG in wild‐type (WT) and Cstb KO P30 mice. Healthy or apoptotic (pyknotic/karyorrhectic) nuclear morphology was visualized with 4,6‐diamidino‐2‐phenylindole (DAPI; white), and microglia were stained for Iba1 (cyan). High‐magnification examples of phagocytic microglia following the typical “ball‐and‐chain” form (upper right panel), with the tip of their processes or phagocytosing with their soma (lower right panel). Arrows point to apoptotic cells engulfed by microglia (Iba1+), and arrowheads point to nonphagocytosed apoptotic cells. B, Number of apoptotic cells (pyknotic/karyorrhectic) in the septal hippocampus (sp hippo; n = 6 animals per condition) showing both phagocytosed (phago) and nonphagocytosed (non‐phago) apoptotic cells. C, Total number of granule cells per septal hippocampus. D, Density of granule cells (per mm3) in the septal hippocampus of both WT and Cstb KO P30 mice. E, Proportion (in mm3) between the volume of the septal and the temporal portions of the hippocampus in WT and Cstb KO P30 mice. F, Phagocytic index (Ph index; in % of apoptotic cells being engulfed by microglia) in the septal hippocampus. G, Weighted phagocytic capacity (Ph capacity) of DG microglia (in parts per unit [ppu]). H, Histogram showing the Ph capacity distribution of DG microglia (in % of microglial cells with 0‐2 phagocytic pouches [Ph]). I, Number of phagocytic (Iba1 + with DAPI inclusions) and nonphagocytic microglial cells (Iba1 + with no DAPI inclusions) per septal hippocampus. J, Phagocytosis/apoptosis (Ph/A; in fold change) in the septal hippocampus. Bars represent the mean ± standard error of the mean. *P < .05, **P < .01, ***P < .001, a P = .052 by one‐tailed Student t test. Scale bars = 40 µm (A, low magnification), 20 µm (A, high magnification); z‐thickness = 7 µm (A, low magnification), 3.5 µm (A, high magnification)
Next, we analyzed microglial phagocytosis by measuring the phagocytic index (Ph index), that is, the percentage of apoptotic cells engulfed by microglia. Microglia in Cstb KO mice engulfed 16.6 ± 1.2% of the apoptotic cells, significantly less than WT mice (84.3 ± 4.1%, P < .001; Figure 1F), suggesting that Cstb KO microglia were altered. To directly test the proportion of microglial cells that were engaged in phagocytosis, we analyzed their phagocytic capacity (Ph capacity), that is, the number of phagocytic pouches containing an apoptotic cell per microglial cell. Cstb KO mice showed an increased Ph capacity compared to WT mice, with fewer nonphagocytic cells and more cells with one phagocytic pouch (Figure 1G,H). Microglial numbers were also increased in Cstb KO mice compared to WT mice (Figure 1I), leading to an increase in net phagocytosis (number of microglia multiplied by the Ph capacity). Nonetheless, this increase in net phagocytosis was insufficient to compensate for the increase in apoptosis, leading to a reduction in the phagocytosis/apoptosis coupling (ratio between net phagocytosis and total number of apoptotic cells; Figure 1J). Thus, despite the attempt to increase microglial numbers and Ph capacity, microglia were not able to remove the increasing numbers of dead cells in Cstb KO at P30.
As we had observed an increase in the total number of microglia (Figure 1I), we then analyzed whether this effect was due to microglial proliferation using Ki67, a marker for proliferating cells that is expressed during all active phases of the cell cycle (Figure 2A,B). 25 We observed a significant increase in proliferating microglia in Cstb KO mice compared to WT mice at P30 (Figure 2C). In addition, Cstb KO microglia presented a hypertrophic morphology (Figures 1A and 2A), which was reminiscent of the ameboidlike, multinucleated phenotype of microglia observed in the kainic acid (KA) model of epilepsy. 15 , 26 To assess whether Cstb KO microglia also showed multinuclearity, we quantified the percentage of cells with more than one nucleus (Figure 1D). We found that whereas multinucleated cells were absent in control mice, they represented 13.1% ± 1.5% of microglia in Cstb KO mice, similar to what we observed 3 days and 7 days after KA injection. 15
FIGURE 2.
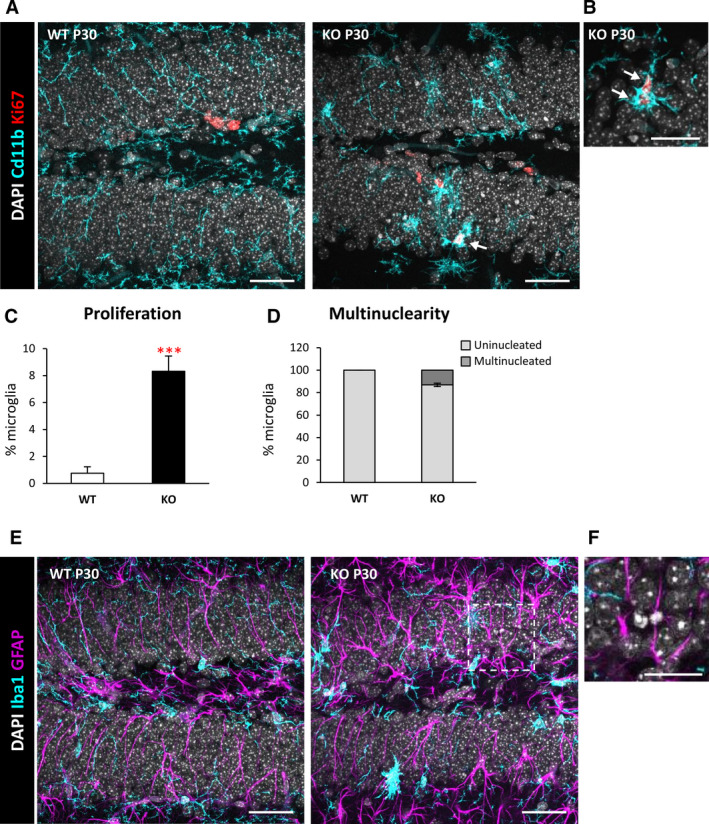
Increased proliferation and multinuclearity in the dentate gyrus (DG) of postnatal day 30 (P30) Cstb knockout (KO) mice. A, DG general view of both wild‐type (WT) and Cstb KO P30 mice. Proliferating cells were stained for Ki67 (marker for all the active phases of the cell cycle), nuclei (4,6‐diamidino‐2‐phenylindole [DAPI]), and microglia (CD11b). B, High‐magnification image of proliferating Ki67+ microglia in P30 Cstb KO mice. Arrows in A and B point to Ki67+ microglia. C, Percentage of proliferating microglia assessed by the marker Ki67. D, Proportion (in %) between uninucleated and multinucleated microglia in the septal hippocampus of WT and Cstb KO P30 mice. E, Representative images of the DG in WT and Cstb KO mice stained for DAPI (nuclei), microglia (Iba1), and astrocytes (glial fibrillary acidic protein [GFAP]). F, High‐magnification image of an apoptotic cell not phagocytosed by either microglia or astrocytes in Cstb KO mice. Bars represent the mean ± standard error of the mean. ***P < .001 by one‐tailed Student t test. Scale bars = 40 µm (A), 20 µm (B), 40 µm (E), 20 µm (F); z‐thickness = 7 μm (A, E), 3.5 μm (B)
Finally, we addressed whether the impairment in microglial phagocytosis was compensated by recruitment of other cell types for phagocytosis. Microglia are specialized phagocytic cells; however, other cell types, such as astrocytes, can also perform phagocytosis under certain conditions. 16 To characterize whether astrocytes contributed to the phagocytosis of apoptotic cells, we immunostained the DG for the astrocytic marker glial fibrillary acidic protein, as well as CD11b for microglia and DAPI for the nuclei (Figure 2E,F). We observed a notable morphology change in astrocytes, with cells showing thicker processes, in agreement with previously reported reactive astrogliosis observed in the Cstb KO at P30, the onset of myoclonus, 11 but no evidence of their involvement in phagocytosis. Overall, these results demonstrate microglial phagocytosis was impaired and not compensated by astrocytes in Cstb KO mice.
3.2. Cstb knockdown in microglia does not alter phagocytosis in vitro
To determine a possible cell‐autonomous effect of Cstb on microglial phagocytosis, we first addressed whether microglia expressed Cstb in vivo. For this purpose, we used FACS (fluorescent activated cell sorting) to purify hippocampal microglia from fms‐EGFP mice (Figure 3A), in which the enhanced green fluorescent protein (EGFP) is driven by the fms gene, which encodes for the Macrophage Colony Stimulating Factor Receptor 1 (MCSF‐1), 27 , 28 allowing the discrimination of microglia from other cell types. We analyzed the mRNA expression of Cstb as well as downstream cathepsins B, L, and S by quantitative reverse transcription polymerase chain reaction (RT‐qPCR), because Cstb mutations are related to increased cysteine protease expression 29 and activity. 30 , 31 We found that Cstb and cathepsins B and L were expressed by both microglia and nonmicroglia cells, whereas cathepsin S was solely expressed by microglia (Figure 3B). Although Cstb was not enriched in microglia compared to other cell types, its robust expression suggested that the phagocytosis impairment could be the direct consequence of microglia lacking Cstb per se.
FIGURE 3.
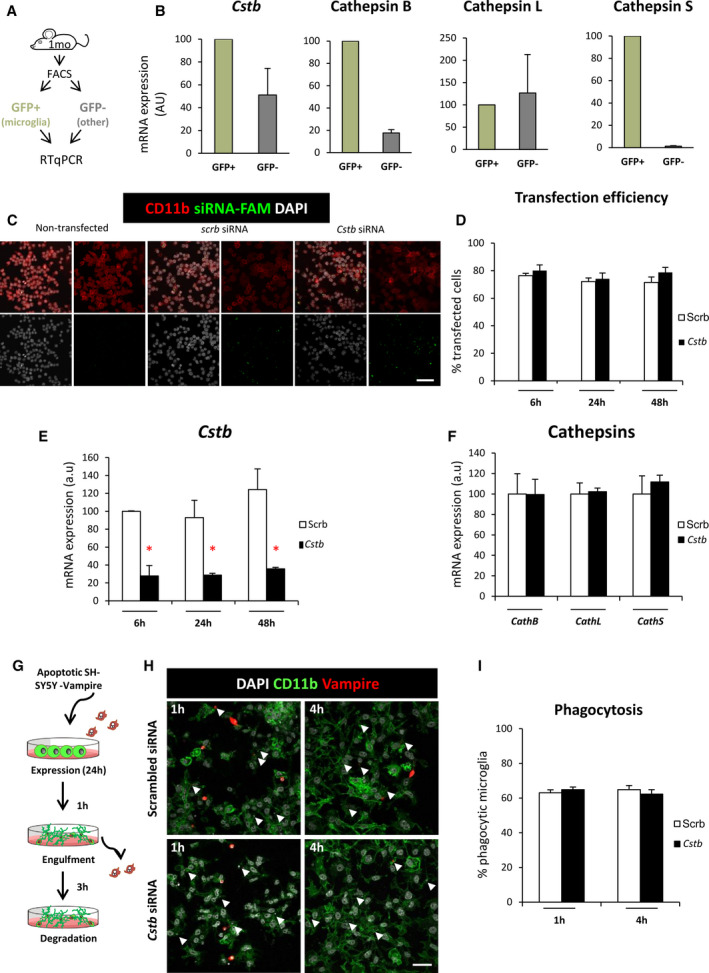
Cstb knockdown in microglia does not alter phagocytosis in vitro. A, Experimental design used to isolate microglia (green fluorescent protein [GFP]+) from nonmicroglial cells (GFP−) for the hippocampi of 1‐month‐old mice using flow cytometry and quantitative reverse transcription polymerase chain reaction (RT‐qPCR) for gene expression analysis. B, Expression of CSTB gene and cathepsins B, L, and S in microglia (GFP+) versus nonmicroglial cells (GFP−) in flow‐activated cell sorting (FACS)‐sorted cells from fms‐EGFP mice, in which the promoter of the fms gene, encoding for the macrophage colony stimulating factor receptor 1, drives the expression of the enhanced green fluorescent protein mouse hippocampi. Ornithine decarboxylase antizyme 1 (OAZ1) was selected as a reference gene. C, Representative confocal images of nontransfected (left panels) and scrambled/Cstb siRNA transfected BV2 microglia (middle and right panels). Nuclei are stained with 4,6‐diamidino‐2‐phenylindole (DAPI; white), BV2 microglia were stained for CD11b (red), and siRNA transfection was assessed by 6‐carboxyfluorescein (green) labeling. D, Percentage of scrambled/Cstb siRNA transfected cells along a time course (6, 24, and 48 hours). E, RT‐qPCR Cstb gene expression in BV2 cells after Cstb siRNA silencing through a time course (6, 24, and 48 hours), using OAZ1 as a reference gene. F, RT‐qPCR cathepsins B, L, and S gene expression in BV2 cells 24 hours after siRNA Cstb silencing, using OAZ1 as a reference gene. G, Experimental design of the phagocytosis assay performed 24 hours after BV2 siRNA transfection. Knockdown BV2 cells are fed for 1 and 4 hours with apoptotic SH‐SY5Y vampire neurons. H, Representative confocal images of scrambled and Cstb siRNA transfected BV2 cells (CD11b staining, green) fed with apoptotic SH‐SY5Y vampire neurons (red) for 1 and 4 hours. Arrowheads show phagocytosed SH‐SY5Y vampire fragments or full cells. I, Percentage of phagocytic BV2 cells after 1 and 4 hours of phagocytosis. Only particles fully enclosed by BV2 pouches were identified as phagocytosis. Bars represent the mean ± standard error of the mean. *P < .05 by two‐way analysis of variance. Scale bars = 60 µm (C), 40 µm (H)
To directly assess the effect of microglial Cstb on phagocytosis, we set up an in vitro model of Cstb knockdown in the microglial cell line BV2 (Figure 3C‐I). We transfected BV2 microglia with 6‐carboxyfluorescein–labeled siRNAs against Cstb or a scrambled siRNA as a control (Figure 3C). We obtained a high transfection efficiency through a time course of 6, 24, and 48 hours (Figure 3D) and validated the transcription downregulation of the Cstb gene by RT‐qPCR through the time course. We observed that the expression of Cstb was greatly reduced up to 48 hours (Figure 3E), whereas the expression of the related cathepsins was not affected by Cstb siRNA treatment (Figure 3F). Control and Cstb KO microglia were then fed with apoptotic SHSY5Y‐vampire neurons for 1 and 4 hours, in which apoptosis was previously induced (staurosporine 3 µmol·L–1, 4 hours; Figure 3G). 15 Microglia were stained with CD11b, apoptotic neurons constitutively expressed the red fluorescent protein, and nuclei were stained with DAPI (Figure 3H). We found no differences in phagocytosis in microglia treated with either scrambled or Cstb siRNA (Figure 3I). Our in vitro model, nonetheless, does not fully recapitulate the in vivo situation, because Cstb KO mice have Cstb chronic depletion that is accompanied by modulation of cathepsin activity and/or expression. 29 , 31 Nonetheless, these results suggest that the cell‐autonomous acute Cstb deficiency in microglia is not sufficient to induce the phagocytosis impairment observed in vivo. We therefore searched for alternative mechanisms to explain the reduced phagocytosis in Cstb KO mice. As we had previously shown that seizures interfere with phagocytosis in a mouse model of MTLE, our next step was to determine whether the phagocytosis blockage was related to seizures by analyzing an early developmental stage, P14.
3.3. Phagocytosis impairment is specific to the granule cell layer in Cstb KO mice at P14
At P14, Cstb KO mice are asymptomatic and do not have seizures, 11 and in agreement we did not observe any changes in the number of active neurons, labeled with the immediate early gene cFos, whose expression is rapidly induced upon depolarization (Figure 4A‐C). 32 cFos+ neurons were found in similar numbers in both WT and Cstb KO mice at P14, and they were located exclusively in the granular layer (GL) and not in the SGZ, where radial neural stem cells and their immature progeny reside (Figure 4A,C). However, we did notice that whereas in WT mice most apoptotic cells were found in the SGZ, in Cstb KO mice apoptotic cells were found mostly in the GL and in very close proximity to cFos+ neurons (Figure 4B), prompting us to analyze separately GL and SGZ apoptosis and phagocytosis.
FIGURE 4.
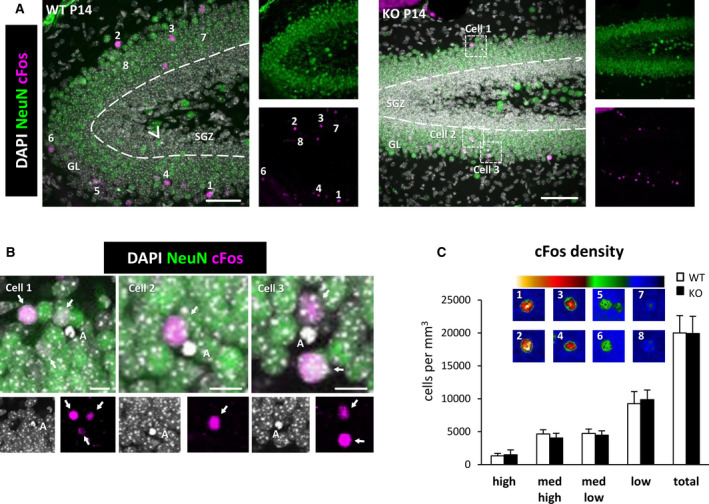
cFos+ cells in the dentate gyrus (DG) of wild‐type (WT) and Cstb knockout (KO) postnatal day 14 (P14) mice. A, Representative confocal images of the DG of WT and Cstb KO mice. Healthy or apoptotic (pyknotic/karyorrhectic) nuclear morphology was visualized with 4,6‐diamidino‐2‐phenylindole (DAPI; white), neurons were identified with the neuronal marker NeuN (green), and activated neurons were stained for the early expression gene cFos (magenta). Arrowhead points to an apoptotic cell in the subgranular zone (SGZ) in WT mice; framed apoptotic cells in Cstb KO mice are shown in B. Numbered cFos+ cells are shown in C, as cells with high (1, 2), medium‐high (3, 4), medium‐low (5, 6), and low (7,8) cFos intensity. GL, granular layer. (B) High‐magnification examples showing the close proximity between apoptotic cells (DAPI, white) and cFos+ neurons (magenta) in Cstb KO mice. Granular neurons are stained with NeuN (green). Arrows point to cFos+ neurons. C, Distribution of cFos+ cells in WT and Cstb KO mice (per mm3). The color code indicates the classification criteria of the cFos+ cells based on their intensity (high, medium‐high, medium‐low, low). A total of 1046 cells for WT P14 mice and 713 cells for Cstb KO mice were quantified and classified according to their cFos expression. No significant differences were found. Scale bars = 50 μm (A), 10 μm (B); z‐thickness = 28 µm (A, WT), 17.5 µm (A, KO)
In the SGZ, we did not observe any significant difference in apoptosis or phagocytosis between WT and Cstb KO mice at P14 (Figure 5A‐F). In contrast, in the GL there were few apoptotic cells in WT mice and a 10‐fold increase in Cstb KO mice (Figure 5D,E). The majority of these GL apoptotic cells were not phagocytosed, resulting in a reduced GL Ph index in Cstb KO mice compared to WT mice at P14 (Figure 5F). Similar to the global effect we had observed before in the whole DG at P30, we found that GL microglia increased their Ph capacity in Cstb KO mice compared to WT mice, although this effect was insufficient to cope with the increased number of apoptotic cells (Figure 5G,H). In this early stage, we found no obvious morphological changes or increases in microglial numbers (Figure 5I). Overall, we found that the increase in apoptotic cells was not compensated by a sufficient increase in phagocytosis, resulting in an uncoupling between apoptosis and phagocytosis (Figure 5J). Overall, these results demonstrate that the microglial phagocytosis impairment was specific to the GL and preceded seizure development in Cstb KO mice at P14. These data suggested that there could be functional differences between GL and SGZ that could explain the GL‐specific increase in apoptosis and microglial phagocytosis impairment.
FIGURE 5.
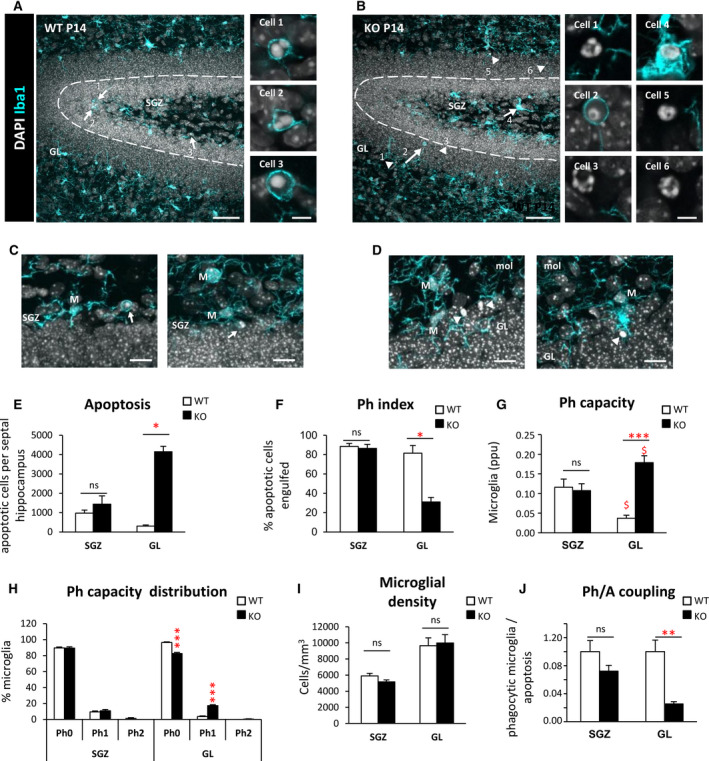
Phagocytosis impairment is specific to the granule cell layer in Cstb knockout (KO) mice at postnatal day 14 (P14). A, B, Dentate gyrus (DG) general view of both wild‐type (WT) and Cstb KO P14 mice. Nuclei are stained with 4,6‐diamidino‐2‐phenylindole (DAPI; white) and microglia with Iba1 (cyan). Closeup images show phagocytosed and nonphagocytosed apoptotic cells in WT and Cstb KO P14 mice. GL, granular layer; SGZ, subgranular zone. C, D, Representative images of apoptotic cells (condensed DAPI) engulfed by microglia (M; cyan) in the SGZ of WT P14 mice (C) and nonphagocytosed cells in the GL of Cstb KO P14 mice (D). Arrows point to phagocytosed apoptotic cells and arrowheads to nonphagocytosed apoptotic cells (A‐D). E, Number of apoptotic cells (pyknotic/karyorrhectic) both in the SGZ and GL, per septal hippocampus (n = 12 animal for each condition). F, Phagocytic index (Ph index; in % of apoptotic cells being engulfed by microglia) in the SGZ and GL of the septal hippocampus in WT and Cstb KO P14 mice. G, Histogram showing the phagocytic capacity (Ph capacity) distribution of DG microglia (in % of microglial cells) in the SGZ and GL. H, Weighted Ph capacity of DG microglia (in parts per unit). I, Microglial density (cells/mm3) per septal hippocampus in both WT and Cstb KO P14 mice, distinguishing between SGZ and GL. J, Phagocytosis/apoptosis (Ph/A; in fold change) in the SGZ and GL of the septal hippocampus in WT and Cstb KO P14 mice. Bars represent the mean ± standard error of the mean. *P < .05, **P < .01, ***P < .001 by Student t test comparing WT versus KO. ns, not significant. Scale bars = 50 μm (A, B), 5 μm (inserts in A, B), 30 μm (C, D); z‐thickness = 18.9 µm (A, B), 9.8 µm (C left), 16.1 (C right), 11.2 µm (D left), 12.6 µm (D right). $ means P < .005 comparing GL vs SGL
3.4. Apoptotic cells are in close proximity to active cFos+ neurons
The close vicinity of apoptotic cells to GL cFos+ neurons in P14 mice (Figure 4B) suggested that the phagocytosis efficiency of GL microglia could be related to neuronal activity, as neuronal hyperactivity during seizures prevents microglia from targeting apoptotic cells. 15 To address whether the proximity of cFos+ neurons had an impact on apoptosis and phagocytosis, we directly estimated the distance to each phagocytosed and nonphagocytosed cell to the closest cFos+ neuron (nearest neighbor [NN]; Figure 6A). Because of the limitation imposed by the relatively small thickness of our z‐stacks (border effect; see Materials and Methods for more detail), we only had a sufficient number of apoptotic cells in KO mice (n = 15 phagocytosed and n = 5 nonphagocytosed apoptotic cells from n = 9 mice, from an initial set of 295 cells).
FIGURE 6.
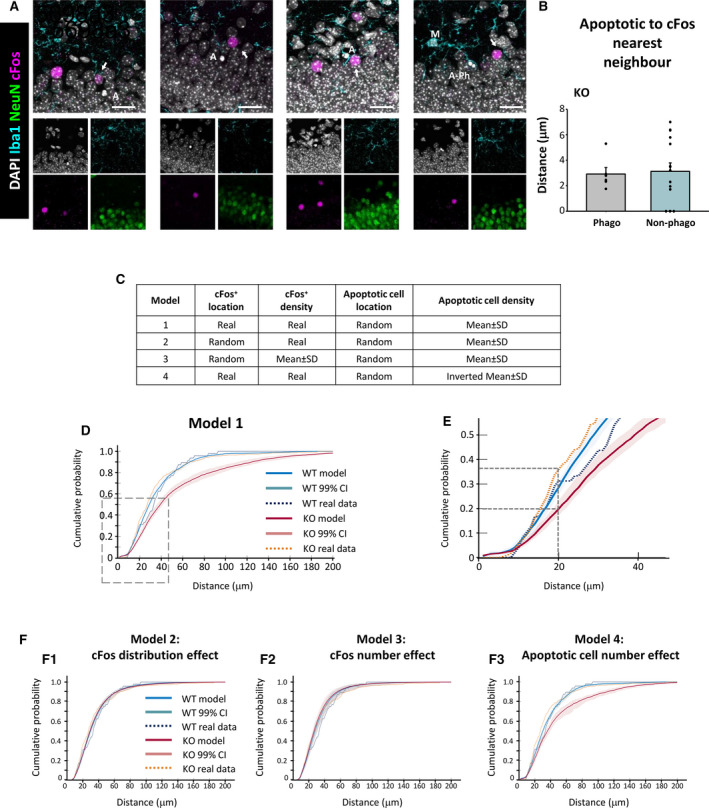
Proximal relationship between apoptotic cells and cFos+ neurons in the granule cell layer of Cstb knockout (KO) mice. A, Representative confocal images of the granular layer of Cstb KO mice. Healthy or apoptotic (pyknotic/karyorrhectic) nuclear morphology was visualized with 4,6‐diamidino‐2‐phenylindole (DAPI; white), neurons were identified with the neuronal marker NeuN (green), microglia (M) were identified with Iba1 (cyan), and activated neurons were stained for the early expression gene cFos (magenta). Both phagocytosed (A‐Ph) and nonphagocytosed apoptotic cells (A) were close to cFos+ neurons (arrows). B, Quantification of distance from phagocytosed (Phago) and nonphagocytosed (Non‐phago) apoptotic cells to the cFos+ nearest neighbor (NN), for those cells that met the inclusion criteria (see Materials and Methods). C, Summary of the different simulation models based on the location and density of cFos+ and apoptotic cells. D, Cumulative probability of the distances between apoptotic cells and NN cFos+ neurons for wild‐type (WT; blue) and Cstb KO mice (red), resulting from 10 000 simulations of a virtual three‐dimensional (3D) model, indicating the 99% confidence interval (CI). The cumulative probability for real (measured) data is shown for WT (dotted blue) and Cstb KO (dotted orange). E, Amplification of the area shown in D. F, Cumulative probabilities of the distances between apoptotic cells and NN cFos+ neurons for WT (blue) and Cstb KO mice (red), resulting from 10 000 simulations of the indicated virtual 3D model. Scale bars indicate 20 μm. z‐thickness = 9.1 µm, 7.7 µm, 16.8 µm, 9.1 µm (A, from left to right)
In Cstb KO mice, both phagocytosed and nonphagocytosed cells were found very close to the cFos+ NN (3.0 ± 0.5 and 3.0 ± 0.6 μm, respectively; nonsignificant difference; Figure 6B). To determine whether this short distance would be expected if cells were homogenously distributed through the thickness of our z‐stacks, we calculate that 50% of the cells should be located at one‐quarter distance to either z‐border (olive green‐shaded area in Figure S1), that is, at 5.9 ± 0.2 and 6.0 ± 0.1 μm from the cFos+ NN for phagocytosed and nonphagocytosed cells, respectively (Figure S1). Thus, both phagocytosed and nonphagocytosed apoptotic cells in Cstb KO mice seem to be much closer (3 μm) to active neurons than would be expected. Although we could not detect significant differences between phagocytosed and nonphagocytosed cells in Cstb KO mice, these results do suggest an unexpected relationship between neuronal activity and apoptosis.
To strengthen the analysis of distance between apoptotic cells and NN cFos+ neurons in both WT and KO mice and to overcome the limitation of analyzing only 20 cells, we developed a mathematical model (see Materials and Methods for details). We created a virtual three‐dimensional (3D) model of the GL based on the spatial distribution and density of neurons, cFos+ neurons, and apoptotic cells (Figure S1). In this model, we reproduced the same experimental limitations imposed by the limited thickness of the z‐stacks (see Materials and Methods) compared to the size of the XY field of view, and therefore no cells were discarded due to the border effect. Using this virtual 3D model, we compared the entire experimental distribution of NN distance between cFos+ and apoptotic cells to the distribution generated by the 10 000 simulations. Specifically, we developed four different models (Figure 6C).
In the first model, cFos+ neurons were located in their original position (in the z‐stack) and the position of the apoptotic cells was randomized, and their numbers were extrapolated from the experimental cell density (Figure 6C). For each apoptotic cell, we performed 10 000 randomizations and calculated the distance to the NN cFos+ cell. Finally, the cumulative probability to encounter a cFos+ cell within a given distance from an apoptotic cell was computed with its 99% confidence interval in both the WT‐ and KO‐derived v3D grids (Figure 6D,E). In WT mice, the measured apoptotic‐cFos+ NN distance was within the modeled confidence interval up to a distance of 22 μm, which matches the average z‐stack thickness (24.2 ± 0.4 μm) and validates that our model is isotropic, that is, does not depend on the orientation and therefore is within the range of the z‐stack thickness. In contrast, in Cstb KO mice the measured percentage of NN cFos+‐apoptotic cells between 17 and 24 μm was significantly higher than what was observed in both the WT and Cstb KO models. For instance, in the Cstb KO model, 20% of the apoptotic cells were found at <20 μm from the cFos+ NN, whereas in the real data from Cstb KO mice, 20% of the apoptotic cells were found at <16 μm and 35% of the apoptotic cells were found at <20 μm (Figure 6E). This data suggested that as in our direct quantifications (Figure 6B), apoptotic cells were closer than expected to active cFos+ neurons in Cstb KO mice.
Nonetheless, the modeled distance was significantly larger in KO than in WT mice (Figure 6E). To further understand the role of other parameters, specifically cell number or location, in the model, we used other three different models (Figure 6C): Model 2 to test the impact of cFos+ cell location, Model 3 to test cFos+ cell density, and Model 4 to test apoptotic cell density. We first tested the effect of cFos+ cell location (Model 2), by randomly positioning both cFos+ neurons and apoptotic cells (Figure 6F1). In this model, there were no differences between the modeled NN distances between WT and KO. Importantly, in KO mice the difference between the measured and modeled apoptotic‐cFos+ NN distance was strongly reduced, although it was still significant, with a maximum around 20 μm. The reduced difference between the measured and modeled NN distance in KO mice in Model 2 suggest that the differences between the modeled NN distances in Model 1 originated from a differential distribution of cFos+ cells between WT and KO.
We then tested the effect of cFos+ density by creating a Model 3 in which cFos+ cells were positioned randomly in the v3D grid and their number was estimated from the experimentally measured cFos density rescaled by the number of cells in the grid, whereas apoptotic cells were modeled randomly as above (Figure 6F2). The results of Model 3 were similar to Model 2, with no differences between modeled and measured NN distances for WT and KO, suggesting that the number of cFos+ cells was not related to the differential effect found in Model 1. Finally, we tested the effect of different density of apoptotic cells in the WT‐ and KO‐modeled distances in Model 4. For this, we went back to use the real distribution of cFos+ cells (as in Model 1) and positioned the apoptotic cells randomly but with an inverted density: the WT model using KO apoptotic cell density and vice versa (Figure 6F3). The results of Model 4 were similar to Model 1; the modeled NN distance was larger in KO than in WT, and apoptotic cells were found closer to active neurons than expected in KO mice. Overall, these data suggests that although Cstb KO mice have similar cFos+ density and intensity at P14 (Figure 4C) and do not yet have seizures, 11 they have an abnormal distribution of cFos+ active neurons that results in a closer distance to apoptotic cells (Figure 6B,C). These results suggest a very close relationship between abnormal neuronal activity in Cstb KO mice and apoptotic cells. Indirectly, they also suggest a relationship with the phagocytosis impairment found in Ctsb KO mice, because the number of apoptotic cells observed is the net result between apoptosis induction and phagocytosis removal.
4. DISCUSSION
In this paper, we show for the first time that microglial phagocytosis of apoptotic cells is impaired in Cstb KO mice, a model that recapitulates the main features of clinical EPM1. Additionally, we provide evidence on the possible mechanisms that underlie the phagocytic disruption associated with CSTB deficiency, based on the following findings. First, microglial phagocytosis of apoptotic cells was reduced in the hippocampus of symptomatic Cstb KO mice at P30. Second, the cell‐autonomous lack of Cstb in microglia did not drive phagocytosis impairment, because Cstb downregulation in pure microglial cultures did not alter phagocytosis efficiency. Third, microglial phagocytosis impairment was already present in the hippocampus of Cstb KO mice at P14, prior to the onset of seizures, suggesting that microglial phagocytosis impairment appears at early stages of disease and independent of seizures. Fourth, the impairment was specific for the GL, whereas the neurogenic niche of the SGZ was spared. Fifth, a virtual 3D model of the hippocampal GL in young CSTB KO mice (P14) predicted an aberrant distribution of cFos active neurons that results in a closer distance to apoptotic cells. These results suggest that local neuronal activity may alter apoptosis dynamics in Cstb KO mice, which may explain, at least in part, the reported impairment on microglial phagocytosis associated with CSTB deficiency. Here, we will first discuss the pathological effects of Cstb deficiency in the hippocampus, including atrophy, apoptosis, and phagocytosis impairment. Then, we will speculate on the impact of the microglial phagocytosis deficiency for the pathology of epilepsy. Finally, we will examine possible mechanisms underlying this impairment, including a cell‐autonomous effect of Cstb on microglia and environment‐related factors such as seizures and local neuronal activity.
4.1. Early symptomatic Cstb KO mice exhibit slight atrophy of the hippocampus
EPM1 is the most common type of progressive myoclonus epilepsy, a heterogeneous group of inherited diseases that concur with myoclonus, epilepsy, and progressive neuronal degeneration. 33 Most EPM1 patients are homozygous for a promoter region repeat expansion mutation resulting in significantly reduced CSTB expression. 34 Patients with total lack of CSTB display an early infantile onset rapidly progressing encephalopathy. 35 , 36 Progressive loss of brain volume affecting cerebellum, cortex, and hippocampus has been reported in patients with CSTB mutations 5 , 6 , 36 , 37 as well as in Cstb KO mice. 3 , 10 , 11 , 38 Accordingly, EPM1 patients have progressive motor deficits and mild cognitive impairment. 39
Here, we show earlier effects in hippocampal atrophy than previously described starting at 2 months of age using magnetic resonance imaging. 38 We found that at 1 month (P30), Cstb KO mice already contained fewer hippocampal granule cells and a tendency to atrophy in the septal hippocampus, with no changes in the temporal region. Furthermore, we found increased granule neuron apoptosis as early as P14. These developmental defects suggest that damage associated with CSTB mutations starts earlier than previously suggested, with atrophy developing from 1 to 6 months in cerebellum, cortex, and hippocampus in Cstb KO mice. 38 However, more subtle effects are found as early as P7, when the cerebellum begins to show decreased inhibition and enhanced excitation. 40 Therefore, it is possible that, like the hippocampus, cerebellum and cortex also experience pathophysiological changes such as apoptosis and microglial phagocytosis impairment in the first postnatal days.
4.2. Microglial phagocytosis of apoptotic cells is impaired in early symptomatic Cstb KO mice
In addition to the neuronal damage, other cell types have also been involved in the pathology of EPM1. Some of the earliest changes pointed toward an altered inflammatory response by microglia, the brain resident macrophages. 11 , 13 Here we focused on another aspect of microglia: their phagocytic function. Microglia are very efficient phagocytes in the adult hippocampus in physiological conditions, where unchallenged microglia rapidly clear the excess newborn cells produced in the neurogenic niche, 22 actively participating in the regulation of neurogenesis. 17 After stressful stimuli such as inflammation and excitotoxicity, microglia use different strategies to enhance phagocytosis and match the increased apoptosis levels 15 : recruiting more cells to become phagocytic (theoretically, up to 100%), increasing the phagocytic capacity of each microglial cell (at least up to seven pouches per cell), and proliferating. Combined, these strategies make microglia a very powerful phagocyte.
However, the phagocytic potential of microglia was not fully summoned in the hippocampus of young adult Cstb KO mice, at the age when they start to manifest clinical myoclonus (P30). Although microglia tried to compensate for increased apoptosis by (1) increasing the number of apoptotic cells cleared by each microglial cell and (2) increasing their numbers through proliferation, net phagocytosis did not match increased apoptosis in Cstb KO mice. These results are in line with previous mouse and human data on MTLE, where microglial phagocytosis was blocked, leading to accumulation of apoptotic cells in the hippocampus. 15 Accumulation of apoptotic cells has also been observed in the cerebellum as early as 2 months. 7 As an efficient phagocytosis involves rapid clearance of apoptotic cells (around 90 minutes in the hippocampus), 22 these data indirectly suggest microglial phagocytosis dysfunction in the cerebellum of Cstb KO mice, a hypothesis that needs to be tested.
4.3. Phagocytosis dysfunction in epilepsy
What are the consequences of impaired phagocytosis? The most obvious one is the accumulation of apoptotic cells. As executor caspases are activated in their cytoplasm, when not removed through phagocytosis, apoptotic cells evolve into secondary necrotic cells. 41 Their permeable membrane allows the release of toxic intracellular contents and contributes to further damaging healthy surrounding neurons, which may further contribute to altering the hippocampal connectivity and to cognitive impairment. In addition, phagocytosis is immunomodulatory, 15 and in a mouse model of MTLE, phagocytosis impairment correlates with the development of an inflammatory response. 15 In Cstb KO mice, microglia exhibited a hypertrophic morphology and multinuclearity, suggestive of microglial dysfunction and inflammation. 15 , 42 These results are also in agreement with the early abnormal morphology and expression of the inflammatory protein F4/80 in microglia prior to gross neurodegeneration in Cstb KO mice, 11 although the inflammatory profile of nonphagocytic microglia needs to be directly determined. Inflammation is a common feature of many types of epilepsies and epilepsy models, including Cstb KO mice. 12 , 13 , 40 In agreement, inflammatory mediators such as interleukin 1 beta potentiate seizures. 43 , 44 Our results suggest that in both MTLE 15 and EPM1 (here), microglial inflammation may arise at least in part due to the lack of control resulting from dysfunctional phagocytosis. Therefore, developing novel tools to manipulate microglial phagocytosis may serve to control secondary neuronal damage, inflammation, and their impact on seizures in epilepsy patients.
4.4. Microglial phagocytosis disruption is not due to the cell‐autonomous lack of CSTB
The microglial phagocytosis impairment could be mechanistically related to the lack of Cstb in microglia and/or in other brain cell types. We here show that Cstb expression was not restricted to microglia in the brain, suggesting that cell‐autonomous (lack of Cstb in microglia) or environment‐driven (lack of Cstb in other cell types) mechanisms could underlie the phagocytosis impairment. However, we observed no alterations in an in vitro model of phagocytosis using siRNA to deplete microglial Cstb. Nonetheless, this in vitro model does not fully recapitulate the full extent of CSTB deficiency in EPM1 patients, which includes a complex modulation of activity and mRNA expression of target cathepsins of CSTB. 30 , 38 Neither does it fully mimic the complexity of in vivo recognition and engulfment of apoptotic cells, which requires the release of “find‐me” signals from apoptotic cells and microglial process motility to engulf the cells (whereas in vitro apoptotic cells are simply dumped on top of microglia). 45 Despite the model's limitations, these data suggest that the lack of Cstb in microglia per se does not impact the clearance of apoptotic cells and suggests that the phagocytosis impairment could be related to environmental factors associated with the lack of Cstb, including its main pathological feature, seizures.
4.5. Microglial phagocytosis impairment is independent of seizure activity
Neuronal hyperactivity during seizures alters the microglial phagocytic response to apoptotic cells. For example, seizure‐induced widespread release of ATP in MTLE masks the microgradients of the “find‐me” signal ATP used by microglia to find apoptotic cells. 15 To analyze the impact of seizures in microglial phagocytosis, we took advantage of young Cstb KO mice (P14), which do not present clinical seizures. Using cFos as a marker of neuronal depolarization, 32 we did not find significant changes in the number of cFos+ neurons but, nonetheless, our virtual 3D model indirectly suggested that they were abnormally distributed in Cstb KO mice at P14, which is likely reflected in the seizures that appear at later stages. In addition, it is possible that more subtle alterations in the hippocampal circuit are already present at P14. Interestingly, cFos active neurons, increased numbers of apoptotic cells, and microglial phagocytosis impairment were selectively observed in the GL of the hippocampus and spared the SGZ, illuminating an unexpected relationship between neuronal activity, cell death, and microglial phagocytosis.
4.6. Local neuronal activity in the GL may contribute to the microglial phagocytosis impairment
In the GL, apoptotic cells were frequently positioned near active neurons labeled with cFos in Cstb KO mice, at an average distance of 3 μm. In agreement, our virtual 3D model suggested that apoptotic cells were located closer to the active neurons than would be expected based on their relative cell densities. The number of apoptotic cells at a given time point is the net result of apoptosis induction (input) minus phagocytosis (output), and thus these results indirectly suggest that local neuronal activity in the GL could alter apoptosis and phagocytosis dynamics in Cstb KO mice. Neuronal activity–dependent molecules such as ATP exert their functions locally due to limited diffusion to short distances in the tortuous brain parenchyma. 46 For instance, the effective diffusivity of a small molecule such as sucrose (0.342 kDa, close to the 0.551 kDa of ATP) is 310 μm/s, whereas that of larger molecules such as nerve growth factor (26.5 kDa) is 2.95 μm/s. Therefore, the average 3‐μm distance between apoptotic cells and active cFos neurons found in Cstb KO mice implies that the dead cells are well within the influence area of the active neurons' soma. Thus, both the dead cells and the reaching microglial processes could be affected by the somatic release of modulators by granule neurons, or by the perisynaptic release of modulators by incoming fibers from several afferent systems. 47
The close relationship between apoptotic cells and active neurons in Cstb KO mice is supported by both direct measurements and the virtual 3D model but does not provide evidence that this relationship affects phagocytosis. Nonetheless, it is important to note that the net apoptosis observed is in part the result of the phagocytosis dynamics; the number of apoptotic cells at any given time depends as much on the input (apoptosis induction) as on the output (removal by phagocytosis). 14 Therefore, our data show an overall impairment of microglial phagocytosis in the GL of Cstb KO mice and suggest a complex scenario in which subseizure local neuronal activity may affect the clearance of apoptotic cells by microglia.
In addition, other pathophysiological mechanisms may participate in the phagocytosis impairment observed in Cstb KO mice. For instance, one possibility is that intrinsic differences between GL and SGZ microglia explain why the impairment is restricted to the GL. SGZ microglia are likely “trained” for phagocytosis, as they have been exposed to apoptotic cells during the postnatal period, whereas GL microglia are not. This “training” could result in differences in their phagocytosis potential. For instance, SGZ microglia can reach a Ph capacity around 0.8 parts per unit (ppu) in a lipopolysaccharide model at P30. 22 This number is very far from the 0.18 ppu of GL microglia in Cstb KO mice at P14 reported here. Microglial subpopulations in the CA region of the hippocampus have been recently observed upon seizures induced by pilocarpine, based on the expression of keratan sulfate polysaccharides (epitope 5D4), whose overexpression is related to a higher ex vivo phagocytosis of zymosan particles possibly related to the engulfment of synapses. 48 Another potential candidate is the complement system, which is used by microglia to facilitate recognition (opsonization) of apoptotic cells 49 as well as synapses, 50 and is progressively activated in rodent and human epilepsy, 51 , 52 including Cstb KO mice. 29 , 40 Nonetheless, it is beyond the scope of this paper to test the role of complement or keratan sulfate, and future studies will decipher the underlying mechanisms of microglial phagocytosis impairment in Cstb KO mice.
In summary, we here extend our initial observations that microglial phagocytosis impairment in the hippocampus is an early feature of mouse and human MTLE and show that it also occurs in a genetic model of EPM1 by Cstb deficiency. We also provide an unexpected link between phagocytosis impairment, accumulation of apoptotic cells, and local neuronal activity in these mice that further supports the suggestion that both abnormal local neuronal activity (in presymptomatic Cstb KO mice) and network hyperactivity (in MTLE mice) regulate the efficiency of microglial phagocytosis. Phagocytosis is an essential component of the brain regenerative response, as it removes cell debris and is immunomodulatory. Therefore, future therapies for epilepsy patients should be aimed not only at reducing neuronal death but also at harnessing microglial phagocytosis.
CONFLICT OF INTEREST
None of the authors has any conflict of interest to disclose.
ETHICAL PUBLICATION STATEMENT
We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Supporting information
FigS1
Supinfo
ACKNOWLEDGMENTS
This work was supported by grants from the Spanish Ministry of Science and Innovation (https://www.ciencia.gob.es/), with FEDER funds to A.S. (RTI2018‐099267‐B‐I00, BFU2012‐32089, and RYC‐2013‐12817), to A.S. and J.V. (BFU2015‐66689), and to P.B. (SAF2015‐69484‐R); a Leonardo Award from the BBVA Foundation to A.S. (IN16,_BBM_BAS_0260); a Tatiana Foundation project grant to A.S. (P‐048‐FTPGB 2018); a Basque Government Department of Education project grant to A.S. (PI_2016_1_0011; http://www.euskadi.eus/basque‐government/department‐education/); Ikerbasque startup funds to J.V. and P.B.; and the Folkhälsan Research Foundation (to A.‐E.L.). In addition, V.S.‐T. and O.A. are recipients of a predoctoral fellowship from the Basque Government; A.‐E.L. is a HiLIFE Fellow at the University of Helsinki. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. We thank Victor Sánchez Zafra for technical support. We are very grateful to Eva Benito for thoroughly discussing the manuscript.
Sierra‐Torre V, Plaza‐Zabala A, Bonifazi P, et al. Microglial phagocytosis dysfunction in the dentate gyrus is related to local neuronal activity in a genetic model of epilepsy. Epilepsia. 2020;61:2593–2608. 10.1111/epi.16692
REFERENCES
- 1. Joensuu T, Lehesjoki AE, Kopra O. Molecular background of EPM1‐Unverricht‐Lundborg disease. Epilepsia. 2008;49:557–63. [DOI] [PubMed] [Google Scholar]
- 2. Lalioti MD, Scott HS, Buresi C, Rossier C, Bottani A, Morris MA, et al. Dodecamer repeat expansion in cystatin B gene in progressive myoclonus epilepsy. Nature. 1997;386:847–51. [DOI] [PubMed] [Google Scholar]
- 3. Pennacchio LA, Lehesjoki A‐E, Stone NE, Willour VL, Virtaneva K, Miao J, et al. Mutations in the gene encoding cystatin B in progressive myoclonus epilepsy (EPM1). Science. 1996;271:1731–4. [DOI] [PubMed] [Google Scholar]
- 4. Riccio M, Di Giaimo R, Pianetti S, Palmieri PP, Melli M, Santi S. Nuclear localization of cystatin B, the cathepsin inhibitor implicated in myoclonus epilepsy (EPM1). Exp Cell Res. 2001;262:84–94. [DOI] [PubMed] [Google Scholar]
- 5. Koskenkorva P, Niskanen E, Hyppönen J, Könönen M, Mervaala E, Soininen H, et al. Sensorimotor, visual, and auditory cortical atrophy in Unverricht‐Lundborg disease mapped with cortical thickness analysis. AJNR Am J Neuroradiol. 2012;33:878–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mascalchi M, Michelucci R, Cosottini M, Tessa C, Lolli F, Riguzzi P, et al. Brainstem involvement in Unverricht‐Lundborg disease (EPM1): an MRI and (1)H MRS study. Neurology. 2002;58:1686–9. [DOI] [PubMed] [Google Scholar]
- 7. Pennacchio LA, Bouley DM, Higgins KM, Scott MP, Noebels JL, Myers RM. Progressive ataxia, myoclonic epilepsy and cerebellar apoptosis in cystatin B‐deficient mice. Nat Genet. 1998;20:251–8. [DOI] [PubMed] [Google Scholar]
- 8. Maher K, Jeric Kokelj B, Butinar M, Mikhaylov G, Mancek‐Keber M, Stoka V, et al. A role for stefin B (cystatin B) in inflammation and endotoxemia. J Biol Chem. 2014;289:31736–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Manninen O, Koskenkorva P, Lehtimaki KK, Hypponen J, Kononen M, Laitinen T, et al. White matter degeneration with Unverricht‐Lundborg progressive myoclonus epilepsy: a translational diffusion‐tensor imaging study in patients and cystatin B‐deficient mice. Radiology. 2013;269:232–9. [DOI] [PubMed] [Google Scholar]
- 10. Shannon P, Pennacchio LA, Houseweart MK, Minassian BA, Myers RM. Neuropathological changes in a mouse model of progressive myoclonus epilepsy: cystatin B deficiency and Unverricht‐Lundborg disease. J Neuropathol Exp Neurol. 2002;61:1085–91. [DOI] [PubMed] [Google Scholar]
- 11. Tegelberg S, Kopra O, Joensuu T, Cooper JD, Lehesjoki AE. Early microglial activation precedes neuronal loss in the brain of the Cstb‐/‐ mouse model of progressive myoclonus epilepsy, EPM1. J Neuropathol Exp Neurol. 2012;71:40–53. [DOI] [PubMed] [Google Scholar]
- 12. Korber I, Katayama S, Einarsdottir E, Krjutskov K, Hakala P, Kere J, et al. Gene‐expression profiling suggests impaired signaling via the interferon pathway in Cstb‐/‐ microglia. PLoS One. 2016;11:e0158195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Okuneva O, Korber I, Li Z, Tian L, Joensuu T, Kopra O, et al. Abnormal microglial activation in the Cstb(‐/‐) mouse, a model for progressive myoclonus epilepsy, EPM1. Glia. 2015;63:400–11. [DOI] [PubMed] [Google Scholar]
- 14. Márquez‐Ropero M, Benito E, Plaza‐Zabala A, Sierra A. Microglial corpse clearance: lessons from macrophages. Front Immunol. 2020;11:506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Abiega O, Beccari S, Diaz‐Aparicio I, Nadjar A, Laye S, Leyrolle Q, et al. Neuronal hyperactivity disturbs ATP microgradients, impairs microglial motility, and reduces phagocytic receptor expression triggering apoptosis/microglial phagocytosis uncoupling. PLoS Biol. 2016;14:e1002466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sierra A, Abiega O, Shahraz A, Neumann H. Janus‐faced microglia: beneficial and detrimental consequences of microglial phagocytosis. Front Cell Neurosci. 2013;7:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Diaz‐Aparicio I, Paris I, Sierra‐Torre V, Plaza‐Zabala A, Rodriguez‐Iglesias N, Marquez‐Ropero M, et al. Microglia actively remodel adult hippocampal neurogenesis through the phagocytosis secretome. J Neurosci. 2020;40:1453–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Morioka S, Maueroder C, Ravichandran KS. Living on the edge: efferocytosis at the interface of homeostasis and pathology. Immunity. 2019;50:1149–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lemke G. How macrophages deal with death. Nat Rev Immunol. 2019;19:539–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Koizumi S, Ohsawa K, Inoue K, Kohsaka S. Purinergic receptors in microglia: functional modal shifts of microglia mediated by P2 and P1 receptors. Glia. 2013;61:47–54. [DOI] [PubMed] [Google Scholar]
- 21. Arandjelovic S, Ravichandran KS. Phagocytosis of apoptotic cells in homeostasis. Nat Immunol. 2015;16:907–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sierra A, Encinas JM, Deudero JJ, Chancey JH, Enikolopov G, Overstreet‐Wadiche LS, et al. Microglia shape adult hippocampal neurogenesis through apoptosis‐coupled phagocytosis. Cell Stem Cell. 2010;7:483–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Toda T, Parylak SL, Linker SB, Gage FH. The role of adult hippocampal neurogenesis in brain health and disease. Mol Psychiatry. 2019;24:67–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sierra A, Beccari S, Diaz‐Aparicio I, Encinas JM, Comeau S, Tremblay ME. Surveillance, phagocytosis, and inflammation: how never‐resting microglia influence adult hippocampal neurogenesis. Neural Plast. 2014;2014:610343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Scholzen T, Gerdes J. The Ki‐67 protein: from the known and the unknown. J Cell Physiol. 2000;182:311–22. [DOI] [PubMed] [Google Scholar]
- 26. Christensen RN, Ha BK, Sun F, Bresnahan JC, Beattie MS. Kainate induces rapid redistribution of the actin cytoskeleton in ameboid microglia. J Neurosci Res. 2006;84:170–81. [DOI] [PubMed] [Google Scholar]
- 27. Sasmono RT, Oceandy D, Pollard JW, Tong W, Pavli P, Wainwright BJ, et al. A macrophage colony‐stimulating factor receptor‐green fluorescent protein transgene is expressed throughout the mononuclear phagocyte system of the mouse. Blood. 2003;101:1155–63. [DOI] [PubMed] [Google Scholar]
- 28. Sierra A, Gottfried‐Blackmore AC, McEwen BS, Bulloch K. Microglia derived from aging mice exhibit an altered inflammatory profile. Glia. 2007;55:412–24. [DOI] [PubMed] [Google Scholar]
- 29. Lieuallen K, Pennacchio LA, Park M, Myers RM, Lennon GG. Cystatin B‐deficient mice have increased expression of apoptosis and glial activation genes. Hum Mol Genet. 2001;10:1867–71. [DOI] [PubMed] [Google Scholar]
- 30. Rinne R, Saukko P, Jarvinen M, Lehesjoki AE. Reduced cystatin B activity correlates with enhanced cathepsin activity in progressive myoclonus epilepsy. Ann Med. 2002;34:380–5. [DOI] [PubMed] [Google Scholar]
- 31. Kaur G, Mohan P, Pawlik M, DeRosa S, Fajiculay J, Che S, et al. Cystatin C rescues degenerating neurons in a cystatin B‐knockout mouse model of progressive myoclonus epilepsy. Am J Pathol. 2010;177:2256–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Verma IM, Sassone‐Corsi P. Proto‐oncogene fos: complex but versatile regulation. Cell. 1987;51:513–4. [DOI] [PubMed] [Google Scholar]
- 33. Arielle C, Edoardo F, Silvana F, Pierre G, Riadh G, Reetta K, et al. Unverricht‐Lundborg disease. Epileptic Disord. 2016;18:28–37. [DOI] [PubMed] [Google Scholar]
- 34. Joensuu T, Kuronen M, Alakurtti K, Tegelberg S, Hakala P, Aalto A, et al. Cystatin B: mutation detection, alternative splicing and expression in progressive myclonus epilepsy of Unverricht‐Lundborg type (EPM1) patients. Eur J Hum Genet. 2007;15:185–93. [DOI] [PubMed] [Google Scholar]
- 35. Mancini GM, Schot R, deWit MC, de Coo RF, Oostenbrink R, Bindels‐de Heus K, et al. CSTB null mutation associated with microcephaly, early developmental delay, and severe dyskinesia. Neurology. 2016;86:877–8. [DOI] [PubMed] [Google Scholar]
- 36. O'Brien A, Marshall CR, Blaser S, Ray PN, Yoon G. Severe neurodegeneration, progressive cerebral volume loss and diffuse hypomyelination associated with a homozygous frameshift mutation in CSTB. Eur J Hum Genet. 2017;25:775–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Koskenkorva P, Khyuppenen J, Niskanen E, Könönen M, Bendel P, Mervaala E, et al. Motor cortex and thalamic atrophy in Unverricht‐Lundborg disease: voxel‐based morphometric study. Neurology. 2009;73:606–11. [DOI] [PubMed] [Google Scholar]
- 38. Manninen O, Laitinen T, Lehtimäki KK, Tegelberg S, Lehesjoki A‐E, Gröhn O, et al. Progressive volume loss and white matter degeneration in Cstb‐deficient mice: a diffusion tensor and longitudinal volumetry MRI Study. PLoS One. 2014;9:e90709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lehesjoki AE, Gardiner M. Progressive myoclonus epilepsy: Unverricht‐Lundborg disease and neuronal ceroid lipofuscinoses In: Noebels JL, Avoli M, Rogawski MA, Olsen RW, Delgado‐Escueta AV, eds. Jasper's Basic Mechanisms of the Epilepsies. 4th ed. New York, NY: Oxford University Press; 2012:878–86. [PubMed] [Google Scholar]
- 40. Joensuu T, Tegelberg S, Reinmaa E, Segerstrale M, Hakala P, Pehkonen H, et al. Gene expression alterations in the cerebellum and granule neurons of Cstb(‐/‐) mouse are associated with early synaptic changes and inflammation. PLoS One. 2014;9(2):e89321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Savill J, Dransfield I, Gregory C, Haslett C. A blast from the past: clearance of apoptotic cells regulates immune responses. Nat Rev Immunol. 2002;2:965–75. [DOI] [PubMed] [Google Scholar]
- 42. Hornik TC, Neniskyte U, Brown GC. Inflammation induces multinucleation of microglia via PKC inhibition of cytokinesis, generating highly phagocytic multinucleated giant cells. J Neurochem. 2014;128:650–61. [DOI] [PubMed] [Google Scholar]
- 43. Vezzani A, Viviani B. Neuromodulatory properties of inflammatory cytokines and their impact on neuronal excitability. Neuropharmacology. 2015;96:70–82. [DOI] [PubMed] [Google Scholar]
- 44. Iori V, Iyer AM, Ravizza T, Beltrame L, Paracchini L, Marchini S, et al. Blockade of the IL‐1R1/TLR4 pathway mediates disease‐modification therapeutic effects in a model of acquired epilepsy. Neurobiol Dis. 2017;99:12–23. [DOI] [PubMed] [Google Scholar]
- 45. Beccari S, Diaz‐Aparicio I, Sierra A. Quantifying microglial phagocytosis of apoptotic cells in the brain in health and disease. Curr Protoc Immunol. 2018;e49. [DOI] [PubMed] [Google Scholar]
- 46. Wolak DJ, Thorne RG. Diffusion of macromolecules in the brain: implications for drug delivery. Mol Pharm. 2013;10:1492–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Leranth C, Hajszan T. Extrinsic afferent systems to the dentate gyrus. Prog Brain Res. 2007;163:63–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ohgomori T, Jinno S. The expression of keratan sulfate reveals a unique subset of microglia in the mouse hippocampus after pilocarpine‐induced status epileptics. J Comp Neurol. 2020;528:14–31. [DOI] [PubMed] [Google Scholar]
- 49. Diaz‐Aparicio I, Sierra A. C1q is related to microglial phagocytosis in the hippocampus in physiological conditions. Matters. 2019. https://sciencematters.io/articles/201904000013 [Google Scholar]
- 50. Thion MS, Garel S. Microglia under the spotlight: activity and complement‐dependent engulfment of synapses. Trends Neurosci. 2018;41:332–4. [DOI] [PubMed] [Google Scholar]
- 51. Wyatt‐Johnson SK, Brewster AL. Emerging roles for microglial phagocytic signaling in epilepsy. Epilepsy Curr. 2020;20(1):33–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wyatt SK, Witt T, Barbaro NM, Cohen‐Gadol AA, Brewster AL. Enhanced classical complement pathway activation and altered phagocytosis signaling molecules in human epilepsy. Exp Neurol. 2017;295:184–93. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
FigS1
Supinfo