

Abstract
We have prepared Zn and free‐base porphyrins appended with a fac‐Re(phen)(CO)3Br (where phen is 1,10‐phenanthroline) at the meso position of the porphyrin, and performed photocatalytic CO2 reduction using porphyrin–Re dyads in the presence of either triethylamine (TEA) or 1,3‐dimethyl‐2‐phenyl‐2,3‐dihydro‐1H‐benzo[d]imidazole (BIH) as an electron donor. The Zn porphyrin dyad showed a high turnover number for CO production compared with the free‐base porphyrin dyad, suggesting that the central Zn ion of porphyrin plays an important role in suppressing electron accumulation on the porphyrin part and achieving high durability of the photocatalytic CO2 reduction using both TEA and BIH. The effect of acids on the CO2 reduction was investigated using the Zn porphyrin–Re dyad and BIH. Acetic acid, a relatively strong Brønsted acid, rapidly causes the porphyrin's color to fade upon irradiation and dramatically decreases CO production, whereas proper weak Brønsted acids such as 2,2,2‐trifluoroethanol and phenol enhance the CO2 reduction.
Keywords: CO2 reduction, photocatalysis, photosensitizer, porphyrin, rhenium tricarbonyl complex
The Zn porphyrin and Re complex dyad acted as a good photocatalyst in photocatalytic CO2 reduction using both triethylamine and 1,3‐dimethyl‐2‐phenyl‐2,3‐dihydro‐1H‐benzo[d]imidazole (BIH) as an electron donor. The central Zn ion of the porphyrin played an important role in high durability. The effect of acids on the photocatalytic CO2 reduction was investigated with BIH, showing proper weak Brønsted acids enhanced the CO2 reduction.
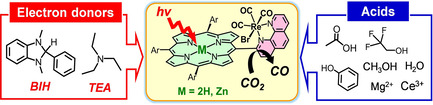
Introduction
Artificial photosynthesis has received much attention because of its great potential for solving environmental challenges such as global warming and ocean acidification, as well as the shortage of fossil fuels. Because sunlight is mainly composed of visible light and the photon flux is low, a photosensitizer is an essential component for efficient collection of visible light in a solar energy conversion system. [1] In natural photosynthesis, a network of chlorophylls efficiently captures even dilute photons from the sun and transfers the absorbed solar energy to produce energy‐rich compounds. [2] As synthetic chlorophyll analogues, porphyrin derivatives have been actively studied to elucidate the high efficiency of energy and electron transfer processes in photosynthetic systems[ 3 , 4 ] and have been used as photosensitizers in photoredox reactions[ 5 , 6 , 7 , 8 , 9 ] because of their absorption bands in the visible region [e.g., ϵ 423nm ≈ 540 000 m −1 cm−1 for zinc tetraphenylporphyrin (ZnTPP)]. [10]
Dyads combining porphyrin and the Re diimine tricarbonyl complex have been developed for photocatalytic CO2 reduction.[ 11 , 12 , 13 , 14 , 15 ] Inoue and co‐workers reported that the photocatalytic CO2 reduction using a dyad, in which a Zn porphyrin and a Re(bpy)(CO)3Br (where bpy is 2,2’‐bipyridine) complex were connected via an amide‐bridged linker, gave CO in the presence of triethylamine (TEA) as an electron donor. The reaction quantum yield (Φ CO) strongly depended on the excitation wavelength, showing 10 % and 0.64 % on exciting at 364 and 428 nm, respectively. [11] Perutz and co‐workers reported that a saturated methylene spacer in the amide‐bridge linker of the dyad significantly improved the CO production to reach a turnover number (TONCO) of 260 by irradiation at λ > 520 nm in the presence of triethanolamine (TEOA) as the electron donor. [12a] Tschierlei, Schwalbe, and co‐workers reported that the dyad, in which a Zn porphyrin and a Re(bpy)(CO)3Cl were linked via a π‐conjugated moiety, gave CO with a TONCO value of 13 using 5 vol % TEA as the electron donor by irradiation at λ > 375 nm. [13a] The dyads using Fe, Co, Cu, and Pd porphyrin instead of Zn porphyrin did not show catalytic activity for the CO2 reduction. Moore, Schwalbe, and co‐workers reported that the rigid xanthene‐bridged dyad of a Zn porphyrin and a Re(bpy)(CO)3Cl showed a TONCO of 5.7 or 195 using either 5 vol % TEA or a mixture of 20 vol % TEOA and 1,3‐dimethyl‐2‐phenyl‐2,3‐dihydro‐1H‐benzo[d]imidazole (BIH) by irradiation at λ > 450 nm. The dyad of a free‐base porphyrin instead of the Zn porphyrin showed no activity for a photocatalytic CO2 reduction using 5 vol % TEA. [13b] Tamiaki and co‐workers reported that irradiation at λ > 540 nm of a dyad composed of a chlorophyll and a Re(bpy)(CO)3Cl complex, gave CO with a TONCO of 18 using a mixture of 17 vol % TEOA and 0.1 m BIH. [14]
We have recently reported that the dyad of a Zn porphyrin and Re(phen)(CO)3Br (where phen is 1,10‐phenanthroline), in which the Re complex is directly connected at the 2‐position of the phen moiety to a meso‐position of the porphyrin, named as ZnP‐phen=Re (Scheme 1), showed a TONCO larger than 1300 by irradiation at λ = 420 nm using BIH as an electron donor[ 13b , 15 , 16 , 17 ] and phenol (PhOH) as a proton source. [15] However, in the previous work, only photocatalytic CO2 reduction using BIH as the electron donor, PhOH as the proton source, and the dyad in which the porphyrin center was a Zn ion, was investigated, and other applicable ranges and conditions were not examined. In the current work, we report three main experiments that supplement our earlier study. First, to investigate the role of the central Zn ion of the porphyrin in the photocatalytic CO2 reduction, we prepared a dyad consisting of a free‐base porphyrin and a fac‐Re(phen)(CO)3Br, named as H2P‐phen=Re (Scheme 1), and compared its catalytic activity with that of ZnP‐phen=Re. Second, to check whether the photocatalytic reaction proceeds with an electron donor other than BIH, we performed photocatalytic CO2 reduction using TEA as the electron donor, which has been more often used in the porphyrin=Re complex dyad systems. Third, we investigated the effect of proton sources (Brønsted acids) and metal cations in the photocatalytic CO2 reduction using the system of ZnP‐phen=Re and BIH.
Scheme 1.
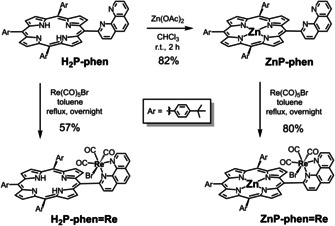
Synthetic routes to ZnP‐phen=Re and H2P‐phen=Re.
Results and Discussion
Syntheses and structures of the dyads
The synthetic procedures are shown in Scheme 1. The dyads, ZnP‐phen=Re and H2P‐phen=Re, were synthesized by the reactions of the corresponding porphyrins [15] with 1 equiv of Re(CO)5Br, respectively. Use of an excess amount (4 equiv) of Re(CO)5Br caused a low yield (<10 %) of the target dyad accompanied with unidentified species that were decomposed in silica‐gel column chromatography. The crude products obtained by the reactions with 1 equiv of Re(CO)5Br were recrystallized from CHCl3/ethanol/hexane to afford the pure dyads, ZnP‐phen=Re and H2P‐phen=Re, each of which shows a single spot on silica‐gel thin layer chromatography (TLC). The chemical compositions of the dyads were identified by elemental analysis.
1H NMR spectra of the purified ZnP‐phen=Re and H2P‐phen=Re in CDCl3 are shown in Figure 1. The signals were assigned with a combination of a 2D NMR (1H‐1H correlation spectroscopy (COSY); Figures S3 and S8 in Supporting Information). The spectrum of H2P‐phen=Re shows an inner NH proton signal at −2.51 ppm, which is a characteristic signal for free‐base porphyrin. Both spectra of ZnP‐phen=Re and H2P‐phen=Re show that one β proton appears at the higher magnetic field, 8.4 and 8.2 ppm, compared with the other seven β protons for ZnP‐phen=Re and H2P‐phen=Re, respectively. In addition, six doublet (J=8 Hz) signals are observed on the ortho protons of the phenylene groups. The above observations indicate that both ZnP‐phen=Re and H2P‐phen=Re have A‐ and A’‐type structures in Figure S9; that is, facial structures. Judging from the facts that the anisotropic magnetic field of the carbonyl group shifts the β 3 proton to the low magnetic field and the β 2 proton to the high magnetic field (Figure S10, top view) and from the correlations in the 1H‐1H COSY, the β protons shifting to higher magnetic fields at 8.4 and 8.2 ppm were assigned to be β 7 protons. The characteristic β 7 protons are magnetically shielded by the nearby Br atom. The formation of the A‐ and A’‐type isomers (Figure S9) is supported by three signals of CO carbons at different positions on the 13C NMR spectra (Figure S5) and the pattern of the CO stretching bands on the infrared (IR) spectra (Figure S11). [15] The free energies of formation of the Zn porphyrin isomers were estimated with density functional theory (DFT) calculations, and showed that the A‐type isomers were more stable than the B‐ and C‐ type isomers that have trans(CO) structures (Figure S12).
Figure 1.
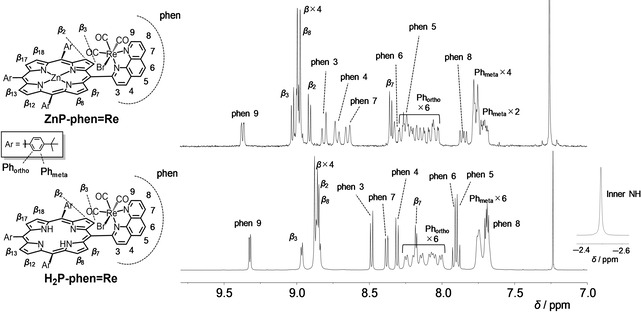
1H NMR spectra of ZnP‐phen=Re (top, 300 MHz) and H2P‐phen=Re (bottom, 500 MHz) in CDCl3. The top Figure corresponds to Figure S7 in ref. [15].
Figure 1 shows the broadened peaks (half maximum of the peak: 5–7 Hz) of the phenylene protons at 8.0–8.3 ppm in H2P‐phen=Re compared with the other β and phen peaks (1–3 Hz), whereas all the peaks including the phenylene protons have similar widths of 2–3 Hz in ZnP‐phen=Re. The conformational flexibility of the porphyrin skeleton is known to depend on the atom(s) coordinated to the central core and increases according to the following order in solution: ZnII < PdII and CuII < free base < NiII. [18] This order is consistent with the degree of distortion of the porphyrin skeleton from planarity in the crystal structures. [18] Thus, it is thought that the broad peaks observed for the phenylene protons in H2P‐phen=Re originate from the flexibility of the free‐base porphyrin.
H2P‐phen=Re complexes sometimes show low reproducibility in the 1H NMR signals even when samples showing a single spot at the same position on the silica‐gel TLC are used (Figure S7). We encountered poor reproducibility even using the same sample. Judging from the peak shapes of phen and β‐pyrrole, aggregations seem to be ruled out. The 1H NMR signals of ZnP‐phen and ZnP‐phen=Re in CDCl3 were significantly affected by the presence of [D4]MeOH (Figures S1 and S2), and a small amount of ethanol was observed at about 1.2 and 3.7 ppm in the 1H NMR spectra of H2P‐phen=Re (Figure S7a and b, insets). [19] Thus, a trace amount of ethanol, which was used as a solvent for recrystallization and was included as a stabilizer in the chloroform used for sample transfer, might be the cause of the low reproducibility in the 1H NMR spectrum.
Photophysical and electrochemical properties
The ultraviolet–visible (UV/Vis) absorption spectra of the porphyrins were measured in N,N‐dimethylacetamide (DMA), which is a suitable solvent for photocatalytic CO2 reduction using the Re complex. [20] DMA is known to act as an axial ligand for Zn porphyrin and causes a red shift in UV/Vis absorption spectra. [21] The absorption spectrum of ZnP‐phen in DMA was red‐shifted compared with that in CHCl3 (Figure S13), indicating that DMA coordinates the Zn porphyrins not only in ZnP‐phen, but also in ZnP‐phen=Re. Porphyrin has intense absorption bands in the visible region. The absorption coefficients of the Soret bands for ZnP‐phen and ZnP‐phen=Re (ϵ > 200 000 m −1 cm−1) are larger by a factor of more than 10 than that of a RuII tris‐diimine complex, [Ru(dmb)3]2+ (dmb = 4,4’‐dimethyl‐2,2’‐bipyridine; Figure 2 a), which is widely used as a photosensitizer in many photocatalytic reactions including CO2 reduction.[ 17 , 22 ] In addition, the porphyrins have relatively intense absorption peaks of the Q‐bands at more than 540 nm (ϵ > 15 000 m −1 cm−1), where the Ru tris‐diimine complexes have almost no absorption bands. The Soret band of ZnP‐phen=Re is significantly broader than that of ZnP‐phen, indicating strong electronic interaction between the Zn porphyrin and the Re complex. [15] H2P‐phen=Re also shows a broader Soret band than that of H2P‐phen (Figure 2 b). In general, monomeric Zn and free‐base meso‐tetraarylporphyrins have no strong absorption band between 450 and 500 nm. However, the dyads of ZnP‐phen=Re and H2P‐phen=Re have sufficient absorption by broadening the Soret bands, giving the potential to use a much wider wavelength range of visible light (450–500 nm) for photocatalytic reactions.
Figure 2.
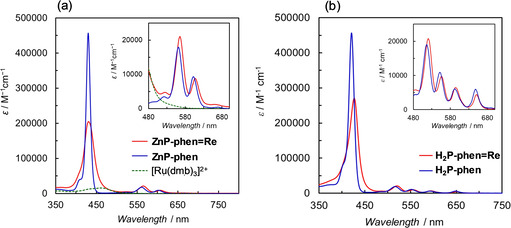
UV/Vis absorption spectra of (a) ZnP‐phen, ZnP‐phen=Re and [Ru(dmb)3]2+ (dmb=4,4’dimethyl‐2,2’‐bipyridine), and (b) H2P‐phen and H2P‐phen=Re in DMA. The insets show the magnifications of the Q‐band region. The spectra of ZnP‐phen and ZnP‐phen=Re correspond to Figure 2 in ref. [15].
The redox potentials of the dyads and the corresponding parts in Ar‐saturated DMA were obtained with cyclic voltammetry (CV, Figure 3) and differential pulse voltammetry (DPV, Figure S15). The anodic signals of H2P‐phen and ZnP‐phen appeared at −1.2 and −1.4 V, respectively, on the reversed scans after sweeping more negative potentials, indicating that the C=C bond hydrogenated species were formed by two‐electron reduction. [23] By contrast, the anodic become reversible on sweeping right after the first reduction waves, indicating that the one‐electron reduced species of the porphyrins are stable in DMA. The first reduction potential of ZnP‐phen=Re appeared at −1.71 V, which is almost the same with that of fac‐Re(phen)(CO)3Br (−1.72 V), indicating that the one‐electron species localizes the electron on the Re part. By contrast, the first reduction signal of H2P‐phen=Re appeared at −1.48 V, similar to that of H2P‐phen (−1.52 V), indicating that the one‐electron species localizes the electron on the porphyrin part. The reduction potentials evaluated from the electrochemical measurements correlated with the lowest unoccupied molecular orbital (LUMO) levels estimated from the DFT calculations; in particular, between ZnP‐phen=Re coordinated by DMA and H2P‐phen=Re (Figure 4). In ZnP‐phen=Re (Figure 4, left and center), the highest occupied molecular orbital (HOMO) level was significantly increased by the DMA coordination (180 mV), but the LUMO level was only slightly increased (50 mV). This reflects that the LUMO mainly distributes not on the porphyrin, but on the Re part in ZnP‐phen=Re.
Figure 3.
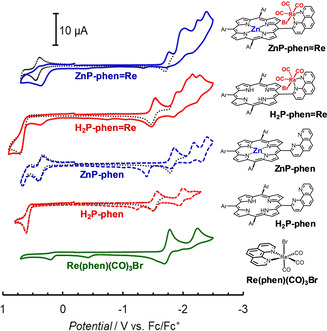
CVs of the dyads, porphyrins and fac‐Re(phen)(CO)3Br (0.5 mm) in Ar‐saturated DMA with 0.1 m nBu4NPF6 as a supporting electrolyte. Scan rate=100 mV s−1.
Figure 4.
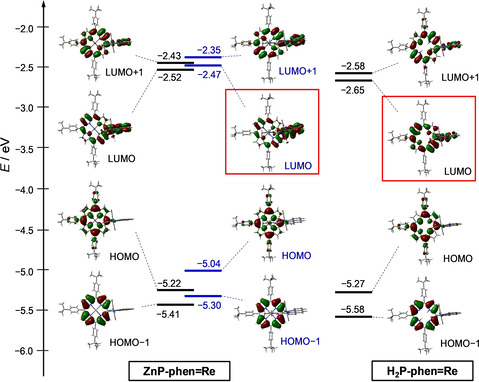
Energy level diagrams and frontier orbitals of ZnP‐phen=Re without (left) and with (center) the axial‐coordinated DMA and H2P‐phen=Re (right). The structures are optimized at the B3LYP/LANL2DZ/6‐31G(d) level using PCM with the default parameter for DMA (isovalue=0.02).
The first reduction signals of ZnP‐phen=Re and H2P‐phen=Re appeared at similar potentials to those of fac‐Re(phen)(CO)3Br and H2P‐phen, respectively. By contrast, the second signals were shifted to the negative side by more than 100 mV compared with the mononuclear units, indicating that there are strong electronic interactions between the porphyrin and the Re complex parts in the dyads.
The fluorescence of ZnP‐phen and H2P‐phen was almost completely quenched by introducing the Re complex parts in DMA (Figure S16). It has been reported that the fluorescence quenching in ZnP‐phen=Re originates not from the intramolecular electron transfer from the Zn porphyrin part to the Re part, but from spin–orbit coupling imposed by the large Re atom to induce a rapid intersystem crossing (ISC) on the porphyrin. [15] The fluorescence quenching in H2P‐phen=Re also comes from the rapid ISC induced by the Re atom, because the intramolecular electron transfer from the free‐base porphyrin part to the Re part is a very endothermic reaction (Figure S17). ZnP‐phen=Re showed phosphorescence from Zn porphyrin in Ar‐saturated DMA even at room temperature because of the rapid ISC caused by the large Re atom. [15] However, we could not observe phosphorescence of H2P‐phen=Re under the same conditions. Quimby and Longo reported that no phosphorescence of free‐base TPP could be unambiguously detected in methylcyclohexane‐isopentane glass even at 77 K, and estimated that the phosphorescent quantum yield of free‐base TPP is less than 1/100 that of ZnTPP. [10b] Thus, no detectable phosphorescence of H2P‐phen=Re could originate from the much lower phosphorescent ability of the free‐base porphyrin.
Photocatalytic CO2 reductions using TEA as the electron donor
TEA is a weaker reductant and is more often used as an electron donor than BIH in porphyrin–Re complex dyad systems.[ 11 , 12 , 13 ] Photocatalytic CO2 reductions were performed using TEA as the electron donor and either ZnP‐phen=Re or H2P‐phen=Re as a photocatalyst. In addition, as a control experiment, the reaction using a mixed system of ZnTPP and fac‐Re(phen)(CO)3Br was also conducted. The photocatalytic CO2 reduction using the dyads gave CO selectively and the time profiles of the CO production under irradiation at 420 nm are shown in Figure 5 a. Irradiation under Ar atmosphere instead of CO2 gave only H2 without formation of CO. The TONCO for ZnP‐phen=Re reached a value of 23 after 60 h and the CO production still continued even after 60 h. The value of TONCO>25 was the highest in the reported dyads composed of a porphyrin and a Re complex in close proximity. [13]
Figure 5.
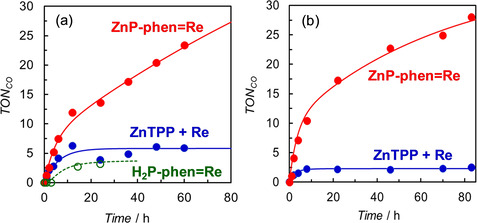
Time dependences of CO formation during irradiation of 2.0 mL of CO2‐saturated DMA solutions containing 5 vol% TEA in the presence of ZnP‐phen=Re (0.05 mm), H2P‐phen=Re (0.05 mm) or a mixed system of ZnTPP (0.05 mm) and fac‐Re(phen)(CO)3Br (0.05 mm); irradiation was performed at (a) 420 nm (30 mW) and (b) 450 nm (40 mW).
ZnP‐phen=Re has a broadened Soret band compared with the Zn porphyrins without the Re unit. The selected wavelength of 450 nm in Figure 5 b is generally the foot of the Soret band of monomeric Zn porphyrin but the photocatalytic reaction of ZnP‐phen=Re proceeds efficiently. Contrary to the behavior of a monomeric Zn porphyrin such as ZnTPP, in ZnP‐phen=Re there is high absorbance even at 450 nm because of the broad Soret band caused by the strong electronic interaction between the porphyrin and the Re complex. Based on their power (30 and 40 mW), the photon number of the 450 nm lamp was estimated to be about 1.4 times that of the 420 nm lamp, and the initial reaction rate of ZnP‐phen=Re in Figure 5 a (5.2 for 4 h) is approximately 1.4 times that in Figure 5 b (7.1 for 4 h). Thus, ZnP‐phen=Re could efficiently use all the irradiated photons with the wavelength where monomeric Zn meso‐tetraarylporphyrins do not have enough absorption.
In contrast to ZnP‐phen=Re, the CO productions for H2P‐phen=Re and the mixed system stop by 10 h and the TONCO values are low. The colors of solutions of both H2P‐phen=Re and the mixed system changed into pale‐yellow by 10 h. The rapid color fading is due to hydrogenation of more than two pyrrole C=C bonds of the porphyrin. The complete bleaching of porphyrin absorption by hydrogenation has been observed in the photocatalytic CO2 reduction using the amide‐bridged dyads. [12] By contrast, the solution color of ZnP‐phen=Re gradually changed from dark‐green to green during the irradiation, but the green color was maintained even after 10 h. The UV/Vis absorption spectral change of ZnP‐phen=Re during the irradiation showed a decrease of the Q‐band of the Zn porphyrin at 565 nm and appearance of a new peak at 629 nm with an isosbestic point (Figure S18). The new peak was assigned to the Zn chlorin formed by hydrogenation of only one pyrrole C=C bond of the porphyrin. [12] The Zn chlorin of the dyad was not hydrogenated further and acted as the photosensitizer after 10 h irradiation (Figure 5), whereas H2P‐phen=Re and the mixed system lose the photocatalytic ability upon further hydrogenation.
Photocatalytic CO2 reductions using BIH as the electron donor
Figure 6 shows the time profiles of the CO production in the photocatalytic CO2 reductions using BIH as the electron donor. In all the reactions using the dyads, only CO was observed as a reduction product of CO2 in both without and with PhOH. Control experiments for the dyad systems showed no detectable CO in the absence of any one of the components, the Re catalyst, light or CO2. A trace amount of CO was observed by using fac‐Re(phen)(CO)3Br in the absence of porphyrin (TONCO = ca. 2 after 24 h). The values of TONCO in the photocatalytic CO2 reduction using the dyads reached values more than an order of magnitude larger than with TEA as the electron donor.
Figure 6.
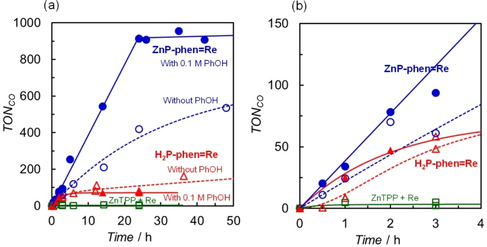
Time dependences of CO formation during irradiation at 420 nm in 2.0 mL of CO2‐saturated DMA solutions containing 0.05 m BIH in the presence of 0.05 mm ZnP‐phen=Re and 0.05 mm H2P‐phen=Re, with 0.1 m PhOH (filled) and without PhOH (open), and a mixed system of 0.05 mm ZnTPP and 0.05 mm fac‐Re(phen)(CO)3Br without PhOH; (a) 0–50 h and (b) 0–4 h. The plots for ZnP‐phen=Re and the mixed system correspond to Figure 3 in ref. [15].
In the absence of PhOH, the TONCO for ZnP‐phen=Re reaches a value of more than 500 (blue open circles), while the mixed system of ZnTPP and fac‐Re(phen)(CO)3Br hardly gave CO (TONCO ≈ 6, green open squares) because ZnTPP in the mixed system was rapidly bleached during irradiation at 420 nm. [15] The CV graphs showed that the two‐electron‐reduced porphyrin species readily caused hydrogenation of the porphyrin skeleton to form chlorin or phlorin,[ 12 , 23 ] whereas the one‐electron‐reduced species of Zn porphyrin was stable in N,N‐dimethylacetamide (Figure 3). Thus, a higher durability in ZnP‐phen=Re would result from suppression of more than two‐electron accumulation on the porphyrin part. As indeed, the photocatalytic CO2 reduction upon irradiation at 560 nm, whose photoinduced electron transfer was slower than that upon irradiation at 420 nm due to the low absorption cross section of Zn porphyrin at 560 nm, gave a linear increase of CO production until all BIH in the system was consumed even in the absence of PhOH (Figure S19).
In our previous report, it was shown that the phosphorescence from the Zn porphyrin in ZnP‐phen=Re is efficiently quenched by BIH under Ar atmosphere, indicating that the photoinduced electron transfer from BIH to the excited porphyrin in ZnP‐phen=Re occurs via the excited triplet state (T1) (Scheme 2). [15] The long‐lived excited state of T1 allows an efficient electron transfer with a high Stern–Volmer constant K SV = 180 000 m −1 in the bimolecular reaction between ZnP‐phen=Re and BIH. The reduction in the excited Zn porphyrin part by BIH is followed by the intramolecular electron transfer from the reduced Zn porphyrin to the Re complex. In the initial stage, the reduction of the Re tricarbonyl Br complex induces dissociation of the Br ligand, an event that starts the catalytic CO2 reduction. [17] The photocatalytic CO2 reduction using ZnP‐phen=Re does not show an induction period for the CO production (Figure 6 b), indicating that an efficient electron transfer to the Re tricarbonyl Br complex occurs. By contrast, the catalysis using H2P‐phen=Re in the absence of PhOH gave an induction period, indicating that intramolecular electron transfer from the reduced free‐base porphyrin to the Re tricarbonyl Br complex is inefficient. The inefficient electron transfer would cause electron accumulation on the porphyrin part, resulting in a low TONCO for H2P‐phen=Re. This is supported by the lower LUMO of the free‐base porphyrin than of the Zn porphyrin (Figure 4) and the energy diagram in which the electron transfer from the one‐electron reduced free‐base porphyrin to the Re part is thermodynamically unfavorable (Figure S20).
Scheme 2.
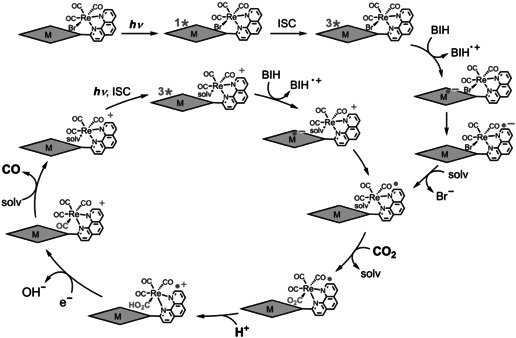
A plausible reaction mechanism in MP‐phen=Re (M=Zn, 2 H).[ 15 , 24 ]
The addition of PhOH, which is a proton source that can promote the reaction with CO2 on the Re complex,[ 24 , 25 ] improves not only the reaction rate, but also the durability of the photocatalytic CO2 reduction in ZnP‐phen=Re (Figure 6 a). The CO production linearly increased to a TONCO value of about 1000, where all BIH was consumed. The addition of PhOH (H+) promotes CO2 conversion into CO on the Re complex, suppressing electron accumulation of ZnP‐phen=Re (Scheme 2). The efficient electron transfer from BIH to the long‐lived T1 of the Zn porphyrin provided a high reaction quantum yield for the CO production reaching 8 % upon both excitations to the S2 (λ ex = 420 nm) and the S1 (λ ex = 560 nm). [15] The reaction quantum yield was almost independent of BIH concentration (>10 mm, Figure S21). While the addition of PhOH did not contribute to improvement in the durability in H2P‐phen=Re, the initial rate of CO production was enhanced (Figure 6 b). The initial rate (<1 h) of H2P‐phen=Re in the presence of PhOH is comparable to that of ZnP‐phen=Re. Considering that the quenching of the excited single state (S1) of free‐base porphyrin of H2P‐phen by BIH was not efficient (η q = 0.69 using [BIH]=0.05 m and K SV = 44 m −1 in Figure S22) and the fluorescence of H2P‐phen=Re was almost completely quenched, the photoinduced electron transfer from BIH to H2P‐phen=Re would also occur via the T1 of the free‐base porphyrin.
As described above, a proton source can enhance the photocatalytic CO2 reduction using ZnP‐phen=Re. The effects of several proton sources (Brønsted acids) having different acidities and of metal cations were examined. [26] Water (pK a(DMSO) = 31.4 in DMSO), methanol (MeOH, pK a(DMSO) = 29.0 in DMSO), 2,2,2‐trifluoroethanol (TFE, pK a(DMSO) = 23.5 in DMSO), PhOH (pK a(DMSO) = 18.0 in DMSO), and acetic acid (AcOH, pK a(DMSO) = 12.3 in DMSO) were selected as the Brønsted acids. [27] Mg(OTf)2 and Ce(OTf)3 were selected as the metal cations. Figure 7 a shows the effects of various acids on the turnover frequencies of CO and H2 up to 2 h. The time dependence of CO formation up to 15 h in the presence of the acids is shown in Figure S23. The TONCO at 15 h is larger for TFE than PhOH, while the linearlity of the CO production is better for PhOH than TFE. No formation of H2 was observed even in the presence of AcOH, which induced H2 production in the electrochemical CO2 reduction using the Re‐bpy complex. [24] The addition of AcOH gives a colorless clear solution rapidly (2 h), indicating that the formation of the Re hydride complex induces the hydrogenation of the porphyrin skeleton instead of hydrogen production. MeOH and water show slight increases in CO production, whereas TFE and PhOH significantly enhanced the catalysis. In contrast, addition of the metal cations suppressed the CO formation. In particular, a trivalent cation, Ce3+, completely deactivated the photocatalysis to produce no reduction products. It is speculated that the adduct of Ce3+ with the Re−CO2 species is too stable, suppressing the formation of the Re−CO species. The plots of turnover frequencies for the CO production against pK a(DMSO) are shown in Figure 7 b, indicating that the proper acidity is important for increasing the CO production in the photocatalytic CO2 reduction. The trend of the initial reaction rates (TFE > PhOH > H2O ≈ MeOH) is in agreement with the trend of the simulated reaction rates in the electrochemical CO2 reduction using the Re–bpy complex reported by Carter et al. (Figure 4 in ref. [28]).
Figure 7.
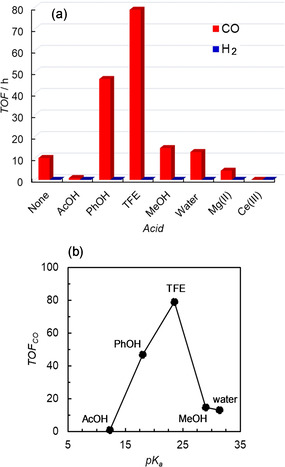
(a) Turnover frequencies (TOFs) of CO and H2 during the irradiation at 420 nm up to 2 h in CO2‐saturated DMA solutions containing 0.05 m BIH and 0.05 mm ZnP‐phen=Re in the presence of acids (0.1 m). (b) pK a‐dependence of the TOF of CO of the photocatalytic CO2 reduction in the presence of Brønsted acids. The pK a used were the values in DMSO. [27]
Ishitani and co‐workers reported that TEOA plays an important role not only in promoting the deprotonation of the oxidized BIH to suppress the back‐electron transfer from the one‐electron reduced photosensitizer to the oxidized BIH, but also in trapping CO2 by forming the Re(bpy)(CO)3(CO2‐TEOA) species.[ 17 , 29 ] The effects of amines (TEOA and TEA) on the photocatalytic CO2 reduction system were examined here using ZnP‐phen=Re and BIH (Figure S24). The time‐course dependences for CO formation did not show improvement in the catalysis by the addition of amine, suggesting that neither the deprotonation of the oxidized BIH nor the CO2 trapping would be the rate‐determining step in the reaction conditions.
Conclusions
We have previously reported that ZnP‐phen=Re acts as a good photocatalyst in the photocatalytic CO2 reduction using BIH as the electron donor. In this work, we demonstrated that ZnP‐phen=Re also acted as a good photocatalyst even when TEA, a weaker electron donor, was used. The photocatalytic CO2 reaction selectively gave CO as the product whose TONCO reaches at least 25 and still continued CO production after irradiation for 60 h. To investigate the effect of the central Zn ion of porphyrin in the catalysis, a dyad composed of free‐base porphyrin and fac‐Re(phen)(CO)3Br, H2P‐phen=Re, was synthesized. The durability of H2P‐phen=Re was lower than that of ZnP‐phen=Re, reflecting ease of decomposition of the porphyrin skeleton by hydrogenation of the C=C bonds. The ease of decomposition is explained by the energy diagram (Figure S20), in which the electron‐transfer process from the one‐electron reduced porphyrin to the Re part is thermodynamically unfavorable for H2P‐phen=Re. Thus, the relationship of the redox potentials between the porphyrin and Re parts is important for developing a highly durable catalyst. The effect of acid on the reaction using ZnP‐phen=Re and BIH was investigated. PhOH and TFE enhanced the CO, but the stronger AcOH acid and a trivalent metal cation almost completely deactivated the catalysis.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was supported by ENEOS Hydrogen Trust Fund. We thank Prof. Akihiko Kudo (Tokyo University of Science) for help with capillary electrophoresis analyses.
Y. Kuramochi, A. Satake, Chem. Eur. J. 2020, 26, 16365.
Contributor Information
Dr. Yusuke Kuramochi, Email: kuramochiy@rs.tus.ac.jp.
Prof. Akiharu Satake, Email: asatake@rs.tus.ac.jp.
References
- 1.
- 1a. Inoue H., Shimada T., Kou Y., Nabetani Y., Masui D., Takagi S., Tachibana H., ChemSusChem 2011, 4, 173–179; [DOI] [PubMed] [Google Scholar]
- 1b. Alstrum-Acevedo J.-H., Brennaman M. K., Meyer T. J., Inorg. Chem. 2005, 44, 6802–6827. [DOI] [PubMed] [Google Scholar]
- 2. Blankenship R. E., Molecular Mechanisms of Photosynthesis, Blackwell Science, Oxford, 2002. [Google Scholar]
- 3. Otsuki J., J. Mater. Chem. A 2018, 6, 6710–6753. [Google Scholar]
- 4.
- 4a. Kuramochi Y., Hashimoto S., Kawakami Y., Asano M. S., Satake A., Photochem. Photobiol. Sci. 2018, 17, 883–888; [DOI] [PubMed] [Google Scholar]
- 4b. Kuramochi Y., Kawakami Y., Satake A., Inorg. Chem. 2017, 56, 11008–11018; [DOI] [PubMed] [Google Scholar]
- 4c. Kuramochi Y., Satake A., Sandanayaka A. S. D., Araki Y., Ito O., Kobuke Y., Inorg. Chem. 2011, 50, 10249–10258; [DOI] [PubMed] [Google Scholar]
- 4d. Satake A., Azuma S., Kuramochi Y., Hirota S., Kobuke Y., Chem. Eur. J. 2011, 17, 855–865; [DOI] [PubMed] [Google Scholar]
- 4e. Kuramochi Y., Sandanayaka A. S. D., Satake A., Araki Y., Ogawa K., Ito O., Kobuke Y., Chem. Eur. J. 2009, 15, 2317–2327; [DOI] [PubMed] [Google Scholar]
- 4f. Nagata N., Kuramochi Y., Kobuke Y., J. Am. Chem. Soc. 2009, 131, 10–11; [DOI] [PubMed] [Google Scholar]
- 4g. Kuramochi Y., Satake A., Itou M., Ogawa K., Araki Y., Ito O., Kobuke Y., Chem. Eur. J. 2008, 14, 2827–2841; [DOI] [PubMed] [Google Scholar]
- 4h. Kuramochi Y., Satake A., Kobuke Y., J. Am. Chem. Soc. 2004, 126, 8668–8669. [DOI] [PubMed] [Google Scholar]
- 5. Rybicka-Jasińska K., Shan W., Zawada K., Kadish K. M., Gryko D., J. Am. Chem. Soc. 2016, 138, 15451–15458. [DOI] [PubMed] [Google Scholar]
- 6. Shanmugam S., Xu J., Boyer C., J. Am. Chem. Soc. 2015, 137, 9174–9185. [DOI] [PubMed] [Google Scholar]
- 7. Lazarides T., Sazanovich I. V., Simaan A. J., Kafentzi M. C., Delor M., Mekmouche Y., Faure B., Reglier M., Weinstein J. A., Coutsolelos A. G., Tron T., J. Am. Chem. Soc. 2013, 135, 3095–3103. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Zhang W., Lai W., Cao R., Chem. Rev. 2017, 117, 3717–3797; [DOI] [PubMed] [Google Scholar]
- 8b. Ladomenoua K., Natalib M., Iengoc E., Charalampidisa G., Scandolab F., Coutsolelos A. G., Coord. Chem. Rev. 2015, 304–305, 38–54. [Google Scholar]
- 9. Kim J. H., Lee S. H., Lee J. S., Lee M., Park C. B., Chem. Commun. 2011, 47, 10227–10229. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Montalti M., Credi A., Prodi L., Gandolfi M. T., Handbook of Photochemistry , 3rd ed., Taylor & Francis, Boca Raton, 2006; [Google Scholar]
- 10b. Quimby D. J., Longo F. R., J. Am. Chem. Soc. 1975, 97, 5111–5117. [Google Scholar]
- 11. Kiyosawa K., Shiraishi N., Shimada T., Masui D., Tachibana H., Takagi S., Ishitani O., Tryk D. A., Inoue H., J. Phys. Chem. C 2009, 113, 11667–11673. [Google Scholar]
- 12.
- 12a. Windle C. D., George M. W., Perutz R. N., Summers P. A., Sun X. Z., Whitwood A. C., Chem. Sci. 2015, 6, 6847–6864; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12b. Windle C. D., Campian M. V., Duhme-Klair A.-K., Gibson E. A., Perutz R. N., Schneider J., Chem. Commun. 2012, 48, 8189–8191. [DOI] [PubMed] [Google Scholar]
- 13.
- 13a. Matlachowski C., Braun B., Tschierlei S., Schwalbe M., Inorg. Chem. 2015, 54, 10351–10360; [DOI] [PubMed] [Google Scholar]
- 13b. Lang P., Pfrunder M., Quach G., Braun-Cula B., Moore E. G., Schwalbe M., Chem. Eur. J. 2019, 25, 4509–4519. [DOI] [PubMed] [Google Scholar]
- 14. Kitagawa Y., Takeda H., Ohashi K., Asatani T., Kosumi D., Hashimoto H., Ishitani O., Tamiaki H., Chem. Lett. 2014, 43, 1383–1385. [Google Scholar]
- 15. Kuramochi Y., Fujisawa Y., Satake A., J. Am. Chem. Soc. 2020, 142, 705–709. [DOI] [PubMed] [Google Scholar]
- 16.
- 16a. Cheung P. L., Kapper S. C., Zeng T., Thompson M. E., Kubiak C. P., J. Am. Chem. Soc. 2019, 141, 14961–14965; [DOI] [PubMed] [Google Scholar]
- 16b. Takeda H., Kamiyama H., Okamoto K., Irimajiri M., Mizutani T., Koike K., Sekine A., Ishitani O., J. Am. Chem. Soc. 2018, 140, 17241–17254; [DOI] [PubMed] [Google Scholar]
- 16c. Lee S. K., Kondo M., Okamura M., Enomoto T., Nakamura G., Masaoka S., J. Am. Chem. Soc. 2018, 140, 16899–16903; [DOI] [PubMed] [Google Scholar]
- 16d. Hong D., Tsukakoshi Y., Kotani H., Ishizuka T., Kojima T., J. Am. Chem. Soc. 2017, 139, 6538–6541; [DOI] [PubMed] [Google Scholar]
- 16e. Guo Z., Cheng S., Cometto C., Anxolabehére-Mallart E., Ng S. M., Ko C. C., Liu G., Chen L., Robert M., Lau T. C., J. Am. Chem. Soc. 2016, 138, 9413–9416; [DOI] [PubMed] [Google Scholar]
- 16f. Tamaki Y., Koike K., Morimoto T., Ishitani O., J. Catal. 2013, 304, 22–28. [Google Scholar]
- 17. Kuramochi Y., Ishitani O., Ishida H., Coord. Chem. Rev. 2018, 373, 333–356. [Google Scholar]
- 18. Freitag R. A., Whitten D. G., J. Phys. Chem. 1983, 87, 3918–3925. [Google Scholar]
- 19.In Figure S7b, the quintet signal was observed at 3.7 ppm. This is expected to be due to the coupling interaction between the CH2 and OH protons: Gottlieb H. E., Kotlyar V., Nudelman A., J. Org. Chem. 1997, 62, 7512–7515. [DOI] [PubMed] [Google Scholar]
- 20.
- 20a. Kuramochi Y., Ishitani O., Inorg. Chem. 2016, 55, 5702–5709; [DOI] [PubMed] [Google Scholar]
- 20b. Kuramochi Y., Kamiya M., Ishida H., Inorg. Chem. 2014, 53, 3326–3332. [DOI] [PubMed] [Google Scholar]
- 21.
- 21a. Lipstman S., Muniappan S., Goldberg I., Acta Crystallogr. Sect. E 2006, 62, m2330–m2332; [DOI] [PubMed] [Google Scholar]
- 21b. Nappa M., Valentine J. S., J. Am. Chem. Soc. 1978, 100, 5075–5080. [Google Scholar]
- 22. Kuramochi Y., Ishitani O., Front. Chem. 2019, 7, 259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.
- 23a. Balducci G., Chottard G., Gueutin C., Lexa D., Savéant J.-M., Inorg. Chem. 1994, 33, 1972–1978; [Google Scholar]
- 23b. Lanese J. G., Wilson G. S., J. Electrochem. Soc. 1972, 119, 1039–1043. [Google Scholar]
- 24. Kou Y., Nabetani Y., Masui D., Shimada T., Takagi S., Tachibana H., Inoue H., J. Am. Chem. Soc. 2014, 136, 6021–6030. [DOI] [PubMed] [Google Scholar]
- 25.
- 25a. Clark M. L., Cheung P. L., Lessio M., Carter E. A., Kubiak C. P., ACS Catal. 2018, 8, 2021–2029; [Google Scholar]
- 25b. Wong K. Y., Chung W. H., Lau C. P., J. Electroanal. Chem. 1998, 453, 161–169. [Google Scholar]
- 26.
- 26a. Hong D., Kawanishi T., Tsukakoshi Y., Kotani H., Ishizuka T., Kojima T., J. Am. Chem. Soc. 2019, 141, 20309–20317; [DOI] [PubMed] [Google Scholar]
- 26b. Sampson M. D., Kubiak C. P., J. Am. Chem. Soc. 2016, 138, 1386–1393; [DOI] [PubMed] [Google Scholar]
- 26c. Hammouche M., Lexa D., Momenteau M., Savéant J.-M., J. Am. Chem. Soc. 1991, 113, 8455–8466. [Google Scholar]
- 27.
- 27a. Bordwell F. G., Acc. Chem. Res. 1988, 21, 456–463; [Google Scholar]
- 27b. Ngo K. T., McKinnon M., Mahanti B., Narayanan R., Grills D. C., Ertem M. Z., Rochford J., J. Am. Chem. Soc. 2017, 139, 2604–2618. [DOI] [PubMed] [Google Scholar]
- 28. Riplinger C., Carter E. A., ACS Catal. 2015, 5, 900–908. [Google Scholar]
- 29.
- 29a. Nakajima T., Tamaki Y., Ueno K., Kato E., Nishikawa T., Ohkubo K., Yamazaki Y., Morimoto T., Ishitani O., J. Am. Chem. Soc. 2016, 138, 13818–13821; [DOI] [PubMed] [Google Scholar]
- 29b. Morimoto T., Nakajima T., Sawa S., Nakanishi R., Imori D., Ishitani O., J. Am. Chem. Soc. 2013, 135, 16825–16828. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary