

Abstract
Mild reaction conditions are highly desirable for bio‐orthogonal side chain derivatizations of amino acids, peptides or proteins due to the sensitivity of these substrates. Transition metal catalysed cross‐couplings such as Suzuki–Miyaura reactions are highly versatile, but usually require unfavourable reaction conditions, in particular, when applied with aryl bromides. Ligand‐free solvent‐stabilised Pd‐nanoparticles represent an efficient and sustainable alternative to conventional phosphine‐based catalysts, because the cross‐coupling can be performed at considerably lower temperature. We report on the application of such a highly reactive heterogeneous catalyst for the Suzuki–Miyaura cross‐coupling of brominated tryptophan derivatives. The solvent‐stabilised Pd‐nanoparticles are even more efficient than the literature‐known ADHP‐Pd precatalyst. Interestingly, the latter also leads to the formation of quasi‐homogeneous Pd‐nanoparticles as the catalytic species. One advantage of our approach is the compatibility with aqueous and aerobic conditions at near‐ambient temperatures and short reaction times of only 2 h. The influence of different N α‐protecting groups, boronic acids as well as the impact of different amino acid side chains in bromotryptophan‐containing peptides has been studied. Notably, a surprising acceleration of the catalysis was observed when palladium‐coordinating side chains were present in proximal positions.
Keywords: bio-orthogonality, halotryptophan, heterogeneous catalysis, oxygen-promoted cross-coupling, Pd nanoparticles
Gentle conditions: Ligand‐free solvent‐stabilised Pd‐nanoparticles serve as a highly efficient catalyst at ambient temperature for a sustainable and biocompatible Suzuki–Miyaura cross‐coupling between aryl bromides and boronic acids under mild, aerobic and aqueous conditions.
Introduction
The Suzuki–Miyaura cross‐coupling is a versatile tool for the selective formation of carbon‐carbon bonds. [1] The stability of the used organoboron compounds under aqueous conditions and their high tolerance towards different functional groups [2] makes the Suzuki–Miyaura cross‐coupling a suitable reaction for bio‐orthogonal late‐stage derivatization of halogenated natural substrates. [3] Latest investigations have demonstrated the applicability of the Suzuki–Miyaura reaction for functionalisation of halogenated or boronated amino acids, peptides and proteins. [4] The modifications of complex natural molecules such as proteins are particularly challenging because gentle reaction conditions (mild temperatures, pH 6–8, aerobic and aqueous conditions) are mandatory to avoid side reactions and degradation. [5] Chalker et al. developed an approach to arylate p‐iodophenylalanine in proteins under biocompatible conditions using a Pd‐pyrimidine precatalyst system. [6] However, aryl iodides are prone to oxidation, and the use of different halogenated aromatic compounds such as bromoaryls is recommended despite their inferior reactivity. Hence, bromotryptophan is an interesting starting material for Pd‐mediated functionalisation as it is readily available on a gram scale by enzymatic halogenation via FAD‐dependent halogenases [7] or by using Trp synthase. [8] Due to their electron‐rich aromatic system, halotryptophans are more challenging substrates for Pd‐mediated cross‐couplings than halophenylalanines. A Suzuki–Miyaura reaction of unprotected halotryptophans in water was first described by Deb Roy et al. who employed an air‐sensitive Pd‐catalyst, namely trisodium 3,3′,3′′‐phosphinetriyltribenzenesulfonate (TPPTS). The use of a water‐soluble phosphine ligand enabled the arylation of 5‐bromotryptophan in fair yields at 40 °C, while other substrates like 7‐bromotryptophan required higher reaction temperatures. [9] The functionalisation of halophenylalanine‐ and halotryptophan‐containing dipeptides in a Suzuki–Miyaura reaction was demonstrated by Willemse et al., who investigated different catalytic systems to assess the compatibility between catalysts and amino acid side chain functionalities. They were able to arylate the base‐sensitive Fmoc‐protected iodophenylalanine at 40 °C without deprotection, albeit with only 80 % conversion after 24 h. [10] Frese et al. and the group of Micklefield independently combined enzymatic halogenation and cross‐coupling in a multi‐step one‐pot reaction without isolation of the halogenated species.[ 11 , 12 ] Based on the investigations of the fluorescence properties of arylated tryptophans by Deb Roy et al., [9] this approach was used by Schnepel et al. to design a high‐throughput fluorescence assay for screening halogenase activity in directed evolution. [13] Gruß et al. utilised a Pd‐mediated Mizoroki–Heck cross‐coupling of unprotected bromotryptophan and styrene derivatives to further improve the fluorogenic properties by red‐shifted excitation wavelength. [14] Kemker et al. were able to use the Suzuki–Miyaura cross‐coupling for fluorescence labelling or side chain‐to‐side chain cyclisation of bromotryptophan containing RGD peptides, thereby increasing stability and selectivity. [15] The Goss group reported on in vivo Suzuki–Miyaura cross‐coupling of bromotryptophan and bromopacidamycin at 37 °C using (2‐dimethylamino)‐4,6‐dihydroxypyrimidine (DMADHP) and trimethyl guanidine (TMG) Pd‐precatalysts which had first been described by the Davis group.[ 6 , 16 , 17 ] They also investigated the limitations of the cross‐coupling at low temperatures due to coordination of the α‐amino group of unprotected bromotryptophan to palladium. Dumas et al. reported poly(d,l‐lactide‐glycolide)‐block‐poly(ethylene glycol) copolymer Pd‐nanoparticles (PLGA‐PEG‐Pd‐NPs) as a catalyst for the Suzuki–Miyaura reaction of N α‐Boc‐protected halophenylalanine under mild conditions in water. With these nanoparticle assemblies in hand, they were able to achieve 98 % conversion at 37 °C after 18 h. [18]
Results and Discussion
We embarked on the search for a more reactive, air‐stable catalyst system to extend the biocompatibility of the Suzuki–Miyaura reaction. As Pd‐nanoparticle catalysed Suzuki–Miyaura reactions are promoted by oxygen, [19] ligand‐free solvent‐stabilised Pd‐nanoparticles described by Kurscheid et al. [20] seemed to be promising candidates for our studies. Here, we report on compatibility studies of the ligand‐free Pd‐nanoparticle catalysed Suzuki–Miyaura reactions and an array of N α‐protected 7‐bromotryptophans or 7‐bromotryptophan containing pentapeptides under mild, aerobic, and aqueous conditions. Our goal was to test a peptide library containing different side chain moieties to make possible predictions for the suitability of Pd‐nanoparticles as a bio‐orthogonal catalyst system.
Synthesis and characterisation of solvent‐stabilised Pd‐nanoparticles
Ligand‐free solvent‐stabilised Pd‐nanoparticles were prepared according to the protocol by Kurscheid et al. [20] Stirring a palladium dichloride suspension in 2‐propanol at room temperature for 16 days gave a red suspension of solvent‐stabilised nanoparticles. We performed the nanoparticle synthesis either under aerobic or inert conditions, while no major difference in catalytic activity could be observed. The Pd‐nanoparticles were characterized by transmission electron microscopy (TEM). The TEM image (Figure 1 a) shows the result of the nanoparticle synthesis. In fact, the formation of bigger clusters of smaller nanoparticles can be observed, but their initial shape is still identifiable. Clustering might occur as a side effect of the sample preparation during the TEM imaging. The amount of dissolved palladium was quantified by atomic absorption spectroscopy (AAS) showing that 65 % of the Pd was in solution. This is in accordance with the results of Kurscheid et al. The Pd‐nanoparticle stock solution was stored in a 2 mL microcentrifuge tube at 6–8 °C without any effort to exclude oxygen. The particles retained catalytic activity even after storing for 15 months.
Figure 1.

(a) Transmission electron microscopy (TEM) image of suspended solvent‐stabilised Pd‐nanoparticles; (b) Fivefold magnification.
The Davis catalyst consists of Pd‐nanoparticles
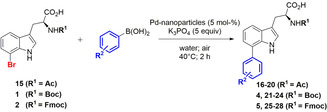
Since biocompatible catalysts for Suzuki–Miyaura cross‐couplings, such as 2‐amino‐4,6‐dihydroxypyrimidine Pd‐precatalysts (ADHP‐Pd), have already been reported by the Davis group, [13] we started our catalytic studies by comparing the ligand‐free Pd‐nanoparticles with one of those (Scheme 1). The same reaction conditions were used as reported by the Davis group. Due to the inhibition of the ADHP‐Pd by the free amino group of tryptophan, [16] we decided to use N α‐Boc‐ (1) or Fmoc‐ (2) protected l‐7‐bromotryptophan as benchmark system. We investigated the influence of inert conditions on the reactivity of the reference catalyst system compared to ligand‐free Pd‐nanoparticles (Figure 2 a).
Scheme 1.
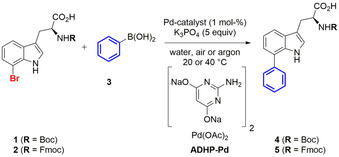
Suzuki–Miyaura reaction of N α‐protected l‐7‐bromotryptophan with both biocompatible ADHP‐Pd and solvent‐stabilised Pd‐nanoparticles.
Figure 2.
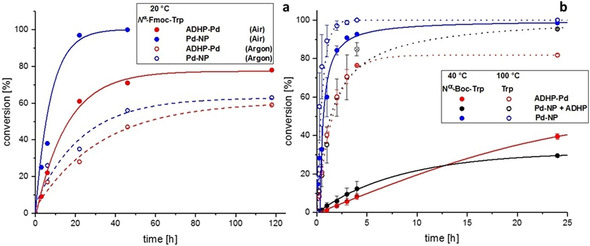
Conversion as function of time in the Suzuki–Miyaura reaction (a) of N α‐Fmoc‐l‐7‐bromotryptophan (20 °C) and phenylboronic acid catalysed by ADHP‐Pd (red) or Pd nanoparticles (Pd‐NP) (blue) under aerobic or inert conditions and (b) of N α‐Boc‐l‐7‐bromotryptophan (40 °C) or unprotected l‐7‐bromotryptophan (100 °C) and phenylboronic acid catalysed by ADHP‐Pd (red), Pd‐NP (blue) or Pd‐NP+ADHP (grey).
l‐7‐Bromotryptophan was generated applying RebH‐containing combined cross‐linked‐enzyme‐aggregates (combiCLEAs) followed by Fmoc‐ or Boc‐protection. [11] The Pd‐nanoparticles show a slightly higher catalytic activity than the ADHP‐Pd (Figure 2). We expected the reactivity of the Pd‐nanoparticles under inert conditions to be decreased compared to oxygen‐promoted catalysis, [19] while the ADHP‐Pd precatalyst system should remain unaffected or even show an increase in activity. Notably, both catalysts were affected in their catalytic activity to a similar extent. Under inert conditions, both catalysts showed nearly the same (lower) catalytic activity compared to aerobic conditions. This phenomenon is usually only observed for nanoparticle‐catalysed reactions (Figure 2 a). Heating to 80 °C led to cleavage of the Fmoc‐protecting group regardless of the catalyst used. At ambient temperatures Pd gets coordinated by the free amino group of unprotected tryptophan. [16]
At higher temperatures the effect of this coordination obviously becomes negligible. Therefore, studies at increased temperature (100 °C) were performed using unprotected l‐7‐bromotryptophan (Figure 2 b). The almost identical response of both catalysts to alterations of the reaction conditions indicates that both catalytic systems might involve a similar reactive species.
We therefore hypothesized that the ADHP‐Pd precatalyst forms ligand‐stabilised Pd‐nanoparticles, too. Hence, during the Suzuki–Miyaura reaction the guanidyl group of the ADHP‐ligand inhibits the catalytically active Pd species due to steric demands of the ligand, by blocking binding sites of the Pd (Figure 4 b), as already reported for the amino group of unprotected tryptophan. [16]
Figure 4.
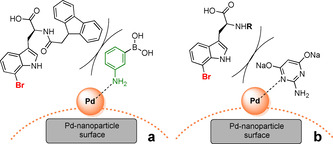
Possible steric hindrance of Pd‐nanoparticles by (a) 3‐aminophenylboronic acid or (b) guanidyl‐ligand ADHP.
Therefore, ligand‐free solvent‐stabilised nanoparticles in 2‐propanol catalyse the reaction without steric hindrance by ligands. This makes these particles much more reactive than the ADHP‐Pd. The ligand ADHP was added to pre‐formed Pd‐nanoparticles to investigate its influence on the performance of the ligand‐free Pd‐nanoparticles. After addition of ADHP, the reactivity of the Pd‐nanoparticles decreased drastically. The conversions of the ADHP‐Pd precatalyst and the Pd‐nanoparticles with ADHP were close to equal during the first 5 h of the reaction.
However, the conversions start to diverge at different reaction temperatures. At 100 °C the reaction catalysed by ADHP‐Pd stagnated at almost 80 % conversion and formation of palladium black was observed, while the Pd‐nanoparticles with added ADHP maintained catalytic activity (Figure 2 b, dotted red and grey curve). We suggest that the presence of 2‐propanol, in which the nanoparticles were suspended, stabilises the nanoparticles and prevents the agglomeration to Pd‐black even at elevated temperatures. [21]
Additionally, 2‐propanol reduces PdII species to Pd0, [22] resulting in a continuous regeneration of catalytically active Pd‐nanoparticles. On the other hand, the ADHP‐Pd kept their catalytic activity at 40 °C after 24 h, whereas conversion in case of the Pd‐nanoparticles with added ADHP stagnated at only 30 % (Figure 2 b red and black curve). This can be explained by different size distributions of the nanoparticles. TEM images of the Davis catalyst (ADHP‐Pd) show clearly defined nanoparticles (Figure 3). This provides final evidence that the ADHP‐Pd precatalyst does not only consist of a single molecular species but also of a heterogeneous, so called quasi‐homogeneous catalytic species.
Figure 3.

Transmission electron microscopy (TEM) image of the Davis catalyst (ADHP‐Pd).
Optimization of reaction conditions
The reaction conditions for the nanoparticle catalysed Suzuki–Miyaura reaction were optimized using commercially available 5‐bromoindole and phenylboronic acid as a benchmark system. Best results were achieved in water, using 5 equiv of boronic acid and potassium phosphate each and a catalyst loading of 5 mol % (Scheme 2). This led to a final 2‐propanol concentration of 0.5 %, since the added catalyst was dissolved in 2‐propanol.
Scheme 2.
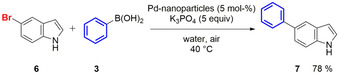
Suzuki–Miyaura cross‐coupling of 5‐bromoindole (6) and phenylboronic acid (3) catalysed by ligand‐free solvent‐stabilised Pd‐nanoparticles (Pd‐NP).
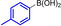
The optimized reaction conditions were then used for the arylation of unprotected bromotryptophan: A solution of l‐7‐bromotryptophan (8), boronic acid and K3PO4 in water (pH 8–9) was warmed to 40 °C. Once all compounds had been dissolved, the catalyst was added. Noteworthy, no effort was taken to exclude oxygen from the reaction‐vessel. The reaction progress was monitored by reversed‐phase high‐performance liquid chromatography (RP‐HPLC) at 220 nm. Using unprotected l‐7‐bromotryptophan (8) at 40 °C in comparison to N α‐protected tryptophan, inhibition of the catalyst was observed and full conversion could not be achieved (Table 1).
Table 1.
Pd‐nanoparticle catalysed Suzuki–Miyaura reaction of unprotected l‐7‐bromotryptophan (8) with different boronic acids under aqueous and aerobic conditions.
|
| ||||
|---|---|---|---|---|
|
Entry |
Boronic acid |
Conversion [%][a] |
Yield [%][b] |
|
|
|
|
40 °C (120 h) |
80 °C (2 h) |
|
|
1 |
|
52 |
55 |
65 |
|
2 |
|
68 |
59 |
69 |
|
3 |
|
78 |
72 |
42 |
|
4 |
|
56 |
95 |
36 |
|
5 |
|
22 |
42 |
41 |
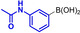
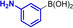
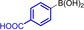
[a] Determined by HPLC (220 nm). [b] Isolated yields after full conversion at 80 °C and purification.
These results are in accordance with the observations of ADHP‐Pd inhibition by unprotected bromotryptophan as reported by the Goss group. [16] Heating to 80 °C increased the conversion, likely due to the weaker coordination of Pd by the free amine at higher temperatures. The reaction was monitored by RP‐HPLC and the products were purified by preparative RP‐HPLC once the reaction had reached completion at 80 °C. Adequate yields of all aryltryptophans were obtained (Table 1). As expected, best conversions at 40 °C were observed for non‐nucleophilic and electron‐rich boronic acids, for example, para‐tolylboronic acid and 3‐acetamidophenylboronic acid, giving 68 % and 78 % conversion after 5 days (Table 1, entries 2 and 3). At higher temperatures the electron‐richer 3‐aminophenylboronic acid gave the best conversion of 95 % after 2 h at 80 °C (entry 4). Remarkably, no precipitating Pd (Pd‐black) was observed in presence of unprotected tryptophan, due to the coordination of Pd by amines. Using the electron‐rich boronic acids 3‐acetamidophenylboronic acid and 3‐aminophenylboronic acid, homocoupling was observed independently of the atmosphere used.
Screening of N α‐derivatized bromotryptophans
The investigation of different N α‐protection groups is of interest beyond bio‐orthogonal reactions which do not require protecting groups, because it broadens the range of available orthogonal protection strategies.
For example, applying the Suzuki–Miyaura cross‐coupling of halogenated amino acids under mild conditions in solid‐phase peptide synthesis (SPPS). In particular, the base sensitive protecting group Fmoc is of great relevance. In addition, the reactivities of N α‐Boc‐ and N α‐acetyl protected l‐7‐bromotryptophan and various functionalised boronic acids were evaluated. Suzuki–Miyaura reactions of N α‐protected bromotryptophans gave promising results at 40 °C and no further heating was necessary to reach full conversion (Table 2). A black precipitate (Pd‐black) was observed almost instantly when the Pd‐nanoparticles were added to the reaction. Exceptions occurred using 3‐aminophenylboronic acid or 4‐carboxyphenylboronic acid as cross‐coupling partners, where no precipitate was observed. Since amines and carboxylic acids coordinate to the surface of the Pd‐nanoparticles, agglomeration of the particles is hindered. The solutions turned slightly yellow without any precipitate. N α‐Acetyl‐l‐7‐bromotryptophan (15) gave best conversions with para‐tolylboronic acid (99 %) and 3‐aminophenylboronic acid (97 %) even after 2 h (Table 2, entries 4 and 10).
Table 2.
Pd‐nanoparticle catalysed Suzuki–Miyaura reaction of N α‐protected l‐7‐bromotryptophans with different boronic acids under aqueous and aerobic conditions.
|
| |||||
|---|---|---|---|---|---|
|
Entry |
R1 |
Boronic acid |
# |
Conv. [%][a] |
Yield [%][b] |
|
1 |
Ac |
|
16 |
82 |
67 |
|
2 |
Boc |
4 |
97 |
51 |
|
|
3 |
Fmoc |
5 |
83 |
36 |
|
|
4 |
Ac |
|
17 |
>99 |
63 |
|
5 |
Boc |
21 |
>99 |
60 |
|
|
6 |
Fmoc |
25 |
>99 |
64 |
|
|
7 |
Ac |
|
18 |
80 |
74 |
|
8 |
Boc |
22 |
>99 |
83 |
|
|
9 |
Fmoc |
26 |
>99 |
97 |
|
|
10 |
Ac |
|
19 |
97 |
73 |
|
11 |
Boc |
23 |
>99 |
82 |
|
|
12 |
Fmoc |
27 |
10[c] |
n.d.[d] |
|
|
13 |
Ac |
|
20 |
72 |
40 |
|
14 |
Boc |
24 |
86 |
75 |
|
|
15 |
Fmoc |
28 |
62 |
55 |
|
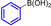
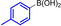
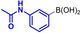

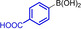
[a] Determined by HPLC (220 nm). [b] Isolated yields after full conversion and purification. [c] Did not reach full conversion. [d] Not determined.
N α‐Boc‐protected bromotryptophan (1) gave the best results among all N α‐protected bromotryptophans, reaching almost full conversion combined with every tested boronic acid in less than 2 h (Table 2). Even with the electron‐poor 4‐carboxyphenylboronic acid 86 % conversion was attained after 2 h (entry 14). Interestingly, catalytic inhibition due to coordination of Pd by 3‐aminophenylboronic acid seems to be negligible in case of N α‐acetyl‐ and Boc‐protected bromotryptophans, since both reactions showed good conversions (Table 2, entry 10 and 11). Since nanoparticles need to be centrifuged off before samples can be purified by preparative RP‐HPLC, hydrophobic precipitated products and those coordinating to palladium might get lost, leading to reduced yields. Under the mild conditions of this nanoparticle catalysis the Fmoc‐protecting group is stable and Fmoc‐protected arylated tryptophans were obtained in good yields (Table 2). Surprisingly, N α‐Fmoc‐l‐7‐bromotryptophan (2) and 3‐aminophenylboronic acid were converted to only 10 % after 2 h at 40 °C (entry 12) while 3‐acetamidophenyl boronic acid showed excellent conversion >99 % (entry 9). Repetition of the experiment led to the same result. This may be assigned to the steric influence of ligands coordinating the Pd‐nanoparticles. To reduce the coordination of the Pd by amines the reaction temperature was elevated to 60 °C, just leading to a fast cleavage of the Fmoc‐group. Hence, 3‐aminophenylboronic acid does not seem to have enough steric hindrance to inhibit the cross couplings with N α‐acetyl‐ or Boc‐ protected tryptophan (entries 10 and 11), while the sterically more demanding Fmoc‐protecting group exerts strong hindrance (entry 12) (Figure 4 a).
Influence of different amino acid side chains in bromotryptophan containing pentapeptides
The Suzuki–Miyaura cross‐coupling in water at 80 °C with different 5‐bromotryptophan containing dipeptides has been reported by Willemse et al. Apart from bromotryptophan, aliphatic and aromatic side chains had been introduced into the peptides.[ 4 , 10 ] An array of l‐7‐bromotryptophan containing pentapeptides were designed to investigate the compatibility of ligand‐free Pd‐nanoparticles with complex substrates such as peptides. The influence of all major functional groups present in a protein should be evaluated. Since the nanoparticles are stored in 2‐propanol and the compatibility of carboxylic acids with the nanoparticles had already been verified (Tables 1 and 2), a sequence containing serine and aspartic acid was designed to ensure sufficient water solubility of the peptides (Table 3). Presence of aliphatic or aromatic side chains did not considerably interfere with the reaction and the cross‐coupled peptides were obtained in good isolated yields after purification by RP‐HPLC (Table 3; entries 1 and 9–11). Willemse et al. had already described the problems of a Suzuki–Miyaura reaction in the presence of primary amide (asparagine) and imidazole (histidine). [10] For imidazoles, the formation of N‐azolyl palladium complexes were postulated by Düfert et al. [23] This might lead to poisoning of the Pd‐nanoparticles by histidine in the pentapeptide sequence resulting in only traces of cross‐coupling product after 2 h at 40 °C (entry 5). Using Pd‐nanoparticles with the Gln containing pentapeptide (29Gln) gave 14 % conversion after 2 h at 40 °C. Increasing the temperature to 80 °C improved conversion (HPLC) to 72 % after 2 h. The 1H NMR of the isolated arylated peptide at high temperatures shows three different species in a ratio of 7:1:1, which could be identified by LC‐MS as the arylated Gln‐pentapeptide (30Gln), the arylated Glu‐pentapeptide (30Glu) and a cyclised species showing a mass shift −17 Da (nominal mass), which could be the pyroglutamate or glutarimide side product (Figure 5).
Table 3.
Pd‐nanoparticle catalysed Suzuki–Miyaura reaction of l‐7‐bromotryptophan‐containing pentapeptides to investigate the influence of different amino acid side chains.
|
| ||||
|---|---|---|---|---|
|
Entry |
Xaa |
Conversion [%][a] |
Yield [%][b] |
|
|
|
|
40 °C (2 h) |
80 °C (2 h) |
|
|
1 |
Ala |
47 |
93 |
50 |
|
2 |
Arg |
88 |
>99 |
86 |
|
3 |
Cys |
n.d. |
0 |
0 |
|
4 |
Gln |
14[c] |
72 |
39[d,f] |
|
5 |
His |
traces[c] |
20[c] |
n.d. |
|
6 |
Lys |
86 |
>99 |
53 |
|
7 |
Lys(Boc) |
54 |
n.d. |
70 |
|
8 |
Met |
>99[e] |
n.d. |
65[e] |
|
9 |
Phe |
56 |
>99 |
77 |
|
10 |
Pro |
61 |
>99 |
65 |
|
11 |
Tyr |
58 |
>99 |
73 |
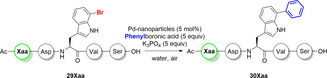
[a] Determined by HPLC (220 nm). [b] Isolated yields after full conversion at 40 °C and purification. [c] Did not reach full conversion. [d] Corrected according to 1H NMR. [e] Full conversion after 5 min. [f] Isolated yield after full conversion at 80 °C and purification.
Figure 5.
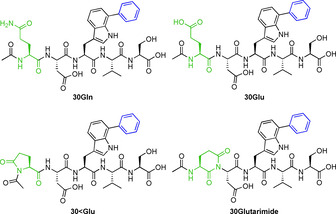
Product and side products of the Suzuki–Miyaura cross coupling using a Gln containing pentapeptide.
The species were inseparable by preparative HPLC giving a corrected isolated yield of 30Gln of 39 % (entry 4). The observed cyclisation is a possible side reaction under basic conditions at elevated temperatures, which occasionally is observed in solid‐phase peptide synthesis. [24] Similar observations had been made by Willemse et al. in Suzuki–Miyaura reactions using ADHP‐Pd. [10] Inhibition of catalysis was expected for pentapeptides comprising lysine or arginine, due to coordination of palladium by amino or guanidino groups. [25] Surprisingly, the catalytic activity increased compared to peptides containing aliphatic or aromatic side chains (entry 1, 9–11) giving 88 % and 86 % HPLC conversion after 2 h at 40 °C (entry 2 and 6). This might occur in virtue of a pre‐coordination of the peptide on the surface of the nanoparticles. Through this pre‐coordination, the proximity of the aryl halide to the palladium could favour oxidative addition (Figure 6).
Figure 6.
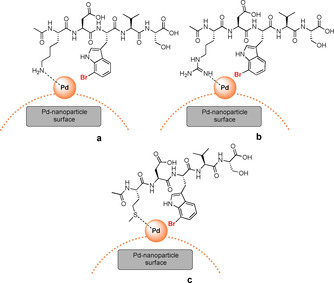
Possible pre‐coordination of Pd‐nanoparticles by certain side chain functionalities of l‐7‐bromotryptophan‐containing pentapeptides.
Since amide formation prevents the coordination to the palladium (Table 2), an N ϵ‐Boc‐protected lysine was introduced into the pentapeptide sequence to prove whether amine‐mediated pre‐coordination of the peptide could be a reason for the accelerated catalytic activity. The Boc‐protected pentapeptide (29Lys(Boc)) showed a conversion (HPLC) of 54 % after 2 hours at 40 °C (entry 7). This value is within the range of the pentapeptides containing aliphatic or aromatic side chains (entry 1, 9–11), letting us draw the conclusion, that a basic, non‐aromatic side chain in the peptide sequence may possibly pre‐coordinate the peptide on the surface of the Pd‐nanoparticles favouring the Suzuki–Miyaura cross‐coupling. Surprisingly, the thioether moiety of methionine accelerates the catalysis as well, causing full conversion at 40 °C after just 5 min (Table 3; entry 8). At the same time, cysteine poisons the catalyst as expected (entry 3). The thioether provides high electron density to the palladium, thus promoting the rate‐limiting oxidative addition step and thereby drastically accelerating the reaction. Adding a methionine to the peptide sequence in the position of the aspartic acid or switching the positions of aspartic acid and methionine led to no conversion of the pentapeptide at all. Thus, poisoning the Pd catalyst. Further investigations on the influence of the position of the thioether moiety to the bromotryptophan for the Suzuki–Miyaura coupling are ongoing. Addition of Boc‐protected methionine as a ligand (5 mol %) or in stochiometric amounts (1.0 equiv.) to the Suzuki–Miyaura reaction of N α‐Boc‐l‐7‐bromotryptophan with phenylboronic acid did not lead to an acceleration but effected inhibition of the catalysis.
We chose N α‐Boc‐bromotryptophan (1), N α‐Fmoc‐bromotryptophan (2) and the pentapeptide 29Lys(Boc) as benchmark candidates to prove the applicability of Pd‐nanoparticles for Suzuki–Miyaura cross‐coupling at room temperature. The cross‐coupling of 1 with phenylboronic acid reached full conversion after 6 h, while full conversion of 2 needed 20 h, giving the arylated tryptophans 4 in 76 % and 5 in 74 % yield after purification by RP‐HPLC. 29Lys(Boc) reached full conversion after 96 h at room temperature giving 30Lys(Boc) in 67 % yield.
Conclusions
Ligand‐free solvent‐stabilised Pd‐nanoparticles were established as a highly efficient catalyst for a biocompatible Suzuki–Miyaura cross‐coupling of different bromotryptophan derivatives under mild, aerobic and aqueous conditions at ambient temperature. In comparison to the commercially available biocompatible Davis‐catalyst (ADHP‐Pd) the ligand‐free nanoparticles displayed increased catalytic activity. With these catalyst systems it is possible to perform cross‐couplings of sensitive substrates at ambient temperature. The catalytically active species of the Davis‐catalyst could be identified as guanidyl‐stabilised quasi‐homogeneous nanoparticles. Evaluation of different N α‐protecting groups for 7‐bromotryptophan identified the Boc‐group as the most favourable one. Under the chosen mild conditions, even the base‐labile Fmoc‐group was stable and compatible with the Pd‐catalyst. Screenings of different 7‐bromotryptophan containing pentapeptides proved the compatibility of the nanoparticles to many amino acid side chain functionalities. Only catalyst‐poisoning moieties like the cysteine thiol or the histidine imidazole compromised the reaction. Surprisingly, some coordinating side chains (lysine, arginine or methionine), which were expected to hinder the reaction, accelerated the catalysis due to a proposed pre‐coordination of the molecule on the nanoparticle surface. Furthermore, the accompanying electron‐donation seemingly promotes the oxidative addition.
Experimental Section
Analytical HPLC was performed on a Shimadzu NexeraXR 20A System with autosampler, degasser, column oven, diode array detector and a Phenomenex Luna C18 column (2.9 μm, 50×2.1 mm) with a gradient (in 5.5 min from 5 % B to 95 % B, 0.5 min 95 % B and back to 5 % B in 3 min, total run time 9 min) at a flow rate of 650 μL min−1 and column oven temperature of 40 °C. HPLC solvent A consists of 99.9 % water and 0.1 % TFA, solvent B of 99.9 % acetonitrile and 0.1 % TFA.
Analytical LC‐MS was performed on an Agilent 6220 TOF‐MS with a Dual ESI‐source, 1200 HPLC system with autosampler, degasser, binary pump, column oven, diode array detector and a Hypersil Gold C18 column (1.9 μm, 50×2.1 mm) with a gradient (in 11 min from 0 % B to 98 % B, back to 0 % B in 0.5 min, total run time 15 min) at a flow rate of 300 μL min−1 and column oven temperature of 40 °C. HPLC solvent A consists of 94.9 % water, 5 % acetonitrile and 0.1 % formic acid, solvent B of 5 % water, 94.9 % acetonitrile and 0.1 % formic acid. ESI mass spectra were recorded after sample injection via 1200 HPLC system in extended dynamic range mode equipped with a Dual‐ESI source, operating with a spray voltage of 2.5 kV.
NMR spectra were recorded on a Bruker Avance III 500 HD (1H: 500 MHz, 13C: 126 MHz, 19F: 471 MHz) or Avance 600 (1H: 600 MHz, 13C: 151 MHz). Chemical shifts δ [ppm] are reported relative to residual solvent signal ([D6]DMSO, 1H: 2.50 ppm, 13C: 39.5 ppm). 2D spectra (COSY, HMQC, HMBC) and DEPT‐135 spectra were used for signal assignment.
High resolution ESI mass spectra were recorded using an Agilent 6220 time‐of‐flight mass spectrometer (Agilent Technologies, Santa Clara, CA, USA) in extended dynamic range mode equipped with a Dual‐ESI source, operating with a spray voltage of 2.5 kV. Nitrogen served both as the nebuliser gas and the dry gas. Nitrogen was generated by a nitrogen generator NGM 11. Samples are introduced with a 1200 HPLC system consisting of an autosampler, degasser, binary pump, column oven and diode array detector (Agilent Technologies, Santa Clara, CA, USA) using a C18 Hypersil Gold column (length: 50 mm, diameter: 2.1 mm, particle size: 1,9 μm) with a short isocratic flow (60 % B for 5 min) at a flow rate of 250 μL min−1 and column oven temperature of 40 °C. HPLC solvent A consists of 94.9 % water, 5 % acetonitrile and 0.1 % formic acid, solvent B of 5 % water, 94.9 % acetonitrile and 0.1 % formic acid. The mass axis was externally calibrated with ESI‐L Tuning Mix (Agilent Technologies, Santa Clara, CA, USA) as calibration standard. The mass spectra are recorded in both profile and centroid mode with the MassHunter Workstation Acquisition B.04.00 software (Agilent Technologies, Santa Clara, CA, USA). MassHunter Qualitative Analysis B.07.00 software (Agilent Technologies, Santa Clara, CA, USA) was used for processing and averaging of several single spectra.
Transmission electron microscopy (TEM) was performed on carbon coated copper grids (mesh 200, Science Service GmbH) which were treated with an oxygen plasma before use (Zepto, Diener electronix GmbH). Pictures were generated on a Philips CM100 PW6021 with a Tungsten Emission source and a voltage of 80 kV or on a JOEL 2200FS with a field emission source and a voltage of 200 kV.
Preparation of ligand‐free solvent‐stabilised Pd‐nanoparticles: Stirring a 100 mm suspension of PdCl2 in 2‐propanol for 16 days under air at room temperature gave a dark red solution with a brown to black precipitate. This stock‐solution was stored at 6–8 °C. Before use the solution was mixed thorough.
Preparation of ADHP‐Pd: On benchtop, 2‐amino‐4,6‐dihydroxypyrimidine (2.0 equiv.) was dissolved in an aqueous 100 mm NaOH solution (4.0 equiv.) and heated to 65 °C over 5 min, followed by addition of Pd(OAc)2 (1.0 equiv.). The mixture was stirred for 30 min, cooled to room temperature followed by addition of water giving an orange solution with a final palladium concentration of 10 mm.
General procedure for Suzuki–Miyaura cross coupling using ligand‐free Pd‐nanoparticles on a preparative scale (GP1): Cross‐couplings were performed on benchtop in a flask equipped with a stirring bar. Therefore, aryl halide (50 μmol, 1.0 equiv.), boronic acid (5.0 equiv.) and K3PO4 (5.0 equiv.) were dissolved in water (5.0 mL), giving a final aryl halide concentration of 10 mm and heated to the desired reaction temperature. The Pd‐nanoparticles dissolved in 2‐Propanol (25 μL; 100 mm; 5 mol %) were added, giving a final 2‐Propanol concentration of 0.5 % (v/v). The reaction progress was monitored by RP‐HPLC at 220 nm. After completion the Pd‐nanoparticles were removed by centrifugation (10 000 rpm; 10 min) and the reaction mixture was directly purified by preparative RP‐HPLC.
General procedure for Solid‐Phase Peptide Synthesis (GP2): All peptides were synthesized on 2‐chlorotrityl chloride resin using the Fmoc/tBu‐strategy. The resin was loaded with Fmoc‐Ser(tBu)‐OH (4 equiv.) and DIEA (8 equiv.) in DCM at room temperature, shaken for 2 h and remaining binding sites were capped by adding MeOH (15 equiv.). Fmoc‐deprotection was performed by addition of 20 % piperidine and 100 mm HOBt in DMF to the resin and shaking for 15 min at room temperature; this procedure was repeated twice. After deprotection, the resin was washed with DMF, DCM, DMF (3×1 min each). Natural Fmoc‐protected amino acids were coupled to the N α‐deprotected peptide by addition of a mixture of amino acid (4 equiv.), TBTU (4 equiv.) and DIEA (8 equiv.) in DMF to the resin and shaking for 2 h at room temperature. After coupling, the resin was washed with DMF, DCM, 2‐propanol and MTBE (3×1 min each) and dried in vacuo. Full conversion was verified by Kaiser‐test. l‐Fmoc‐7‐bromotryptophan (1.1 equiv.) was coupled to the Nα‐deprotected peptide on resin with HATU (1.1 equiv.) and DIEA (2.2 equiv.) in DMF at room temperature for 2 h. Full conversion was verified by a test cleavage and analytical LC‐MS. Before final cleavage, the peptide was N‐acetylated by addition of a solution of acetic anhydride (10 equiv.) and pyridine (10 equiv.) in DMF to the resin. Cleavage and side chain deprotection were performed by addition of a mixture of TFA/H2O/TIS (95:2.5:2.5) to the resin (2×1.5 h) followed by peptide precipitation overnight in MTBE at −20 °C. This mixture was spun down (4000 rpm; 4 °C; 5 min), the MTBE layer discarded, the residue dissolved in water and freeze dried. If necessary, the peptide was purified by RP‐HPLC.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We acknowledge financial support from Deutsche Forschungsgemeinschaft (SE 609/16‐1). The TEM equipment was funded by Deutsche Forschungsgemeinschaft (INST215/444‐1). Open access funding enabled and organized by Projekt DEAL.
S. Dachwitz, D. H. Duwe, Y. H. Wang, H. Gruß, Y. Hannappel, T. Hellweg, N. Sewald, Chem. Eur. J. 2020, 26, 16357.
References
- 1.
- 1a. Miyaura N., Suzuki A., Chem. Rev. 1995, 95, 2457–2483; [Google Scholar]
- 1b. Suzuki A., Angew. Chem. Int. Ed. 2011, 50, 6722–6737; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 6854–6869. [Google Scholar]
- 2. Lennox A. J. J., Lloyd-Jones G. C., Chem. Soc. Rev. 2014, 43, 412–443. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Ronson T. O., Taylor R. J. K., Fairlamb I. J. S., Tetrahedron 2015, 71, 989–1009; [Google Scholar]
- 3b. Deb Roy A., Grüschow S., Cairns N., Goss R. J. M., J. Am. Chem. Soc. 2010, 132, 12243–12245; [DOI] [PubMed] [Google Scholar]
- 3c. Runguphan W., O'Connor S. E., Org. Lett. 2013, 15, 2850–2853; [DOI] [PubMed] [Google Scholar]
- 3d. Durak L. J., Payne J. T., Lewis J. C., ACS Catal. 2016, 6, 1451–1454; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3e. Gruß H., Sewald N., Chem. Eur. J. 2020, 26, 5328–5340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Willemse T., Schepens W., Vlijmen H., Maes B., Ballet S., Catalysts 2017, 7, 74–106. [Google Scholar]
- 5.
- 5a. Spicer C. D., Davis B. G., Nat. Commun. 2014, 5, 4740–4753; [DOI] [PubMed] [Google Scholar]
- 5b. Sletten E. M., Bertozzi C. R., Angew. Chem. Int. Ed. 2009, 48, 6974–6998; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 7108–7133; [Google Scholar]
- 5c. de Graaf A. J., Kooijman M., Hennink W. E., Mastrobattista E., Bioconjugate Chem. 2009, 20, 1281–1295. [DOI] [PubMed] [Google Scholar]
- 6. Chalker J. M., Wood C. S. C., Davis B. G., J. Am. Chem. Soc. 2009, 131, 16346–16347. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Frese M., Sewald N., Angew. Chem. Int. Ed. 2015, 54, 298–301; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 302–305; [Google Scholar]
- 7b. Schnepel C., Sewald N., Chem. Eur. J. 2017, 23, 12064–12086. [DOI] [PubMed] [Google Scholar]
- 8. Smith D. R. M., Willemse T., Gkotsi D. S., Schepens W., Maes B. U. W., Ballet S., Goss R. J. M., Org. Lett. 2014, 16, 2622–2625. [DOI] [PubMed] [Google Scholar]
- 9. Roy A. D., Goss R. J. M., Wagner G. K., Winn M., Chem. Commun. 2008, 4831–4833. [DOI] [PubMed] [Google Scholar]
- 10. Willemse T., van Imp K., Goss R. J. M., van Vlijmen H. W. T., Schepens W., Maes B. U. W., Ballet S., ChemCatChem 2015, 7, 2055–2070. [Google Scholar]
- 11. Frese M., Schnepel C., Minges H., Voß H., Feiner R., Sewald N., ChemCatChem 2016, 8, 1799–1803. [Google Scholar]
- 12. Latham J., Henry J.-M., Sharif H. H., Menon B. R. K., Shepherd S. A., Greaney M. F., Micklefield J., Nat. Commun. 2016, 7, 11873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Schnepel C., Minges H., Frese M., Sewald N., Angew. Chem. Int. Ed. 2016, 55, 14159–14163; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 14365–14369. [Google Scholar]
- 14. Gruß H., Belu C., Bernhard L. M., Merschel A., Sewald N., Chem. Eur. J. 2019, 25, 5880–5883. [DOI] [PubMed] [Google Scholar]
- 15.
- 15a. Kemker I., Feiner R. C., Müller K. M., Sewald N., ChemBioChem 2020, 21, 496–499; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15b. Kemker I., Schnepel C., Schröder D. C., Marion A., Sewald N., J. Med. Chem. 2019, 62, 7417–7430. [DOI] [PubMed] [Google Scholar]
- 16. Sharma S. V., Tong X., Pubill-Ulldemolins C., Cartmell C., Bogosyan E. J. A., Rackham E. J., Marelli E., Hamed R. B., Goss R. J. M., Nat. Commun. 2017, 8, 229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gao Z., Gouverneur V., Davis B. G., J. Am. Chem. Soc. 2013, 135, 13612–13615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dumas A., Peramo A., Desmaële D., Couvreur P., Chimia 2016, 70, 252–257. [DOI] [PubMed] [Google Scholar]
- 19. Liu C., Li X., Chem. Rec. 2016, 16, 84–97. [DOI] [PubMed] [Google Scholar]
- 20. Kurscheid B., Belkoura L., Hoge B., Organometallics 2012, 31, 1329–1334. [Google Scholar]
- 21.
- 21a. Chowdhury S. R., Roy P. S., Bhattacharya S. K., Nano-Struct. Nano-Objects 2018, 14, 11–18; [Google Scholar]
- 21b. Cárdenas-Triviño G., Segura R. A., Reyes-Gasga J., Colloid Polym. Sci. 2004, 282, 1206–1212. [Google Scholar]
- 22. Düfert M. A., Billingsley K. L., Buchwald S. L., J. Am. Chem. Soc. 2013, 135, 12877–12885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.
- 23a. Sewald N., Peptides: Chemistry and Biology, 1st ed., Wiley-VCH, Weinheim, 2002; [Google Scholar]
- 23b. Yang Y. in Side Reactions in Peptide Synthesis, Elsevier/Academic Press, London, 2016, pp. 119–161. [Google Scholar]
- 24. Michels T., Dölling R., Haberkorn U., Mier W., Org. Lett. 2012, 14, 5218–5221. [DOI] [PubMed] [Google Scholar]
- 25. Khalaf M. S., Oakley S. H., Coles M. P., Hitchcock P. B., Dalton Trans. 2010, 39, 1635–1642. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary