

Abstract
Nitrogenases reduce N2, the most abundant element in Earth's atmosphere that is otherwise resistant to chemical conversions due to its stable triple bond. Vanadium nitrogenase stands out in that it additionally processes carbon monoxide, a known inhibitor of the reduction of all substrates other than H+. The reduction of CO leads to the formation of hydrocarbon products, holding the potential for biotechnological applications in analogy to the industrial Fischer–Tropsch process. Here we report the most highly resolved structure of vanadium nitrogenase to date at 1.0 Å resolution, with CO bound to the active site cofactor after catalytic turnover. CO bridges iron ions Fe2 and Fe6, replacing sulfide S2B, in a binding mode that is in line with previous reports on the CO complex of molybdenum nitrogenase. We discuss the structural consequences of continued turnover when CO is removed, which involve the replacement of CO possibly by OH−, the movement of Q176D and K361D, the return of sulfide and the emergence of two additional water molecules that are absent in the CO‐bound state.
Keywords: CO reduction, iron-sulfur proteins, nitrogen fixation, nitrogenase, X-ray crystallography
Carbon monoxide is a non‐competitive inhibitor of biological nitrogen fixation by the enzyme nitrogenase, but also a substrate for the vanadium‐dependent variant of the enzyme, leading to a formation of hydrocarbon products that involve C−C bond formation. A 1.0 Å resolution crystal structure of the enzyme with CO bound to the active site cofactor reveals the binding mode of the ligand and the reversible replacement of a μ2‐bridging sulfide.
Nitrogenases have intrigued researchers for decades, as their ability to reduce chemically inert dinitrogen is unique among known enzymes. In breaking the N2 triple bond the enzyme expends substantial metabolic energy in the form of ATP, and it also requires low‐potential electrons provided through an elaborate array of iron‐sulfur clusters. [1] Unsurprisingly, an enzyme capable of reducing N2 also converts other, chemically similar molecules. Alternative substrates include acetylene, [2] hydrocyanic acid, [3] azide [4] and carbon monoxide, [5] although the latter is a strong, but non‐competitive inhibitor for all other substrates other than protons. [5] Nitrogenases can be subdivided into three groups with respect to their metal requirement: a molybdenum‐dependent enzyme (encoded by a nif gene cluster), a vanadium‐dependent variant (vnf) and a third class relying solely on iron (anf). Nitrogenases are two‐component enzymes that require a reductase component, the Fe protein, which couples electron transfer to ATP hydrolysis. [1a] Mo nitrogenase is the best‐studied variant and shows the highest efficiency of electron utilization, diverting fewer electrons towards an undesired formation of H2 (i.e. the reduction of protons) than in the alternative enzymes. [6] The crystal structures of the MoFe protein NifD2K2 (Figure 1 A) and the cognate Fe protein NifH2 were reported by Kim and Rees and Giorgiadis and Rees in 1992, [7] while the architecture of the vanadium nitrogenase VFe protein VnfD2K2G2 (Figure 1 B) and the Fe protein VnfH2 were elucidated only recently. [8] For the iron‐only nitrogenase system, no structural data is available to date.
Figure 1.
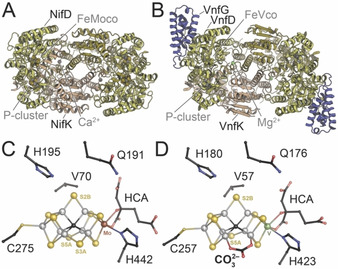
The Mo‐ and V‐dependent nitrogenases of Azotobacter vinelandii. A) Cartoon representation of MoFe protein (PDB 3U7Q). Subunits and clusters are labelled in one half of the NifD2K2 heterotetramer. B) Cartoon representation of VFe protein (PDB 5N6Y). The additional subunit VnfG is shown in purple, subunits and clusters are labelled in one half of the VnfD2K2G2 heterohexamer. C) FeMo cofactor within NifD of MoFe protein. D) FeV cofactor within VnfD of VFe protein. HCA: homocitrate.
The finding that VFe protein reduces CO sparked substantial interest in the enzyme. [5] As a substrate, CO is isoelectronic to N2 and is likely metabolized along a similar mechanistic route. Reminiscent of the industrial Fischer–Tropsch process its reduction leads to hydrocarbons, with ethylene as the predominant product (>93 % yield), but longer‐chain hydrocarbons such as propane and propylene were also detected. [9] These products require C−C bond formation, implying the existence of at least two sites for CO binding in vanadium nitrogenase, [10] but the exact location of these sites remained elusive. Indeed, the binding of two CO ligands to Mo nitrogenase was shown earlier by 13C ENDOR spectroscopy, [11] but the significance of this discovery was lost since CO at the time was considered solely as an inhibitor. In fact, the slow reduction of CO was recently also found for Mo nitrogenase, albeit at an 800‐fold lower rate than in V nitrogenase. [9] CO reduction by VFe protein holds promise for future biotechnological applications, and while the C−C coupling involved in ethylene formation from CO is intriguing by itself, it might also provide crucial clues for the mechanism of N2‐reduction by nitrogenases.
While the dinitrogenases MoFe protein and VFe protein are encoded by distinct structural genes, their active site cofactors (FeMoco and FeVco, respectively) are similar (Figure 1 C,D).[ 8b , 12 ] Both metal clusters comprise 7 irons, which are biosynthetically derived from two [4Fe:4S] clusters that get rearranged into the core L‐cluster, a [8Fe:9S:C] moiety, by the radical‐SAM enzyme NifB. [13] During further cluster maturation, an apical iron is replaced by a heterometal (either Mo or V) that is complexed by a homocitrate ligand. Both cofactors are only coordinated by a cysteine and a histidine residue within their respective D subunits, H442D and C275D in the MoFe protein (Figure 1 C) and H423D and C257D in the VFe protein (Figure 1 D) of Azotobacter vinelandii. Furthermore, in FeVco a novel ligand replaces one of the three μ2‐“belt” sulfides, S3B. [8b] This μ‐1,3‐bridging ligand was assigned as a carbonate based on crystallographic data, [8b] as supported by recent theoretical studies. [14] Its role remains unclear, but there is no indication that the ligand turns over or is even transiently displaced at any time during catalysis. Apart from this, the core of the active site, where six iron ions form a trigonal prism around a central carbide, [15] is preserved in both nitrogenases and here is where N2‐binding and reduction are suggested to take place. [16] The catalytic model by Lowe and Thorneley describes N2 reduction by nitrogenase as an 8‐electron process with mandatory H2 evolution (intermediates E0 to E7). [17] However, crystals of MoFe protein consistently only showed the active site in the resting state, E0. For vanadium nitrogenase, the same resting state was found in the initial crystal structure, [8b] but X‐ray crystallography then also identified an alternative state with an unknown light atom bound to the active site, replacing the bridging sulfide S2B. [18] The displaced sulfur was not lost, but migrated to a nearby holding site 7 Å away that was formed by the rearrangement of residue Q176D. This alternative turnover state was tentatively assigned to the E6 state in the catalytic cycle, representing a state in which N2 has been partly reduced and one molecule of the product ammonia has already been released. [18] The structural data strongly suggests the reversible displacement of sulfide S2B to be part of an activation mechanism. [19] In fact, S2B displacement is not unprecedented in nitrogenases. In MoFe protein, this same sulfide was replaced by carbon monoxide, resulting in an inhibited state of the enzyme that was reversible upon continuous turnover in the absence of CO. [20] Also, after turnover with selenocyanate, the S2B position was the first to be occupied by selenide, followed by a slow migration of Se into the other two belt positions. [21] Other than in V nitrogenase during N2 fixation, the S2B sulfide was absent from the ligand‐bound structures, with no satisfying explanation for its fate. With CO being an inhibitor and SeCN an artificial substrate, deviations from the physiologically relevant states could not be ruled out. Nevertheless, in both cases the lost S2B was recovered after additional cycles of substrate turnover, indicating that its transient removal might be an actual feature of the cofactor rather than a mere oddity or experimental artifact.
To date, both Mo‐ and V‐nitrogenase have exhibited reactivity at analogous dinuclear sites between Fe2 and Fe6. This position becomes chemically accessible due to the exceptional lability of sulfide S2B within an otherwise rigid cofactor scaffold. According to an assignment of relative oxidation states in FeMoco by spatially resolved anomalous dispersion analysis (SpReAD), Fe2 and Fe6 are also the most highly oxidized metal sites in the clusters’ resting state and thus the likely sites for reduction when an electron is transferred from Fe protein. [22]
For V‐nitrogenase, a possible physiological relevance of CO reduction was suggested in the form of a secondary metabolism by V‐ nitrogenases in the bacterial cell. [23] However, as a substrate CO should not form long‐lived and stable adducts with FeVco. Here we show that V‐nitrogenase nevertheless can be isolated and crystallized with CO bound. We report the most detailed crystal structure of vanadium nitrogenase to date at 1.0 Å resolution with bound CO and describe the structural changes that occur when CO‐bound protein is turned over further in the absence of substrate or inhibitor (Figure S1).
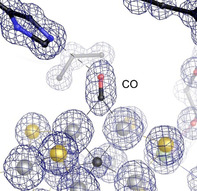
VFe protein, with its active site predominantly in the turnover state (as verified by X‐ray crystallography), [18] was turned over in vitro and crystallized immediately afterwards. When nitrogen, acetylene or cyanide were used as substrates, conformational mixtures were obtained around the belt sulfide S2B and residue Q176D, and modeled as such (data not shown). The labile sulfide S2B partially occupied the resting state position, as in the published structure of the resting state E0, [8b] but some sulfur anomalous density also remained at the nearby holding site of the turnover state. [18] In contrast, under a 100 % CO atmosphere during turnover a single state was obtained that significantly deviated from the previously known V‐nitrogenase structures. In spite of reports to the opposite, [10a] CO pressurization of VFe protein crystals without concomitant turnover did not result in the same structural features. As for MoFe protein, turnover conditions thus seem to be a strict prerequisite for obtaining the CO‐bound state of VFe protein. The atomic‐resolution crystal structure of CO‐bound vanadium nitrogenase reveals a distinct, elongated electron density feature at the Fe2‐Fe6 edge of FeV cofactor, well in line with a diatomic carbon monoxide ligand bound in a μ2‐bridging manner (Figure 2) and significantly different for the densities of carbonate and the remaining S5A (Figure S2). The coordination of CO to the Fe2‐Fe6 edge shows a slight asymmetry, with an average distance to Fe2 of 2.03 Å and to Fe6 of 1.94 Å.
Figure 2.
The CO adduct of FeV cofactor. A 2 Fo‐Fc electron density map at 1.0 Å resolution (contoured at the 3 σ level) reveals the replacement of the μ2‐bridging sulfide S2B with an elongated CO molecule as a bridging carbonyl.
For the CO adduct of FeMo cofactor both distances were reported as identical at 1.86 Å, but the lower resolution of this analysis at 1.50 Å implies that comparisons should be made with caution. [24] Incorporation of CO into FeV cofactor leads to a significant decrease of the Fe2‐Fe6 distance from 2.61 Å in the resting state to 2.54 Å in the CO‐bound state (Figure 3 A). This contraction of the core is not compensated by an increase of the distance to the outer metals. Instead, the distances of Fe1‐Fe2 at 2.67 Å and of Fe6‐V at 2.75 Å are also shortened slightly, and the movement of the inner core is translated along the pseudo‐threefold axis of the cofactor, decreasing the distance between the apical metals Fe1 and V from 7.15 Å to 7.07 Å. The cofactor contraction caused by CO binding in FeVco amounts to double of what was found previously for FeMoco (Figure 3 B,C). [24] Such increased flexibility of FeVco may arise from the widening of the core induced by the carbonate ligand. Notably, the contracted CO‐FeV cofactor still exceeds the length of FeMoco in its resting state, which features a Fe1‐Mo distance of only 7.00 Å. [15a]
Figure 3.
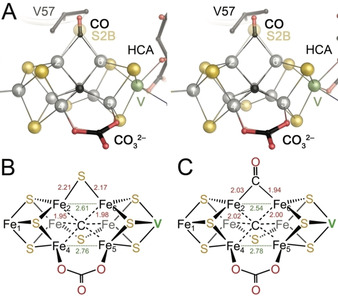
Geometric distortions in FeV cofactor upon binding of CO. A) Stereo representation of a superposition of the CO‐bound state of FeV cofactor with the resting state (transparent). HCA: homocitrate. B) Important bond distances in the resting state of FeV cofactor. C) In the CO‐bound state, the bond lengths from Fe2 and Fe6 to the carbon of CO are substantially shorter than to S2B in the resting state, leading to a contraction of the cluster and a significant shortening of the Fe2‐Fe6 distance by 0.07 Å.
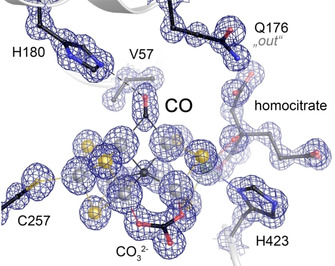
In the structure, CO replaced sulfide S2B of FeV cofactor, but unlike in the turnover state this did not trigger a rotation of the sidechain of residue Q176D toward the cluster, as the diatomic CO ligand would interfere with the γ‐amide of the amino acid. Q176D was consequently found in its out‐conformation, facing away from FeV cofactor, very much in line with the conformation observed for the CO adduct of FeMoco (Figure 2). [24] This of course excluded the displaced sulfide S2B from its holding side, and X‐ray diffraction data collected at 6200 eV provided an anomalous difference electron density map that evidenced the absence of sulfur at both of the previously identified locations for sulfide S2B (Figure S3). No additional anomalous difference peaks that might account for the missing sulfur were found in the map. Hence, the total amount of sulfur atoms in this state of the cofactor is reduced to seven, with only a single sulfide, S5A, retained in the cofactor belt. After CO capture on FeV cofactor was verified by X‐ray crystallography, the remaining sample was desalted and subsequently reused for a second in vitro turnover assay, in which the CO headspace was replaced by Ar. Under these conditions only the remaining bound CO and protons could be reduced. Subsequent crystallization led to a further structure at 1.05 Å resolution from which CO was again absent. Interestingly, this structure did not correspond to the previously reported resting state E0, in which S2B would have been reinstated at Fe2 and Fe6. Instead, a monoatomic ligand was found to replace CO (Figure 4), triggering the same conformational change of Q176D that was found in the turnover state. [18]
Figure 4.
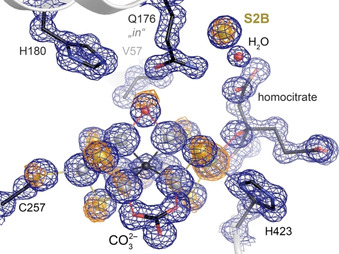
Nitrogenase FeVco after removal of CO at 1.05 Å resolution. The enzyme was found predominantly in the turnover state configuration with a light atom bound at Fe2 and Fe6, now likely OH−. A sulfur anomalous electron density map collected at 6200 eV (orange, contoured at 5σ) indicates small amounts of residual resting state conformation.
In contrast to the published turnover state that was obtained with protein isolated from cells grown under N2‐fixing conditions, the light atom in the present structure cannot be rationalized as a nitrogen species. Its electron density maximum is in line with C, N or O in a protonated form, and it might arise as a product of CO reduction or from water. Indeed, the earlier assignment of the ligand in the reported E6 state as NH was recently contested based on DFT calculations, [14] and a preferential stabilization of a bridging hydroxyl ligand that occurs in FeV cofactor, but not in the structurally related FeMo cofactor, might be related to the observed differences in reactivity. Note that the present structure could not be obtained without additional turnover under Ar atmosphere, as CO‐bound VFe‐protein crystals were unexpectedly stable, with no observable decay of the CO‐associated electron density after storage for weeks at room temperature. We conclude that turnover conditions are not only required for CO capture on FeVco, but also for its release, likely requiring a catalytic reduction of the ligand that does not occur merely upon addition of reductant.
Adjacent to the substrate binding site, the movement of residue Q176D once more opened up the sulfur storage site upon removal of CO. A new electron density maximum appeared at this position, and X‐ray diffraction data collected at 6200 eV showed a distinct peak in an anomalous difference density map, in line with the presence of sulfur. This recurrence of the sulfur signal is not unprecedented, as the same was observed for Mo nitrogenase. [24] However, FeMoco returned to its resting state with all three belt sulfides in place, while in FeVco the returning sulfide occupied the holding site, albeit with a minor amount of reinstated S2B according to the anomalous electron density map (Figure 4). [18] Vanadium nitrogenase obviously has a higher propensity to retain bound ligands at the Fe2‐Fe6 edge of its active site cofactor, stabilized through interactions with the side chain of Q176D. The origin of the returning sulfide nevertheless remains enigmatic. Excess dithionite was suggested as a possible source, [24] as its disproportionation in water leads to the formation of sulfite, which was previously hypothesized to be a sulfur‐donor during FeMoco biogenesis. [25] Notably, both the anomalous signal at 6200 eV and the identical number of electrons would render HS− and Cl− indistinguishable in all analyses of nitrogenase cofactors reported to date. This distinction is relevant with respect to the proposed reaction mechanism of nitrogenases. [19] Here, removal of sulfide S2B likely requires its protonation, which would not occur as straightforwardly with chloride. Also, while it might occupy the sulfide holding site, chloride should not bind to the cofactor as a bridging belt ligand. If the storage site holds sulfide, then the data reported here suggests a high affinity, as it even recruits external sulfide from the environment when Q176D is in its inward‐facing conformation. CO blocks this rotation of the glutamine, but upon its removal the site quickly regains its function. In Mo nitrogenase, the corresponding Q191D was always observed in the outward‐facing state. This residue is highly conserved in all nitrogenases and is essential for N2 fixation. [26] We assume that the sulfide storage site can also be formed close to FeMoco, but that the reincorporation of the displaced sulfur into the cofactor is more favorable, possibly due to an increased stability of a resting state with three belt sulfides, as opposed to only two in FeVco, where carbonate breaks symmetry.
In spite of the known differences in reactivity that distinguish molybdenum‐ and vanadium‐dependent nitrogenases, the present study shows that the mode of interaction with CO under turnover conditions is largely identical in both. This may indicate that the reasons originate from the electronic structure of the respective clusters, FeMoco or FeVco, themselves and may be more subtle than expected.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
The authors thank the staff at beam lines X06SA and X06DA, Swiss Light Source, Villigen, CH, for excellent assistance with data collection. This work was supported by the European Research Council (grant 310656) and Deutsche Forschungsgemeinschaft (RTG 1976, project no. 235777276 and PP 1927, project no. 311061829). Coordinates and structure factors have been deposited with the Protein Data Bank at http://www.pdb.org with accession codes 7ADR (CO adduct) and 7ADY (CO‐removed). Open access funding enabled and organized by Projekt DEAL.
M. Rohde, K. Grunau, O. Einsle, Angew. Chem. Int. Ed. 2020, 59, 23626.
References
- 1.
- 1a. Rees D. C., Tezcan F. A., Haynes C. A., Walton M. Y., Andrade S., Einsle O., Howard J. B., Philos. Trans. R. Soc. London Ser. A 2005, 363, 971–984; [DOI] [PubMed] [Google Scholar]
- 1b.O. Einsle, D. C. Rees, Chem. Rev 2020. [DOI] [PMC free article] [PubMed]
- 2. Moustafa E., Mortenson L. E., Nature 1967, 216, 1241–1242. [DOI] [PubMed] [Google Scholar]
- 3. Li J. G., Burgess B. K., Corbin J. L., Biochemistry 1982, 21, 4393–4402. [DOI] [PubMed] [Google Scholar]
- 4. Fisher K., Dilworth M. J., Newton W. E., Biochemistry 2006, 45, 4190–4198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lee C. C., Hu Y. L., Ribbe M. W., Science 2010, 329, 642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Harris D. F., Lukoyanov D. A., Kallas H., Trncik C., Yang Z. Y., Compton P., Kelleher N., Einsle O., Dean D. R., Hoffman B. M., Seefeldt L. C., Biochemistry 2019, 58, 3293–3301. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Georgiadis M. M., Komiya H., Chakrabarti P., Woo D., Kornuc J. J., Rees D. C., Science 1992, 257, 1653–1659; [DOI] [PubMed] [Google Scholar]
- 7b. Kim J. S., Rees D. C., Nature 1992, 360, 553–560. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Rohde M., Trncik C., Sippel D., Gerhardt S., Einsle O., J. Biol. Inorg. Chem. 2018, 23, 1049–1056; [DOI] [PubMed] [Google Scholar]
- 8b. Sippel D., Einsle O., Nat. Chem. Biol. 2017, 13, 956–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hu Y. L., Lee C. C., Ribbe M. W., Science 2011, 333, 753–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.
- 10a. Lee C. C., Fay A. W., Weng T. C., Krest C. M., Hedman B., Hodgson K. O., Hu Y. L., Ribbe M. W., Proc. Natl. Acad. Sci. USA 2015, 112, 13845–13849; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10b. Lee C. C., Wilcoxen J., Hiller C. J., Britt R. D., Hu Y. L., Angew. Chem. Int. Ed. 2018, 57, 3411–3414; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 3469–3472; [Google Scholar]
- 10c. Hiller C. J., Lee C. C., Stiebritz M. T., Rettberg L. A., Hu Y. L., Chem. Eur. J. 2019, 25, 2389–2395. [DOI] [PubMed] [Google Scholar]
- 11. Pollock R. C., Lee H. I., Cameron L. M., Derose V. J., Hales B. J., Orme-Johnson W. H., Hoffman B. M., J. Am. Chem. Soc. 1995, 117, 8686–8687. [Google Scholar]
- 12. Einsle O., Tezcan F. A., Andrade S. L. A., Schmid B., Yoshida M., Howard J. B., Rees D. C., Science 2002, 297, 1696–1700. [DOI] [PubMed] [Google Scholar]
- 13. Hu Y., Ribbe M. W., Annu. Rev. Biochem. 2016, 85, 455–483. [DOI] [PubMed] [Google Scholar]
- 14. Benediktsson B., Thorhallsson A. T., Björnsson R., Chem. Commun. 2018, 54, 7310–7313. [DOI] [PubMed] [Google Scholar]
- 15.
- 15a. Spatzal T., Aksoyoğlu M., Zhang L. M., Andrade S. L. A., Schleicher E., Weber S., Rees D. C., Einsle O., Science 2011, 334, 940; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15b. Lancaster K. M., Roemelt M., Ettenhuber P., Hu Y. L., Ribbe M. W., Neese F., Bergmann U., DeBeer S., Science 2011, 334, 974–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hoffman B. M., Lukoyanov D., Dean D. R., Seefeldt L. C., Acc. Chem. Res. 2013, 46, 587–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Thorneley R. N. F., Lowe D. J., in Molybdenum Enzymes, Vol. 1 (Ed.: Spiro T. G.), Wiley-Interscience, New York, 1985, pp. 221–284. [Google Scholar]
- 18. Sippel D., Rohde M., Netzer J., Trncik C., Gies J., Grunau K., Djurdjevic I., Decamps L., Andrade S. L. A., Einsle O., Science 2018, 359, 1484–1489. [DOI] [PubMed] [Google Scholar]
- 19. Rohde M., Sippel D., Trncik C., Andrade S. L. A., Einsle O., Biochemistry 2018, 57, 5497–5504. [DOI] [PubMed] [Google Scholar]
- 20. Bjornsson R., Lima F. A., Spatzal T., Weyhermüller T., Glatzel P., Bill E., Einsle O., Neese F., DeBeer S., Chem. Sci. 2014, 5, 3096–3103. [Google Scholar]
- 21. Spatzal T., Perez K. A., Howard J. B., Rees D. C., eLife 2015, 4, e11620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Spatzal T., Schlesier J., Burger E. M., Sippel D., Zhang L. M., Andrade S. L. A., Rees D. C., Einsle O., Nat. Commun. 2016, 7, 10902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rebelein J. G., Lee C. C., Hu Y. L., Ribbe M. W., Nat. Commun. 2016, 7, 13641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Spatzal T., Perez K. A., Einsle O., Howard J. B., Rees D. C., Science 2014, 345, 1620–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tanifuji K., Lee C. C., Sickerman N. S., Tatsumi K., Ohki Y., Hu Y. L., Ribbe M. W., Nat. Chem. 2018, 10, 568–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Scott D. J., May H. D., Newton W. E., Brigle K. E., Dean D. R., Nature 1990, 343, 188–190. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary