

Abstract
The cannabinoid (CB) receptors (CB1R and CB2R) represent a promising therapeutic target for several indications such as nociception and obesity. The ligands with non-selectivity can be traced to the high similarity in the binding sites of both cannabinoid receptors. Therefore, the need for selectivity, potency, and G-protein coupling bias has further complicated the design of desired compounds. Currently studied cannabinoid agonists seldomly investigate their bias, and those that do exhibit bias are typically non-selective. However, certain long-chain endocannabinoids represent a class of selective and potent CB1R agonists. The binding mode for this class of compounds has remained elusive, limiting the implementation of its binding features to currently studied agonists. Hence, in the present study, the binding poses for these long-chain cannabinoids, along with other interesting ligands, with the receptors have been determined, by using a combination of molecular docking and molecular dynamics (MD) simulations along with molecular mechanics-Poisson-Boltzmann surface area (MM-PBSA) binding free energy calculations. The binding poses for the long-chain cannabinoids implicate that a site surrounded by the transmembrane (TM)2, TM7, and extracellular loop (ECL)2 is vital for providing the long-chain ligands with the selectivity for CB1R, especially I267 of CB1R (corresponding to L182 of CB2R). Based on the obtained binding modes, the calculated relative binding free energies and selectivity are all in good agreement with the corresponding experimental data, suggesting that the determined binding poses are reasonable. The computational strategy used in this study may also prove fruitful in applications with other GPCRs or membrane-bound proteins.
Keywords: Cannabinoid receptor, selectivity, endocannabinoid, drug design strategy, modeling
Graphical Abstract
Introduction
G-protein coupled receptors (GPCRs) are a well-studied class of proteins, with significant viability as therapeutic targets due to their functions in critical physiological pathways.1 Currently, there are over four hundred approved drugs targeting these receptors, with over a hundred targets represented amongst them.1 The cannabinoid (CB) receptors are a subset of the GPCRs, consisting of seven transmembrane (TM) regions divided into two subtypes: CB1 receptor (CB1R) and CB2 receptor (CB2R). Each of these subtypes has characteristic functions related to their distribution in the body. While both CB1R and CB2R are found within the brain, CB2R is additionally expressed in the periphery, most prominently within the spleen.2 These receptors’ primary function within the body is the regulation of adenylyl cyclase, which is implicated in nociception.3-6 This implication has led to a levy of studies performed to determine the cannabinoids’ abilities to produce analgesic effects as a potential replacement for non-steroidal anti-inflammatory drugs (NSAIDs) and opioids, which both come with a set of noxious side effects.4, 5, 7-14 Additionally, the knock-out (KO) of CB1R has been implicated in the protection against obesity due to the CB receptors’ functions in regulating metabolism.15 These relationships have made the CB receptors a promising target for several therapeutic indications. However, there has yet to be an FDA-approved selective cannabinoid agonist for either CB1R or CB2R, an advantageous property that would allow for the limiting of off target effects induced by these previously approved non-selective therapeutics.16, 17
Current drug design targeting CB1R or CB2R primarily falls within the small-molecule space, with the molecules studied utilizing the same binding site as the phytocannabinoid class, e.g. (−)-trans-Δ9-tetrahydrocannabinol (THC), cannabidiol (CBD), and cannabigerol (CBG).18 This binding site has been studied extensively and has previously had several star compounds (e.g. rimonabant and WIN-55,212) tailored to it.19-22 While the cannabinoid agonists targeting this site can exhibit high potency towards the CB receptors, many are ultimately unable to exhibit high selectivity towards either receptor.23, 24 Interestingly, the endogenous cannabinoids (endocannabinoids) with a long carbon chain structural scaffold (Figure 1) exhibit considerable selectivity towards CB1R, with comparable potency to many of these previously reported compounds.18, 25, 26 With minimal modifications to these endogenous ligands, analogs of these endocannabinoids exhibit greater selectivity and potency than their precursors (Figure 1).27 However, these long-chain ligands have been far less studied concerning their potential as CB receptor agonist therapeutics in comparison to compounds with structural similarity to the phytocannabinoids. The indole quinuclidinone (IQD) compounds, PNR-4-20 and PNR-4-02, were the only reported G-protein biased agonists of CB2R (Figure 2).28 Incorporating the binding modes of these long-chain endocannabinoids with the recently published new agonists, including a G-protein biased agonist, could be a viable path towards the rational design of the desirable CB1R-specific and G-protein biased agonists.29-33 Hence, it is crucial for rational design of CB1R-specific ligands to first determine how these compounds (Figure 1 and 2) bind with both CB1R and CB2R and to understand the nature of their selectivity.
Figure 1.
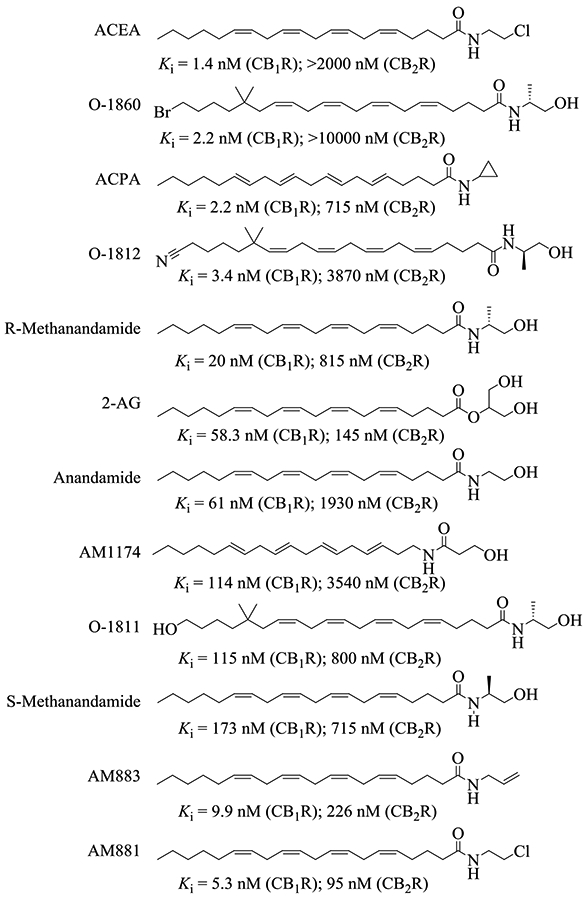
Representative long-chain endocannabinoids and their synthetic analogs. The experimental binding affinities come from the references indicated in Table 2.
Figure 2.
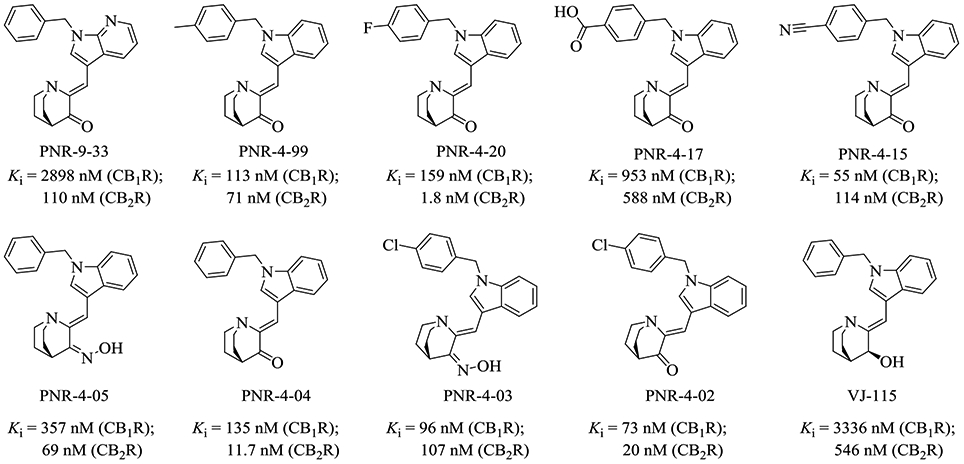
Molecular structures of indole quinuclidinone (IQD) series cannabinoid receptor agonists, along with the experimental binding affinities.28
There have been various reports of computational and experimental studies on CB1R and CB2R binding with various ligands. Reported cryogenic electron microscopy (Cryo-EM) structures of CB1R or CB2R binding with ligands revealed a commonly available binding site, i.e. the phytocannabinoid binding site, for a few ligands examined so far.21, 22, 34-37 Previously reported computational studies have been focused on the phytocannabinoid binding site.19, 20, 22, 38-40 None of the previously reported computational studies examined the possible binding poses of the IQD series of compounds (Figure 2), and there have been few reports concerning the binding poses of the long-chain endocannabinoids.19, 20, 22
Particularly, studies concerning the long-chain endocannabinoids and their analogs have primarily been in vitro based, using structure-activity relationships (SAR) to discover productive modification for these agonists.25, 27, 41-43 Through these studies, several important factors have been determined such as the necessary aliphatic chain length for CB1R binding, and the importance of double bonds within the aliphatic chain.41 Previous in silico attempts at explaining the binding mode of these compounds have relied on homology-based methods of the cannabinoid receptors which were inaccurate in comparison to the recently published structures.44 One attempt to elucidate the binding mode of these endocannabinoids came from McAllister et al. which proposed a folded anandamide structure within a homology-modeled CB1R.44 Similar binding mode was also reported by other groups using molecular docking to homology-modeled CB1R.37 However, the recently published Cryo-EM structures of CB1R and CB2R revealed that the phytocannabinoid binding sites within these receptors possess significant similarity within the pocket proposed for anandamide, eliminating the possibility for CB1R selectivity.21, 22, 35, 36 These new Cryo-EM studies also attempted to computationally place long-chain cannabinoids within the resolved structures of CB1R. While these binding poses were stable when subjected to short-timescale MD simulations, they did not comment on the selectivity of these compounds; additionally, in their proposed binding modes for these compounds at CB1R relied heavily on the central channel binding pocket, thus lacking any major differences between the two receptors that could be used to determine their selectivity for CB1R.21, 22 With this similarity of the central binding pocket in mind, it is critical to explore possible binding modes for these endocannabinoids that do not solely rely on the typical phytocannabinoid binding pocket to decide their selectivity.
Recent advancement in computational power and support for GPU-accelerated hardware have made long-timescale molecular dynamics (MD) simulation (0.1-1 μs) more feasible for large-scale systems, including lipid bilayers with proteins embedded within them.45 Through a combination of the MD simulations and molecular mechanics–Poisson-Boltzmann surface area (MM-PBSA) binding free energy calculations, compounds binding with a given protein can be ranked accurately in correlation with their experimental binding free energy (ΔGexp) values.45, 46 Our group has previously been successful in developing and utilizing various computational approaches for modeling ligands binding with the transmembrane proteins to elucidate subtype selectivity.47-51 Hence, in the present study, we employed a variety of computational methods, including molecular docking, MD, and MM-PBSA, to explore the binding poses of these interesting compounds (Figures 1 and 2) with CB1R and CB2R and reveal the binding and selective mechanism. According to the computational data, the IQD series of compounds bind to the receptors in the known traditional binding site (phytocannabinoid binding site) of the receptors, whereas the long-chain cannabinoids bind to the receptors in a binding mode which is more favorable for CB1R. These computationally determined binding modes show excellent correlation with the empirically obtained binding data, suggesting that these binding modes are reasonable for these receptors.
Results and Discussion
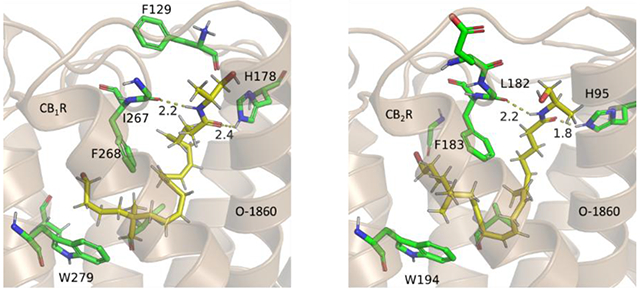
Due to the high binding selectivity and potency of O-1860 with CB1R, its binding mode was studied closely, in the present study, to determine potential unique binding features through molecular docking and MD simulations (Figure 3A and 3B). The obtained binding mode of O-1860 reveal a previously unused binding site in proximity to the extracellular interface of the receptor. As opposed to the previously suggested binding modes concerning these compounds, the hydrophilic binding site within the extracellular interface of CB1R provides pivotal interactions with the long-chain endocannabinoids. Within this region, there are additional critical residue substitutions within CB2R that change the binding pocket’s ability to receive these long chain endocannabinoids. The critical change between the two receptors is from a change in I267 in CB1R to L182 in CB2R (Figure S1 in Supporting Information). This change causes a steric clash with the long-chain cannabinoids (Figure S3 to S14), increasing the hydrogen bond-related distance with L182 and the ligand (Figure 3B) and reducing van der Waals (vdW) interactions with the receptor (Table 1). Conversely, the stricter binding site from these residues in the extracellular interface creates a method to induce CB1R selectivity. The size of the agonist supported by the binding pocket in CB2R will be decreased due to these residue changes from CB1R to CB2R. From these initial results concerning the binding free energy of O-1860, one can see the large differences in the experimental binding affinity be quantitatively validated through the combined MD and MM-PBSA binding free energy calculations (Table 1). Through the decomposition of the per-residue contributions to the binding energy, it was revealed that the residues 177, 267, 268, 279, 376, 379, and 380 within CB1R had greater binding affinities with O-1860 (Figure 3D) than their corresponding analogs in CB2R.
Figure 3.

(A) Room-mean-squares deviation (RMSD) of backbone atoms (black) of CB1R and heavy atoms of O-1860 (red) along with two crucial distances (r1 and r2) indicated in panel C in the MD-simulated CB1R binding with O-1860. (B) RMSD of backbone atoms (black) of CB2R and heavy atoms of O-1860 (red) along with two crucial distances (r1 and r2) indicated in panel D in the MD-simulated CB2R binding with O-1860. (C) A snapshot of the MD-simulated structure of CB1R binding with O-1860 after 100 ns. (D) A snapshot of the MD-simulated structure of CB2R binding with O-1860 after 100 ns. (E) Decomposed per-residue binding energies for residues surrounding O-1860.
Table 1.
The binding free energies (kcal/mol) of O-1860 with CB1R and CB2R based on the combined MD simulation and MM-PBSA calculations.
| CBRs | ΔEele | ΔEvdw | ΔEgas | ΔEpbsol | ΔEpb | −TΔS | ΔGPB |
|---|---|---|---|---|---|---|---|
| CB1R | −27.54 | −70.34 | −97.88 | 38.06 | −59.82 | 22.04 | −37.78 |
| CB2R | −16.37 | −66.42 | −82.79 | 39.75 | −43.03 | 23.70 | −19.33 |
The binding mode of long-chain molecules
To further verify our binding model, twelve additional long-chain molecules with known Ki values for the CB receptors were collected (Figure 1), and their binding free energies were estimated via the MD/MM-PBSA methodology. For each of the long-chain agonists examined in this study, according to the computational data (Table 2) the binding affinity is shown to be higher with the CB1R over the CB2R, this is consistent with previously obtained experimental data.27, 52-55 This selectivity for CB1R extends to anandamide, which has previously been erroneously reported as a non-selective CB agonist.56 However, these previous reports were performed in tissue-based assays, which contained amidohydrolases that degraded anandamide.57-60 When these confounding enzymes are inhibited using phenylmethylsulfonyl fluoride (PMSF), the Ki of anandamide decreased considerably (543 nM against CB1R w/o PMSF vs 90 nM against CB1R w/ PMSF), revealing its true selectivity for CB1R over CB2R (Ki = 1980 nM against CB2R w/ PMSF).56, 57 The computational results obtained have a high correlation with the experimental data, with an R2 of 0.8318 (Figure S2). When compared to O-1860, these long-chain endocannabinoids have similar per-residue contributions to their binding free energies, suggesting that these compounds bind similarly to O-1860 with the CB receptors. (Figure 3E)
Table 2.
Calculated binding free energies (kcal/mol) compared to the corresponding experimental binding free energies (kcal/mol, derived from the experimental Ki shown in Figure 1 or 2).
| Liganda | ΔG(CB1R) | ΔG(CB2R) | ΔG(CB1R)-ΔG(CB2R) | |||||
|---|---|---|---|---|---|---|---|---|
| ΔGPBb | ΔGcorrc | ΔGexpd | ΔGPBb | ΔGcorrc | ΔGexpd | ΔΔGcorrc,e | ΔΔGexpf | |
| ACEA52 | −39.07 | −12.08 | −12.15 | −18.19 | −7.60 | −7.82 | −4.98 | −4.33 |
| O-186027 | −38.44 | −11.97 | −11.88 | −19.30 | −7.96 | −6.86 | −4.54 | −5.02 |
| ACPA53 | −36.78 | −11.67 | −11.88 | −24.95 | −9.79 | −8.44 | −2.68 | −3.45 |
| O-181227 | −35.54 | −11.44 | −11.62 | −18.06 | −7.56 | −7.43 | −4.12 | −4.20 |
| AM88153 | −35.45 | −11.43 | −11.36 | −26.78 | −10.38 | −9.64 | −1.87 | −1.72 |
| AM88354 | −35.10 | −11.36 | −10.99 | −25.48 | −9.96 | −9.12 | −2.11 | −1.86 |
| VJ-11528 | −21.05 | −8.82 | −7.52 | −25.19 | −9.86 | −8.60 | 1.39 | 1.08 |
| R-Methanandamide53 | −29.66 | −10.38 | −10.57 | −22.18 | −8.89 | −8.36 | −1.57 | −2.21 |
| 2-AG53 | −29.28 | −10.31 | −9.93 | −24.23 | −9.55 | −9.39 | −0.95 | −0.54 |
| Anandamide53 | −27.69 | −10.02 | −9.90 | −19.37 | −7.98 | −7.84 | −1.79 | −2.06 |
| AM117455 | −25.09 | −9.55 | −9.53 | −16.34 | −7.00 | −7.48 | −1.89 | −2.05 |
| O-181127 | −24.13 | −9.38 | −9.52 | −19.74 | −8.10 | −8.37 | −0.79 | −1.16 |
| S-Methanandamide25 | −23.83 | −9.32 | −9.28 | −19.40 | −7.99 | −6.98 | −0.80 | −2.30 |
| PNR-4-2028 | −25.79 | −9.67 | −9.33 | −26.52 | −10.29 | −12.00 | 0.52 | 2.67 |
| PNR-4-0428 | −25.22 | −9.57 | −9.43 | −25.84 | −10.07 | −10.89 | 0.49 | 1.46 |
| PNR-4-0228 | −27.64 | −10.01 | −9.80 | −27.78 | −10.70 | −10.57 | 0.37 | 0.77 |
| PNR-4-0528 | −21.95 | −8.98 | −8.85 | −24.10 | −9.51 | −9.83 | 0.88 | 0.98 |
| PNR-4-9928 | −25.28 | −9.58 | −9.54 | −25.78 | −10.06 | −9.81 | 0.46 | 0.28 |
| PNR-9-3328 | −16.18 | −7.93 | −7.60 | −23.26 | −9.24 | −9.55 | 2.13 | 1.95 |
| PNR-4-0328 | −28.42 | −10.15 | −9.63 | −23.26 | −9.24 | −9.57 | −0.98 | −0.06 |
| PNR-4-1528 | −28.18 | −10.11 | −9.96 | −20.72 | −8.42 | −9.53 | −1.57 | −0.43 |
| PNR-4-1728 | −16.52 | −7.99 | −8.26 | −19.31 | −7.96 | −8.55 | 1.04 | 0.29 |
| RMSD (kcal/mol) | 0.35 | 0.75 | 0.75 | |||||
The subscript after the ligand name refers to the reference for the experimental Ki (shown in Figure 1 or 2) used to derive the experimental binding free energy.
Calculated binding free energies obtained from the MM-PBSA calculations without any empirical correction.
The experimental binding free energy was converted from the experimental Ki using the well-known thermodynamic equation: ΔGexp = −RTln (Ki).
ΔΔGcorr is the corrected binding free energy difference (ΔG(CB1R) −ΔG(CB2R)).
ΔΔGexp = −RTln (Ki (CB1R)/Ki(CB2R)) = ΔGexp(CB1R) −ΔGexp(CB2R).
Structure-activity and structure-selectivity correlation relationships of endocannabinoids
O-1860 represents the culmination of a series of additions to the endogenous cannabinoid anandamide, each step of which incrementally increases the binding affinity with CB1R. The first change to O-1860 from anandamide involves the substitution of two additional methyl groups onto carbon-17, turning it into a neopentane moiety. These additional methyl groups allow for additional vdW interactions with the primarily hydrophobic pocket formed by TM4 and TM5 including F268 and W279 (Figure 3C). Additionally, the substitution of the terminal hydroxyl group with a halogen increases the favorable vdW interactions with the mainly hydrophobic pocket between TM3 and TM4 (Figure 3C) over the endocannabinoid (R)-methanandamide (Figure 4A). The second modification of anandamide (Figure 4B) comes from an additional methyl group placed one carbon from the terminal hydroxyl group (Figure 4A) allowing for additional vdW interactions with I267.
Figure 4.

The binding modes of representative long-chain compounds in CB1R and CB2R. (A) CB1R binding with (R)-methanandamide. (B) CB1R binding with anandamide. The binding mode of O-1860 with CB1R consists of two hydrogen bonds with H178 and I267. Additionally, favorable hydrophobic interactions are created with F268 and W279. The removal of the neo-pentane and bromide on the long aliphatic chain further reduces the binding affinity with the CB1R receptor for (R)-methanandamide. The removal of the methyl group adjacent to the amide head group further removes the selectivity and potency to the original endocannabinoid anandamide. (C) Binding pose of PNR-4-20 with CB1R. (D) Binding pose of PNR-4-20 with CB2R. PNR-4-20 binds within the central binding pocket of the receptors, surrounded by several hydrophobic residues including F268 and W279 in CB1R and F183 and W194 in CB2R. The carbonyl formed hydrogen bond with His178/95 of CB1R/CB2R. A slight rotation in the central indole ring is the only major difference between these two binding poses, demonstrating the similarity in the binding pockets between the two receptors.
The binding mode of the IQD series of compounds
Furthermore, we used the IQD derivatives shown in Figure 2 to test our models. It is gratifying that the calculated results were in good agreement to the experimental data, suggesting that these models also can be used to the drug design for this series of compounds. Key to the binding of these compounds to each receptor is the carbonyl group on the quinuclidine ring accepting a hydrogen bond from the nearby H178/95 residue in CB1R/CB2R. Additionally, these compounds have strong hydrophobic interactions in both CB1R and CB2R with nearby F268 & W279 in CB1R (F183 & W194 in CB2R) (Figure S15 to S22). PNR-4-20 (Figure 4C and D) represents both a G-protein biased and potent CB1R agonist, only lacking in selectivity for one of the receptors (or limited selectivity for CB2R). This lack in selectivity for CB1R is readily apparent when one looks at the binding mode comparison (Figure 4C and D) where the only major change in position is a slight rotation in the central indole ring. The similarity of the central binding pocket for the CB receptors is the main hinderance towards developing selective CB receptor agonists. The binding poses proposed for the endocannabinoids and their analogs, shown in the above, give clues as to how these compounds could be modified to allow for CB receptor selectivity.
The correction of binding free energies
Summarized in Table 2 are the binding free energies obtained from the MM-PBSA calculations in comparison with the corresponding binding free energies for CB1R and CB2R with all of the 23 ligands shown in Figure 1 and 2. As seen in Table 2, our computational protocol performs similarly well with both the long-chain cannabinoids and the typical IQD derivatives. The relative magnitudes of the MM-PBSA binding free energy (ΔGPB) values are qualitatively consistent with the relative experimental binding free energies in terms of the structure-activity relationship (SAR) for each receptor and the receptor selectivity between CB1R and CB2R (Figure 5).
Figure 5.
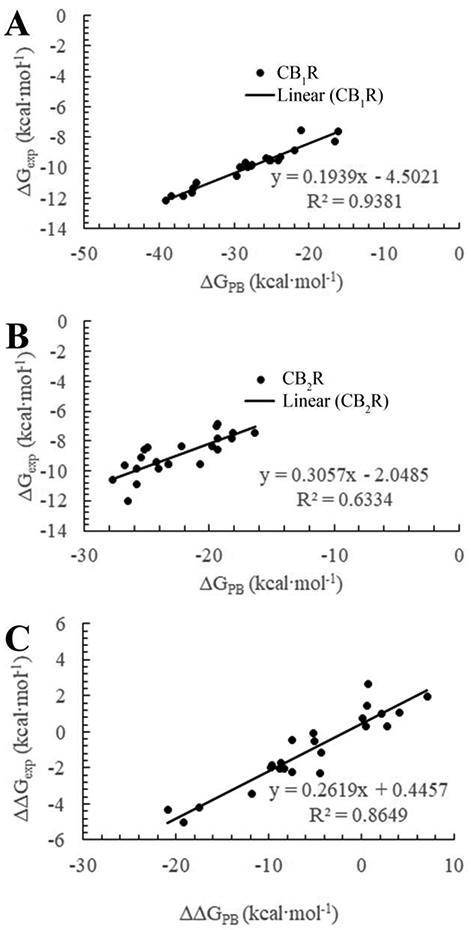
(A) Calculated binding free energy vs experimental binding free energy of ligands with CB1R. (B) Calculated binding free energy vs experimental binding free energy of ligands with CB2R. (C) Difference in experimental binding free energy vs the calculated binding free energy difference between CB1R and CB2R. These measures indicate that there is a strong correlation between the difference in the calculated and experimental binding free energy, allowing us to successfully predict the selectivity for a given ligand towards CB1R or CB2R as well as determine their relative affinity.
In terms of the absolute binding free energies, it is not surprising to note that the MM-PBSA calculations systematically overestimated the binding affinities of the ligands with both CB1R and CB2R. Nevertheless, the empirical linear correlation relationships indicated in Figure 5 may be used to empirically correct the calculated binding free energies. Particularly, for CB1R binding with ligands, we have
| (1) |
with a correlation coefficient (R2) of 0.9381 and a root-mean-squares deviation (RMSD) of 0.36 kcal/mol. For CB2R binding with ligands, we have
| (2) |
with R2 = 0.6334 and RMSD = 0.75 kcal/mol. In addition, for the difference in the binding free energy between CB1R and CB2R, we have
| (3) |
with R2 = 0.8649 and RMSD = 0.69 kcal/mol. In Eq. (3), ΔΔGPB = ΔGPB(CB1R) – ΔGPB(CB2R), and ΔΔGcorr is the corrected binding free energy difference reflecting the selectivity between CB1R and CB2R. In all of these equations, ΔGPB represents the binding free energy obtained directly from the MM-PBSA calculation, whereas ΔGcorr refers to the corrected binding free energy.
As seen in Table 2, the empirically corrected binding free energies with both CB1R and CB2R, as well as the difference between them, are all in excellent agreement with the corresponding experimental data, suggesting that the binding modes determined in this study are reasonable. Furthermore, our models are expected to be valuable for the rational drug design in the future.
In summary, using molecular docking, MD simulation, and MM-PBSA binding free energy calculations, we have been able to determine the binding modes of CB1R and CB2R interacting with both the IQD series of ligands and long-chain cannabinoids (including their synthetic analogs). Based on the obtained binding poses, the calculated relative binding free energies are in good agreement with the corresponding experimental binding affinity data in terms of the SAR for each receptor (CB1R or CB2R) and the receptor selectivity between CB1R and CB2R. The binding poses for the long-chain cannabinoids and their synthetic analogs implicate the site surrounded by the TM2, TM7, and ECL-2 regions being vital for providing the long-chain ligands with the selectivity for CB1R, especially the I267/L182 of CB1R/CB2R. Considering this computational insight, the future rational design of new selective ligands for CB1R and CB2R may be focused on favorable interactions with this site. Particularly, as we have also determined the binding poses of the IQD compounds including the unique G-protein biased agonist PNR-4-20 and PNR-4-02, new compounds may be designed that bring the features of both classes of molecules together, creating selective, potent, and G-protein biased IQD/endocannabinoid hybrids. The similar computational strategy used in this study may also prove fruitful in applications with other GCPRs or membrane-bound proteins.
Methods
Multiple sequences alignment
The amino-acid sequences of CB1R and CB2R were downloaded from the Uniprot database.61 The multiple sequence alignment was performed using the MUSCLE software, which also was used to calculate sequence identity.62 The figure for sequence alignment was generated using the ESPript 3.0 software.63
Docking and molecular dynamics simulations
The cannabinoid receptors (PDB ID: 5XRA for CB1R and 6PT0 for CB2R) were prepared using the PDB2PQR module to fix any potential errors with the models, and to additionally assign the ff14SB force field parameters to the constituent atoms.22, 35 The structures of ligands were built and energy-minimized using the SYBYL v2.0 software (Tripos Inc., St. Louis, USA). Initial poses for the ligands were predicted using the AutoDock 4.2 software with the default parameters.64 To improve the efficiency of calculation, the CB2R model was superimposed to the CB1R model. The grid size was set to 60 × 60 × 60, and the grid center was designated at −43.616, 164.787, 306.920. For each ligand, 265 possible binding poses were generated for further study. Each ligand had its atomic charges calculated through the AM1-BCC method in the Antechamber module and was subsequently energy-minimized through the Sander module of AMBER16 program before being placed within the binding site of each receptor.65-67
For the compound O-1860 binding with each receptor, the complexes were inserted into a POPC lipid bilayer through the use of the Membrane-Builder module of CHARMM-GUI.68 Then the complex structures were energy-minimized over five steps: an initial energy minimization where only hydrogens and lipid bilayer were energy-minimized, followed by energy minimization of the ligand, receptor hydrogens, and lipid bilayer, followed by sidechains of the receptor, residues within 6 Å of the ligand, and finally the entire system. Each of these steps consisted of a total of 3000 energy minimization steps. Then the MD simulation of the energy-minimized complex was performed using the CUDA accelerated PMEMD module of AMBER.65 After energy minimization and heating of the lipid/complex system, 100 ns of MD simulations were performed. One hundred snapshots of the last nanosecond of the system were used within the MM-PBSA module of the AMBER to make sure that the binding free energy based on the final snapshot is reasonably close to the average of binding free energies associated with the one hundred snapshots. The energy decomposition was performed using the decomposition option within the MMPBSA module of the AMBER16.
The last snapshot of the MD-simulated complex with O-1860 was subsequently used as the receptor with O-1860 and lipid bilayer removed. Each ligand (including O-1860) was then placed into the receptor based on the previously obtained binding poses from the AutoDock software.64 The whole complex structure was then energy-minimized, and had its binding free energy calculated using the same MM-PBSA methodology.
Supplementary Material
Acknowledgments
This work was supported in part by the funding of the Molecular Modeling and Biopharmaceutical Center at the University of Kentucky College of Pharmacy, the National Science Foundation (NSF grant CHE-1111761), and the National Institutes of Health (P20 GM130456, UL1TR001998, T32 DA016176, and R01 DA039143). The authors also acknowledge the Computer Center at University of Kentucky for supercomputing time on a Dell Supercomputer Cluster consisting of 388 nodes or 4,816 processors.
Footnotes
Supporting Information
Additional figures for the sequence alignment of human, mouse, and rat CB receptors, the correction of MM-PBSA binding free energies with the corresponding experimental binding free energies for the long-chain ligands, and the modeled binding structures of CB1R and CB2R with various ligands.
References
- 1.Hauser AS, Attwood MM, Rask-Andersen M, Schiöth HB, and Gloriam DE (2017) Trends in GPCR Drug Discovery: New Agents, Targets and Indications. Nature Reviews Drug Discovery 16 (12), 829–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Basu S, and Dittel BN (2011) Unraveling the Complexities of Cannabinoid Receptor 2 (CB2) Immune Regulation in Health and Disease. Immunol. Res 51 (1), 26–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baron EP, Lucas P, Eades J, and Hogue O (2018) Patterns of Medicinal Cannabis Use, Strain Analysis, and Substitution Effect among Patients with Migraine, Headache, Arthritis, and Chronic Pain in a Medicinal Cannabis Cohort. J. Headache Pain 19 (1), 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vučković S, Srebro D, Vujović KS, Vučetić Č, and Prostran M (2018) Cannabinoids and Pain: New Insights from Old Molecules. Front. Pharmacol. 9, 1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Abrams DI, Couey P, Shade SB, Kelly ME, and Benowitz NL (2011) Cannabinoid-Opioid Interaction in Chronic Pain. Clin. Pharmacol. Ther 90 (6), 844–851. [DOI] [PubMed] [Google Scholar]
- 6.Cichewicz DL, and McCarthy EA (2003) Antinociceptive Synergy between Delta(9)-Tetrahydrocannabinol and Opioids after Oral Administration. J. Pharmacol. Exp. Ther 304 (3), 1010–1015. [DOI] [PubMed] [Google Scholar]
- 7.Lötsch J, Weyer‐Menkhoff I, and Tegeder I (2018) Current Evidence of Cannabinoid‐Based Analgesia Obtained in Preclinical and Human Experimental Settings. Eur. J. Pain 22 (3), 471–484. [DOI] [PubMed] [Google Scholar]
- 8.Benedetti F, Amanzio M, Rosato R, and Blanchard C (2011) Nonopioid Placebo Analgesia Is Mediated by CB1 Cannabinoid Receptors. Nat. Med 17 (10), 1228–1230. [DOI] [PubMed] [Google Scholar]
- 9.Agarwal N, Pacher P, Tegeder I, Amaya F, Constantin CE, Brenner GJ, Rubino T, Michalski CW, Marsicano G, and Monory K (2007) Cannabinoids Mediate Analgesia Largely Via Peripheral Type 1 Cannabinoid Receptors in Nociceptors. Nat. Neurosci 10 (7), 870–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Burns TL, and Ineck JR (2006) Cannabinoid Analgesia as a Potential New Therapeutic Option in the Treatment of Chronic Pain. Ann. Pharmacother 40 (2), 251–260. [DOI] [PubMed] [Google Scholar]
- 11.Walker JM, and Huang SM (2002) Cannabinoid Analgesia. Pharmacol. Ther. 95 (2), 127–135. [DOI] [PubMed] [Google Scholar]
- 12.Abdallah FW, Hussain N, Weaver T, and Brull R (2020) Analgesic Efficacy of Cannabinoids for Acute Pain Management after Surgery: A Systematic Review and Meta-Analysis. Reg. Anesth. Pain Med 45 (7), 509–519. [DOI] [PubMed] [Google Scholar]
- 13.Wiese B, and Wilson-Poe AR (2018) Emerging Evidence for Cannabis’ Role in Opioid Use Disorder. Cannabis Cannabinoid Res. 3 (1), 179–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rice A (2001) Cannabinoids and Pain. Curr. Opin. Investig. Drugs 2 (3), 399. [PubMed] [Google Scholar]
- 15.Rossi F, Punzo F, Umano GR, Argenziano M, and Miraglia Del Giudice E (2018) Role of Cannabinoids in Obesity. Int. J. Mol. Sci 19 (9), 2690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pertwee RG (2012) Targeting the Endocannabinoid System with Cannabinoid Receptor Agonists: Pharmacological Strategies and Therapeutic Possibilities. Philos. Trans. R. Soc. Lond., B, Biol. Sci 367 (1607), 3353–3363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pertwee RG (2009) Emerging Strategies for Exploiting Cannabinoid Receptor Agonists as Medicines. Br. J. Pharmacol 156 (3), 397–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shahbazi F, Grandi V, Banerjee A, and Trant JF (2020) Cannabinoids and Cannabinoid Receptors: The Story So Far. iScience, 101301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Song Z-H, Slowey C-A, Hurst DP, and Reggio PH (1999) The Difference between the CB1 and CB2cannabinoid Receptors at Position 5.46 Is Crucial for the Selectivity of WIN55212-2 for CB2. Molecular pharmacology 56 (4), 834–840. [PubMed] [Google Scholar]
- 20.Tuccinardi T, Ferrarini PL, Manera C, Ortore G, Saccomanni G, and Martinelli A (2006) Cannabinoid CB2/CB1 Selectivity. Receptor Modeling and Automated Docking Analysis. J. Med. Chem 49 (3), 984–994. [DOI] [PubMed] [Google Scholar]
- 21.Hua T, Vemuri K, Nikas SP, Laprairie RB, Wu Y, Qu L, Pu M, Korde A, Jiang S, and Ho J-H (2017) Crystal Structures of Agonist-Bound Human Cannabinoid Receptor CB1. Nature 547 (7664), 468–471. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 22.Hua T, Vemuri K, Pu M, Qu L, Han GW, Wu Y, Zhao S, Shui W, Li S, and Korde A (2016) Crystal Structure of the Human Cannabinoid Receptor CB1. Cell 167 (3), 750–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Herzberg U, Eliav E, Bennett G, and Kopin IJ (1997) The Analgesic Effects of R (+)-Win 55,212–2 Mesylate, a High Affinity Cannabinoid Agonist, in a Rat Model of Neuropathic Pain. Neurosci. Lett 221 (2-3), 157–160. [DOI] [PubMed] [Google Scholar]
- 24.Diaz P, Phatak SS, Xu J, Astruc-Diaz F, Cavasotto CN, and Naguib M (2009) 6-Methoxy-N-Alkyl Isatin Acylhydrazone Derivatives as a Novel Series of Potent Selective Cannabinoid Receptor 2 Inverse Agonists: Design, Synthesis, and Binding Mode Prediction. J. Med. Chem 52 (2), 433–444. [DOI] [PubMed] [Google Scholar]
- 25.Vemuri VK, and Makriyannis A, Endocannabinoids and Their Synthetic Analogs In The Cannabinoid Receptors, Springer: 2009; pp 21–48. [Google Scholar]
- 26.ME PRHAA, Elphick ASDMV, and Kunos MGPHHG Mackie K 2010. International Union of Basic and Clinical Pharmacology. Lxxix. Cannabinoid Receptors and Their Ligands: Beyond CB1 and CB2. Pharmacol. Rev 62, 588–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Di Marzo V, Bisogno T, De Petrocellis L, Brandi I, Jefferson R, Winckler R, Davis J, Dasse O, Mahadevan A, and Razdan R (2001) Highly Selective CB1 Cannabinoid Receptor Ligands and Novel CB1/Vr1 Vanilloid Receptor “Hybrid” Ligands. Biochem. Biophys. Res. Commun 281 (2), 444–451. [DOI] [PubMed] [Google Scholar]
- 28.Madadi NR, Penthala NR, Brents LK, Ford BM, Prather PL, and Crooks PA (2013) Evaluation of (Z)-2-((1-Benzyl-1h-Indol-3-Yl)Methylene)-Quinuclidin-3-One Analogues as Novel, High Affinity Ligands for CB1 and CB2 Cannabinoid Receptors. Bioorg. Med. Chem. Lett 23 (7), 2019–2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ford BM, Franks LN, Tai S, Fantegrossi WE, Stahl EL, Berquist MD, Cabanlong CV, Wilson CD, Penthala NR, and Crooks PA (2017) Characterization of Structurally Novel G Protein Biased CB1 Agonists: Implications for Drug Development. Pharmacol. Res 125, 161–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kendall D, Alexander S, Priestley R, Asghar M, and von Mentzer B (2017) Biased Agonism at Cannabinoid CB1 and Δ-Opioid Receptors; Therapeutic Potential. Neuropeptides 65, 131. [Google Scholar]
- 31.Newman-Tancredi A (2011) Biased Agonism at Serotonin 5-Ht1a Receptors: Preferential Postsynaptic Activity for Improved Therapy of Cns Disorders. Neuropsychiatry 1 (2), 149. [Google Scholar]
- 32.Rankovic Z, Brust TF, and Bohn LM (2016) Biased Agonism: An Emerging Paradigm in Gpcr Drug Discovery. Bioorg. Med. Chem. Lett 26 (2), 241–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Whalen EJ, Rajagopal S, and Lefkowitz RJ (2011) Therapeutic Potential of Β-Arrestin-and G Protein-Biased Agonists. Trends Mol. Med 17 (3), 126–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hua T, Li X, Wu L, Iliopoulos-Tsoutsouvas C, Wang Y, Wu M, Shen L, Johnston CA, Nikas SP, and Song F (2020) Activation and Signaling Mechanism Revealed by Cannabinoid Receptor-Gi Complex Structures. Cell 180 (4), 655–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xing C, Zhuang Y, Xu T-H, Feng Z, Zhou XE, Chen M, Wang L, Meng X, Xue Y, and Wang J (2020) Cryo-Em Structure of the Human Cannabinoid Receptor CB2-Gi Signaling Complex. Cell 180 (4), 645–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li X, Hua T, Vemuri K, Ho J-H, Wu Y, Wu L, Popov P, Benchama O, Zvonok N, and Qu L (2019) Crystal Structure of the Human Cannabinoid Receptor CB2. Cell 176 (3), 459–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Salo OM, Lahtela-Kakkonen M, Gynther J, Järvinen T, and Poso A (2004) Development of a 3d Model for the Human Cannabinoid CB1 Receptor. J. Med. Chem 47 (12), 3048–3057. [DOI] [PubMed] [Google Scholar]
- 38.Sapundzhi F, and Dzimbova T (2020) A Computational Study of Cannabinoid Receptors and Cannabinoid Ligands. J. Chem. Technol. Metall 55 (5), 959–964. [Google Scholar]
- 39.Reggio PH, and Traore H (2000) Conformational Requirements for Endocannabinoid Interaction with the Cannabinoid Receptors, the Anandamide Transporter and Fatty Acid Amidohydrolase. Chem. Phys. Lipids 108 (1-2), 15–35. [DOI] [PubMed] [Google Scholar]
- 40.Lynch DL, and Reggio PH (2005) Molecular Dynamics Simulations of the Endocannabinoid N-Arachidonoylethanolamine (Anandamide) in a Phospholipid Bilayer: Probing Structure and Dynamics. J. Med. Chem 48 (15), 4824–4833. [DOI] [PubMed] [Google Scholar]
- 41.Reggio PH (2010) Endocannabinoid Binding to the Cannabinoid Receptors: What Is Known and What Remains Unknown. Curr. Med. Chem 17 (14), 1468–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lin S, Khanolkar AD, Fan P, Goutopoulos A, Qin C, Papahadjis D, and Makriyannis A (1998) Novel Analogues of Arachidonylethanolamide (Anandamide): Affinities for the CB1 and CB2 Cannabinoid Receptors and Metabolic Stability. J. Med. Chem. 41 (27), 5353–61. [DOI] [PubMed] [Google Scholar]
- 43.Dainese E, Gasperi V, and Maccarrone M (2005) Partial Qsar Analysis of Some Selected Natural Inhibitors of Faah Suggests a Working Hypothesis for the Development of Endocannabinoid-Based Drugs. CNS Neurol. Disord. Drug Targets 4 (6), 709–714. [DOI] [PubMed] [Google Scholar]
- 44.McAllister SD, Rizvi G, Anavi-Goffer S, Hurst DP, Barnett-Norris J, Lynch DL, Reggio PH, and Abood ME (2003) An Aromatic Microdomain at the Cannabinoid CB1 Receptor Constitutes an Agonist/Inverse Agonist Binding Region. J. Med. Chem 46 (24), 5139–5152. [DOI] [PubMed] [Google Scholar]
- 45.Perez A, Morrone JA, Simmerling C, and Dill KA (2016) Advances in Free-Energy-Based Simulations of Protein Folding and Ligand Binding. Curr. Opin. Struct. Biol 36, 25–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Loo JS, Yong AY, and Yong YN (2020) The Effect of Multiple Simulation Parameters on MM/PBSA Performance for Binding Affinity Prediction of CB1 Cannabinoid Receptor Agonists and Antagonists. Chem. Biol. Drug Des [Online early access]. DOI: 10.1111/cbdd.13733. Published Online: May 27, 2020. https://onlinelibrary.wiley.com/doi/abs/10.1111/cbdd.13733 (accessed Aug 11, 2020). [DOI] [PubMed] [Google Scholar]
- 47.Huang X, Zheng F, and Zhan C-G (2008) Modeling Differential Binding of Α4β2 Nicotinic Acetylcholine Receptor with Agonists and Antagonists. J. Am. Chem. Soc 130 (49), 16691–16696. [DOI] [PubMed] [Google Scholar]
- 48.Huang X, Zheng F, Chen X, Crooks PA, Dwoskin LP, and Zhan C-G (2006) Modeling Subtype-Selective Agonists Binding with Α4β2 and Α7 Nicotinic Acetylcholine Receptors: Effects of Local Binding and Long-Range Electrostatic Interactions. J. Med. Chem 49 (26), 7661–7674. [DOI] [PubMed] [Google Scholar]
- 49.Huang X, Zheng F, Crooks PA, Dwoskin LP, and Zhan C-G (2005) Modeling Multiple Species of Nicotine and Deschloroepibatidine Interacting with Α4β2 Nicotinic Acetylcholine Receptor: From Microscopic Binding to Phenomenological Binding Affinity. J. Am. Chem. Soc 127 (41), 14401–14414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jin Y, Huang X, Papke RL, Jutkiewicz EM, Showalter HD, and Zhan C-G (2017) Design, Synthesis, and Biological Activity of 5′-Phenyl-1, 2, 5, 6-Tetrahydro-3, 3′-Bipyridine Analogues as Potential Antagonists of Nicotinic Acetylcholine Receptors. Bioorg. Med. Chem. Lett 27 (18), 4350–4353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Huang X, Zheng F, Stokes C, Papke RL, and Zhan C-G (2008) Modeling Binding Modes of Α7 Nicotinic Acetylcholine Receptor with Ligands: The Roles of Gln117 and Other Residues of the Receptor in Agonist Binding. J. Med. Chem 51 (20), 6293–6302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hillard CJ, Manna S, Greenberg MJ, DiCamelli R, Ross RA, Stevenson LA, Murphy V, Pertwee RG, and Campbell WB (1999) Synthesis and Characterization of Potent and Selective Agonists of the Neuronal Cannabinoid Receptor (CB1). J. Pharmacol. Exp. Ther 289 (3), 1427–1433. [PubMed] [Google Scholar]
- 53.Thakur GA, Nikas SP, and Makriyannis A (2005) CB1 Cannabinoid Receptor Ligands. Mini Rev. Med. Chem 5 (7), 631–640. [DOI] [PubMed] [Google Scholar]
- 54.Willinsky M Composition and Methods to Improve Stability, Dosing, Pharmacodynamics and Product Shelf Life of Endocannabinoids, Phytocannabinoids and Synthetic Cannabinoids Delivered by Nasal Inhaer. US20180000727A1, 2018. [Google Scholar]
- 55.Lin S, Khanolkar AD, Fan P, Goutopoulos A, Qin C, Papahadjis D, and Makriyannis A (1998) Novel Analogues of Arachidonylethanolamide (Anandamide): Affinities for the CB1 and CB2 Cannabinoid Receptors and Metabolic Stability. J. Med. Chem 41 (27), 5353–5361. [DOI] [PubMed] [Google Scholar]
- 56.Felder CC, Joyce KE, Briley EM, Mansouri J, Mackie K, Blond O, Lai Y, Ma AL, and Mitchell RL (1995) Comparison of the Pharmacology and Signal Transduction of the Human Cannabinoid CB1 and CB2 Receptors. Mol. Pharmacol 48 (3), 443–450. [PubMed] [Google Scholar]
- 57.Adams IB, Ryan W, Singer M, Thomas BF, Compton DR, Razdan RK, and Martin BR (1995) Evaluation of Cannabinoid Receptor Binding and in Vivo Activities for Anandamide Analogs. J. Pharmacol. Exp. Ther 273 (3), 1172–1181. [PubMed] [Google Scholar]
- 58.Wiley JL, Dewey MA, Jefferson RG, Winckler RL, Bridgen DT, Willoughby KA, and Martin BR (2000) Influence of Phenylmethylsulfonyl Fluoride on Anandamide Brain Levels and Pharmacological Effects. Life Sci. 67 (13), 1573–1583. [DOI] [PubMed] [Google Scholar]
- 59.Compton DR, and Martin BR (1997) The Effect of the Enzyme Inhibitor Phenylmethylsulfonyl Fluoride on the Pharmacological Effect of Anandamide in the Mouse Model of Cannabimimetic Activity. J. Pharmacol. Exp. Ther 283 (3), 1138–1143. [PubMed] [Google Scholar]
- 60.Laine K, Järvinen K, Pate DW, Urtti A, and Järvinen T (2002) Effect of the Enzyme Inhibitor, Phenylmethylsulfonyl Fluoride, on the Iop Profiles of Topical Anandamides. Invest. Ophthalmol. Vis. Sci 43 (2), 393–397. [PubMed] [Google Scholar]
- 61.UniProt C (2008) The Universal Protein Resource (Uniprot). Nucleic Acids Res. 36 (Database issue), D190–D195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Edgar RC (2004) Muscle: Multiple Sequence Alignment with High Accuracy and High Throughput. Nucleic Acids Res. 32 (5), 1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Robert X, and Gouet P (2014) Deciphering Key Features in Protein Structures with the New Endscript Server. Nucleic Acids Res. 42 (W1), W320–W324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, and Olson AJ (2009) Autodock4 and Autodocktools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem 30 (16), 2785–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pearlman DA, Case DA, Caldwell JW, Ross WS, Cheatham TE III, DeBolt S, Ferguson D, Seibel G, and Kollman P (1995) Amber, a Package of Computer Programs for Applying Molecular Mechanics, Normal Mode Analysis, Molecular Dynamics and Free Energy Calculations to Simulate the Structural and Energetic Properties of Molecules. Comput. Phys. Commun 91 (1-3), 1–41. [Google Scholar]
- 66.Jakalian A, Jack DB, and Bayly CI (2002) Fast, Efficient Generation of High-Quality Atomic Charges. Am1-Bcc Model: II. Parameterization and Validation. J. Comput. Chem 23 (16), 1623–41. [DOI] [PubMed] [Google Scholar]
- 67.Case DA, Cheatham TE 3rd, Darden T, Gohlke H, Luo R, Merz KM Jr., Onufriev A, Simmerling C, Wang B, and Woods RJ (2005) The Amber Biomolecular Simulation Programs. J. Comput. Chem 26 (16), 1668–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jo S, Kim T, Iyer VG, and Im W (2008) CHARMM-GUI: A Web-Based Graphical User Interface for Charmm. J. Comput. Chem 29 (11), 1859–1865. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.