

Summary
CRISPR-Cas9 genome engineering can be used to functionally investigate the complex mechanisms of immune system regulation. Decades of work have aimed to genetically reprogram innate immunity, but current approaches are inefficient or nonspecific, limiting their use. Here, we detail an optimized strategy for non-viral CRISPR-Cas9 ribonucleoprotein (cRNP) genomic editing of primary innate lymphocytes (ILCs) and myeloid lineage cells, resulting in high-efficiency editing of target gene expression from a single electroporation.
For complete details on the use and execution of this protocol, please refer to Riggan et al. (2020).
Graphical Abstract
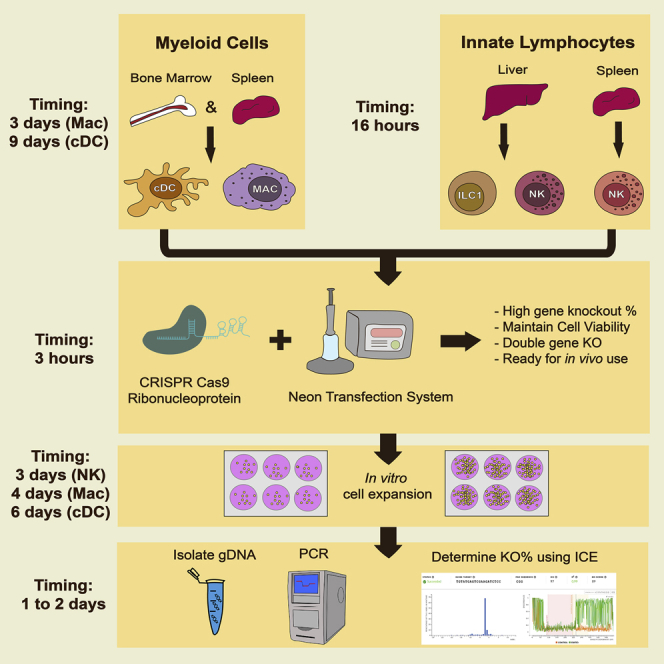
Highlights
-
•
Isolation of precursors and mature innate immune cells from peripheral tissues
-
•
Magnetic purification of group 1 innate lymphoid cells from spleen and liver
-
•
CRISPR RNP electroporation of primary innate immune cells
-
•
Analysis of gene editing efficiency via flow cytometry and Sanger sequencing
CRISPR-Cas9 genome engineering can be used to functionally investigate the complex mechanisms of immune system regulation. Decades of work have aimed to genetically reprogram innate immunity, but current approaches are inefficient or nonspecific, limiting their use. Here, we detail an optimized strategy for non-viral CRISPR-Cas9 ribonucleoprotein (cRNP) genomic editing of primary innate lymphocytes (ILCs) and myeloid lineage cells, resulting in high-efficiency editing of target gene expression from a single electroporation.
Before You Begin
It will be useful to have digital annotations of your gene(s) of interest in a program such as Benchling, SnapGene, or ApE. These platforms can be used to locate your CRISPR guide sequences and the associated primers used to PCR the region surrounding the cut site in order to check the editing efficiency of your reaction later in the protocol. To start, it is useful to source guides from previously published whole genome CRISPR screens (Ex: Wang et al., 2017). We recommend testing 3–5 guide targets from this list per gene until a high-efficiency guide is identified experimentally. Each guide will require a PCR product which flanks the CRISPR cut side by about 300 bp (±100) in both directions. We find it most useful to target exonic regions that contain multiple guide sequences to reduce the number of PCR reactions that must be optimized. Before beginning this protocol, ensure that you have optimized the PCR for your targeted genomic region.
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-Mouse NK1.1 | BioLegend | PK136 Cat# 108727 |
| Anti-Mouse CD3e | BioLegend | 17A2 Cat# 100222 |
| Anti-Mouse CD11b | BioLegend | M1/70 Cat# 101222 |
| Anti-Mouse CD19 | BioLegend | 6D5 Cat# 115530 |
| Anti-Mouse CD49b/DX5 | BioLegend | DX5 Cat# 103517 |
| Anti-Mouse KLRG1 | BioLegend | 2F1 Cat# 138416 |
| Anti-Mouse CD45.2 | BioLegend | 104 Cat# 109821 |
| Anti-Mouse TCRβ | BioLegend | H57-597 Cat# 109220 |
| Anti-Mouse CD200r1 | BioLegend | OX-110 Cat# 123915 |
| Anti-Mouse CD11c | BioLegend | N418 Cat# 117318 |
| Anti-Mouse XCR1 | BioLegend | ZET Cat# 148204 |
| Anti-Mouse MHCII | BioLegend | M5/114.15.2 Cat# 107625 |
| Anti-Mouse CD64 | BioLegend | X54-5/7.1 Cat# 139306 |
| Anti-Cas9 | Cell Signaling Technology | 7A9-3A3 Cat# 35193 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Cas9-NLS | SYNTHEGO | N/A |
| Cas9-NLS | qb3 UC Berkley | N/A |
| Alt-R® Cas9 Electroporation Enhancer, 10 nmol | IDT | Cat# 1075916 |
| Synthetic Guide RNAs | SYNTHEGO | N/A |
| Recombinant mFLT3-L | Peprotech | Cat# 250-31L |
| Recombinant mGM-CSF | Peprotech | Cat# 315-03 |
| Recombinant mM-CSF | Peprotech | Cat# 315-02 |
| Recombinant mIL-15 | Peprotech | Cat# 210-15 |
| RPMI Medium 1640 (Plus L-Glutamine, Plus 25 mM HEPES) | Gibco | Cat# 22400-089 |
| DMEM (1×) (Plus 4.5 g/L D-Glucose) | Gibco | Cat# 11960-044 |
| Heat-Inactivated Fetal Bovine Serum | Gibco | Cat# F4135 |
| L-Glutamine 200 mM (100×) | Gibco | Cat# 25030-081 |
| Sodium Pyruvate (100 mM) | Gibco | Cat# 11360-070 |
| MEM-NEAA (100×) | Gibco | Cat# 11140-050 |
| Penicillin-Streptomycin (100×) | Gibco | Cat# 10378-016 |
| 2-mercaptoethanol (55 mM) | ThermoFisher | Cat# 21985023 |
| Percoll | VWR | Cat# 17-0891-02 |
| 10× HBSS | Gibco | Cat# 14185052 |
| Critical Commercial Assays | ||
| DNeasy Blood & Tissue Kit | QIAGEN | Cat# 69504 |
| EasySep™ Mouse NK Cell Isolation Kit | Stem Cell | Cat# 19855 |
| EasySep™ Buffer | Stem Cell | Cat# 20104 |
| Experimental Models: Organisms/Strain | ||
| Mouse: C57BL/6 (CD45.2) | Jackson Lab | Stock # 000664 |
| Mouse: B6.SJL (CD45.1) | Jackson Lab | Stock # 002114 |
| Oligonucleotides | ||
| sgRNA targeting sequence: CD11c #1 AAGAGCTCTCACCAACAGCC | Wang et al., 2017 | sgItgax_9 |
| sgRNA targeting sequence: CD11b #1 AGTGTGACTACAGCACAAGC | Wang et al., 2017 | sgItgam_3 |
| sgRNA targeting sequence: NK1.1 #2 GAGGAAGGTCAAGCTGACTG | Wang et al., 2017 | sgKlrb1c_2 |
| Software and Algorithms | ||
| FlowJo, Version 9.9.6 | Ashland, OR: Becton, Dickinson and Company | https://www.flowjo.com/solutions/flowjo |
| Prism | GraphPad | https://www.graphpad.com/scientific-software/prism |
| ICE Analysis | SYNTHEGO | https://ice.synthego.com/ |
| Other | ||
| Neon Transfection System | ThermoFisher | Cat# MPK5000 |
| Neon 100μL Transfection Kit | ThermoFisher | Cat# MPK10096 |
| Dounce Homogenizer | Corning | Cat# 1234F37 |
| Porcelain Mortar | FisherScientific | Cat# FB961A |
| Porcelain Pestle | FisherScientific | Cat# FB961K |
| 100μm Nitex mesh | FisherScientific | Cat# NC0486649 |
| FACS tubes | Falcon | Cat# 38007 |
| Falcon Round Bottom Tubes, 14 mL | FisherScientific | Cat# 50-197-4781 |
| EasyEights™ EasySep™ Magnet | Stem Cell | Cat# 18103 |
| Greiner Cell Scrapers | Sigma Aldrich | Cat# C5981-100ea |
| Microscope Slides | VWR | Cat# 89085-399 |
| NanoDrop OneC Microvolume UV-Vis Spectrophotometer | ThermoFisher | Cat# ND-ONE-W |
Materials and Equipment
Media should be kept at 4°C, then warmed to 37°C for use.
Adjusted Percoll should be made on the day of the experiment and should be approximately 22°C upon use.
| Reagent: CR-10 | Final Concentration | Volume (mL) |
|---|---|---|
| RPMI Medium 1640 | n/a | 427 |
| HEPES | 25 mM | n/a |
| Heat-Inactivated Fetal Bovine Serum | 10% | 50 |
| L-Glutamine | 1% | 5 |
| Sodium Pyruvate (200 mM) | 1% | 5 |
| MEM-NEAA | 1% | 5 |
| Penicillin-Streptomycin | 1% | 5 |
| Sodium Bicarbonate | 0.5% | 2.5 |
| 2-mercaptoethanol (55 mM) | 0.01% | 0.5 |
| Total | n/a | 500 |
| Reagent: LHM | Final Concentration | Volume (mL) |
|---|---|---|
| RPMI 1640 | n/a | 427.5 |
| HEPES | 25 mM | n/a |
| Heat-Inactivated Fetal Bovine Serum | 2.5% | 12.5 |
| L-Glutamine | 1% | 5 |
| Penicillin-Streptomycin | 1% | 5 |
| EDTA 10 mM | 1 mM | 50 |
| Total | n/a | 500 |
| Reagent: DC Media | Final Concentration (ng/mL) | Volume (μL) |
|---|---|---|
| CR-10 | n/a | n/a |
| mFLT3-L | 200 | n/a |
| mGM-CSF | 50 | n/a |
| Total | n/a | n/a |
| Reagent: Macrophage Media | Final Concentration | Volume (mL) |
|---|---|---|
| DMEM (1×) | n/a | 435 |
| Heat-Inactivated Fetal Bovine Serum | 10% | 50 |
| L-Glutamine | 1% | 5 |
| Sodium Pyruvate (200 mM) | 1% | 5 |
| Penicillin-Streptomycin | 1% | 5 |
| mM-CSF | 50 ng/mL | n/a |
| Total | n/a | 500 |
| Reagent: NK Media | Final Concentration (ng/mL) | Volume (μL) |
|---|---|---|
| CR-10 media | n/a | n/a |
| mIL-15 | 50 | n/a |
| Total | n/a | n/a |
| Reagent: Adjusted Percoll | Final Concentration | Volume (mL) |
|---|---|---|
| 10× HBSS | n/a | 1 |
| 100% Percoll | n/a | 10 |
| Total | n/a | 11 |
| Reagent: 40% or 60% Liver-prep Percoll | Final Concentration | Volume (mL) |
|---|---|---|
| Adjusted Percoll | 40% or 60% | 4 or 6 |
| 1× PBS | 60% or 40% | 6 or 4 |
| Total | n/a | 10 |
Step-By-Step Method Details
Splenic Natural Killer (NK) Cell Isolation
Timing: 30 min
Before innate immune cell populations can be edited, they must first be isolated ex vivo.
-
1.
Remove spleen(s) from the mice and place in a 6-well plate containing 4 mL LHM on ice.
-
2.
Disrupt spleen within the well using the rough sides of two microscope slides.
-
a)
Push slides together to squeeze out contents of the spleen. Continue until the spleen is no longer visibly red in color.
-
3.
Pipette liquid through 100 μm nitex mesh covering a 15-mL centrifuge tube on ice.
-
4.
Centrifuge at 450 × g for 3 min at 4°C.
-
5.
Aspirate the fluid above the pellet.
-
6.
Add 1 mL RoboSep buffer, pipette up and down to mix well for up to 1 x 108 nucleated cells (~1 spleen).
Note: No need to perform RBC lysis, as the isolation kit removes red blood cells
Liver Group 1 ILC Isolation
Before innate immune cell populations can be edited, they must first be isolated ex vivo.
-
7.
Transfer liver tissue to dounce tissue homogenizer using forceps and add 5 mL of PBS. Dounce tissue slowly and firmly. Transfer homogenized liver into a 15 mL conical tube.
-
8.
Pellet the homogenized liver by centrifugation at 450 × g for 3 min at 22°C and carefully decant supernatant slowly.
-
9.
Resuspend homogenized liver in 5 mL of PBS and pellet the homogenized liver by centrifugation at 450 × g for 3 min at 22°C.
-
10.
Resuspend pellet in 3 mL of liver-prep 40% Percoll and carefully overlay 3 mL of liver prep onto 60% Percoll using a 5 mL serological pipette to create a gradient.
-
11.
Allow gradients to settle for 10–15 min.
-
12.
Centrifuge samples at 800 × g without brake for 15 min at 22°C.
-
13.
Aspirate top layer of suspension, and pipet liver leukocytes at the interphase of the gradient into a new 15 mL conical capped with 100 μm nitex mesh to strain debris from cell suspension.
-
14.
Wash cells with 8 mL PBS and then pellet cells by centrifugation at 800 × g for 5 min.
-
15.
Resuspend liver leukocytes in 1 mL RoboSep buffer and transfer samples to a 5 mL FACS tube (up to 5 livers can be pooled into 1 mL for isolation).
Note: Do not perform RBC lysis
Group 1 ILC Magnetic Purification
This protocol is adapted from the STEM CELL EasySep™ Mouse NK Cell Isolation Kit
-
16.
Obtain 1 EasySep™ Mouse NK Cell Isolation Kit per 10 spleens to be isolated.
-
17.
Use 5 mL FACS tubes for samples of 1–2 mL and 14 mL round bottom tubes for samples of 2–5 mL.
-
18.
Resuspend cell suspension using P1000 pipette and add 50 μL isolation cocktail per 1 mL sample solution. (e.g., 250 μL for 5 mL sample containing 5 spleens).
-
a)
Cap tube and invert to mix
-
b)
Let sit at 22°C for 10 min
-
19.
Vortex RapidSpheres™.
Note: Particles should appear evenly dispersed.
-
20.
Add 100 μL RapidSpheres™ per 1 mL sample solution.
-
a)
Cap tube and invert to mix.
-
b)
Let sit at 22°C for 5 min.
-
c)
Invert every 90 s to keep RapidSpheres™ in solution.
-
21.
Add RoboSep buffer to top up sample to the indicated volume (up to 2.5 mL for 5 mL tube and up to 10 mL for 14 mL tube)
-
a)
Mix by gently pipetting up and down 2 to 3 times
-
22.
Place the tube (without lid) into the EasyEights™ EasySep™ Magnet and incubate for 5 min
-
23.
Carefully pipette using P1000 for 5 mL tubes or 10 mL serological (do not pour) the enriched cell suspension into a new 15 mL conical tube.
Note: Do not repeat isolation protocol for increased cell number, this results in much lower overall purity
-
24.
This tube contains purified Group 1 ILCs. Save 1% of your total volume and check the purity by flow cytometry. Cells should be 85%–90% NK1.1 + at this point. See Figure 1A.
Figure 1.

Analysis of Isolated NK Cell and BM-cDC1 Phenotypes
(A) Representative gating strategy for the identification of purified splenic NK cells.
(B) Representative gating strategy and phenotypic analysis of BM-cDC1.
Group 1 ILC Activation
-
25.
Pellet cells by centrifugation at 450 × g for 5 min.
-
26.
Resuspend in 1 mL pre-warmed CR-10 media containing 50 ng/mL recombinant murine IL-15 using P1000 pipette
-
27.
Add appropriate volume of CR-10 + IL-15 according to Table 1
Table 1.
Culture Conditions for IL-15-Activated NK Cells
| Number of Spleens | Purified Cell Number | Well Plate | Media Volume (mL) |
|---|---|---|---|
| 1 | 5 × 105-1 × 106 | 48 | 1 |
| 2 | 1 × 106–2 × 106 | 24 | 2 |
| 3–6 | 2 × 106–4 × 106 | 12 | 4 |
| 6–8 | 4 × 106–6 × 106 | 6 | 6 |
-
28.
Culture cells for 16–24 h in TC treated plates according to Table 1.
-
29.
Proceed to step 36, cRNP Treatment and Electroporation of 24-h-activated Liver Group 1 ILCs
Bone-Marrow-Derived cDC1 (BM-cDC1) and Macrophage (BMDM) Production
-
30.
Day 0: Harvest Bone Marrow
-
a)
Sacrifice mouse and proceed to isolate tibia, fibula, femur
-
i.
Cut at the ankle joint and at the hip and then place legs into 6 cm petri dish on ice until ready to remove muscle
-
b)
Clean bones prior to crushing
-
i.
Scrape muscle and fat off of leg bones, holding with forceps and scraping with scissors
-
c)
Place cleaned bones in 5 mL LHM in 15 mL conical on ice until all bones are cleaned
-
31.
Process Bone Marrow to single cell suspension
-
a)
Wash bones with 5 mL 70% Ethanol (1×) and 5 mL LHM (2×).
-
b)
Pour into mortar and crush with pestle until bones are white.
-
c)
Aspirate LHM-bone marrow mixture and pass through 100 μm nitex mesh into new 15 mL conical.
-
d)
Add another 5 mL LHM and crush bones again, filtering through 100 μm nitex mesh into the same 15 mL conical as before
-
e)
Centrifuge bone marrow mixture at 450 × g for 3 min at 4°C
-
f)
Resuspend pellet in 1 mL LHM and add 1 mL ACK lysis buffer. Lyse on ice for 2.5 min.
-
g)
Centrifuge mixture at 450 × g for 3 min at 4°C
-
h)
Resuspend pellet in 10 mL DC or Macrophage media depending on the target cell type
-
32.
Plate bone marrow suspension
Note: If making BMDC1, add cytokines to DC media:
-
a)
Final concentration 200 ng/mL FLT3-L
-
b)
Final concentration 5 ng/mL GM-CSF
Note: Macrophage media already contains the necessary cytokines
-
i.
Add 5 mL DC or Macrophage media to two 10 cm non-TC-treated dishes
-
ii.
Add 5 mL resuspended bone marrow mixture to each, making a total of 10 mL in each dish
-
33.
Day 3: Macrophage precursors are ready for RNP treatment.
-
34.
Day 5: For BMDC1 only: Add 5 mL DC media (no cytokines necessary) to each dish.
-
35.
Day 9: For BMDC1 only: Conventional dendritic cell precursors (cDCP) are ready for RNP treatment.
CRITICAL: For either BMDC1 or BMDM production, bone marrow single cell suspensions should spend minimal time in ACK lysis buffer. Quenching with excess LHM will help to minimize cell death, but removal right after centrifugation works fine
cRNP Treatment and Electroporation of Bone-Marrow-Derived cDCP, Macrophages, and 24-h-Activated Group 1 ILCs
This step outlines the cRNP editing of D9 cDCP, D3 BMDMs, and 24-h-activated Group 1 ILCs
-
36.
Make the volume of media and cytokines required:
-
a)
2.5 mL DC media is needed for each guide to be tested.
-
b)
3.5 mL Macrophage media is needed for each guide to be tested.
-
c)
2 mL NK media is needed for each guide to be tested.
-
37.
Place 1 mL warmed media into a 1.5 mL tube for each guide.
-
38.
Thaw sgRNA and resuspend each in 15 μL TE Buffer (Provided by SYNTHEGO).
-
39.
Make sgRNA and Cas9 Master mixes.
-
a)
40 pmol Cas9 MM for each condition.
-
i.
1 μL of 40 pmol/μL Cas9.
-
ii.
5 μL ddH2O per reaction.
-
iii.
Total of 6 μL per sample.
-
b)
For myeloid cell types: 120 pmol sgRNA MM for each condition
-
i.
1.2 μL Guide.
-
ii.
4.8 μL ddH2O per reaction.
-
iii.
Total of 6 μL per sample.
-
c)
For lymphoid cell types: 120 pmol sgRNA MM for each condition
-
i.
1.2 μL Guide.
-
ii.
0.9 μL IDT enhancer.
-
iii.
3.9 μL ddH2O per reaction.
-
iv.
Total of 6 μL per sample.
-
40.
Pipette 6 μL Cas9 master mix to tube containing 6 μL guide master mix to create RNP complex
Note: you can create a large mastermix of Cas9 to use for each individual guide
-
a)
Add Cas9 master mix to sgRNA master mix 1:1 to minimize precipitation
-
41.
Incubate for 10–15 min at 22°C.
Note: RNP complex is stable for up to 1 h at RT
-
42.
Add Cas9 master mix to sgRNA master mix 1:1 to minimize precipitation
-
43.
During complexing step, harvest and count viable cells
-
a)
Harvesting cDCP is done using a serological pipette to remove the non-adherent cells.
-
b)
Harvesting BMDM requires a cell scraper to remove the adherent precursors from the dish.
-
c)
Group 1 ILCs can be harvested using a p1000 pipette.
-
i.
Purity should be > 95% NK1.1+ cells at this point
-
44.
Spin down cells at 450 × g for 3 min at 22°C and resuspend in T Buffer
-
a)
Myeloid cells: 1 x 106 cells/100 μL in T Buffer
-
b)
Lymphoid cells: 5 x 105 cells/100 μL T Buffer
-
45.
Add 100 μL cell suspension in T Buffer to 1.5 mL tubes containing complexed cRNP mixes
Note: If bubbles keep occurring using neon pipette, change total volume to 100 μL cell suspension in T Buffer
-
46.
Draw up mixture into Neon tip and electroporate (see Methods Video S1)
-
a)
1,900 V 1 × 20 ms
-
b)
After electroporation, immediately place cells into filled 1.5 mL tubes and invert slowly to dilute out T Buffer
-
47.
Once all samples have been electroporated, place 1.5 mL tubes into rack and place in 37°C incubator for 90 min
-
a)
Rack can be placed on its side to prevent T buffer and cells from concentrating at bottom of tube
-
b)
Note: If using cells in vivo, rest cells for 10–15 min in 37°C incubator then proceed to remove T buffer via centrifugation followed by resuspension in PBS for injection
-
48.
While samples are incubating, fill non-TC-treated plates with respective media
-
a)
cDCP will be placed into a 24-well plate with 1.5 mL DC media and cytokines.
-
b)
BMDM will be placed into a 12-well plate with 2.5 mL Macrophage media.
-
c)
Group 1 ILCs will be placed into a 48-well plate with 1 mL NK media.
-
49.
Spin down sample tubes at 450 × g for 3 min at 22°C and aspirate supernatant.
-
50.
Resuspend cell pellets and place into plates.
-
51.
Wait until cells are mature before isolating genomic DNA or assessing knockout via flow cytometry.
-
a)
cDCP will have matured by D15 (For phenotypic analysis, see Figure 1B).
-
b)
BMDM will have matured by D7.
-
c)
Group1 ILCs will be ready after 3–4 days in culture.
. Ensuring quick and precise aspiration, electroporation, and ejection of cells will maximize viability.
Analysis of Editing Efficiency via Flow Cytometry
Once electroporation is complete and innate leukocytes have been cultured for the appropriate amount of time, they can be analyzed by flow cytometry and/or by Sanger sequencing. Representative results for flow cytometry are shown in Figure 2.
-
52.
Prepare fluorophore-conjugated antibody cocktail for staining in 1× PBS.
-
53.
Harvest innate leukocytes from plating conditions and transfer to 96-well v-bottom plate.
-
54.
Centrifuge plate at 450 × g for 3 min at 4°C.
-
55.
Flick off supernatant and proceed with surface staining for 30 min at 4°C.
-
56.
Add 200 μL 1× PBS to wash excess antibody cocktail and then centrifuge plate at 450 × g for 3 min at 4°C.
-
57.
Resuspend pellet in desired volume of 1× PBS and record data.
-
58.
Data are analyzed using FlowJo (TreeStar).
Figure 2.

Flow Cytometry Analysis of cRNP-Edited Innate Leukocytes
(A and B) 1 × 106 cDC1 or macrophages were electroporated at 1,900 V with 1× 20 ms pulse in the presence of Itgax (CD11c) or Itgam (CD11b) cRNP complex, respectively.
(A) CD11c expression in BM-cDC1 6 days after electroporation compared to controls electroporated in the presence of Cas9 protein alone.
(B) CD11b expression in BMDM 4 days after electroporation compared to controls electroporated in the presence of Cas9 protein alone.
(C and D) Group 1 ILCs were electroporated at 1,900 V with 1× 20 ms pulse in the presence of Klrb1c (NK1.1) cRNP complex.
(C) NK1.1 expression 3 days after electroporation of 5 × 105 rmIL-15 pre-activated purified splenic NK cells compared to controls electroporated in the presence of Cas9 protein alone.
(D) NK1.1 expression 3 days after electroporation of 2.5 × 105 rmIL-15 pre-activated purified liver ILC1 (TCRβ−CD3ϵ−NK1.1+CD49b−CD200r+) compared to controls electroporated in the presence of Cas9 protein alone. Data are representative of 3 independent experiments of 3 mice per group.
Analysis of Editing Efficiency via Sanger Sequencing
Once electroporation is complete and innate leukocytes have been cultured for the appropriate amount of time, they can be analyzed by flow cytometry and/or by Sanger sequencing.
-
59.
Harvest innate leukocytes from plating conditions and proceed with DNA isolation using the DNeasy Blood and Tissue kit (Qiagen).
-
60.
After DNA is eluted, check purity/concentration using the NanoDrop OneC Microvolume UV-Vis Spectrophotometer (Thermo Scientific).
-
61.
Dilute genomic DNA to 50 ng/μL and then proceed with PCR amplification of cRNP-targeted genomic regions of approximately 500–1,000 base pairs.
-
a)
PCR conditions will be specific for each primer pair.
-
b)
Remember to include non-edited genomic DNA as to use as a control for sequencing
-
62.
Confirm successful PCR reaction by visualizing results via Gel Electrophoresis before submitting for Sanger sequencing (GENEWIZ).
-
63.
Once sequencing has been performed, retrieve .ab1 files and analyze InDel Percentage and Knockout score using ICE analysis (SYNTHEGO). Representative results for Sanger sequencing are shown in Figure 3.
Figure 3.

Analysis of Editing Efficiency of cRNP-Edited Innate Leukocytes Using Sanger Sequencing and ICE Analysis
(A–C) SYNTHEGO ICE analysis on Sanger sequencing results from PCR region surrounding the Klrb1c locus 3 days after electroporation of 5 × 105 rmIL-15 pre-activated purified splenic NK cells.
(A) Alignment plot showing control (orange) and edited (green) sequences.
(B) Indel plot displaying the predicted range of insertions and deletions in the edited gene locus.
(C) Traces from control and edited DNA files. The guide sequence is underlined in black, PAM sequence in red, and expected cut site in vertical dashed line.
Expected Outcomes
DNA isolation of cRNP-edited innate leukocytes typically yields 70–200 ng/μL genomic DNA which can be used for analysis of gene knockout using PCR followed by Sanger sequencing. If the Cas9:sgRNA RNP complex is efficient in generating indels, analysis of cRNP-edited innate leukocytes should yield between 80%–98% knockout of the targeted gene by either sequencing or analysis by flow cytometry. If analysis yields a low knockout score/percentage, the protocol will need to be repeated with new Cas9;sgRNA complexes.
Limitations
While these conditions are suitable for use on the cell types described, there are some important limitations that should be considered:
Different leukocytes will need their own optimized electroporation conditions. Primarily, the pulse code should be optimized for each cell type of interest. We have not extensively tested this procedure on cell types not listed in this protocol and editing efficiency may differ for given guide RNA sequences.
We have observed that the maximum amount of intracellular Cas9 in Group 1 ILCs following electroporation is about 90%. As such, maximum editing efficiency in Group 1 ILCs is 90%, while myeloid cells can be edited to near 100% efficiency for certain genes.
Editing efficiency of the same gRNA can be variable between cell lineages. As such, each gRNA sequence should be tested in cell types of interest to determine cell type specific knockout efficiencies.
The procedure outlined in this protocol has been optimized for 5 x 105 to 1 x 106 cells. We have noticed that both viability and Cas9 uptake can be affected by the number of cells electroporated. Thus, the cRNP procedure will need to be optimized for lower or higher cell numbers.
Troubleshooting
Problem 1
Electroporation Errors due to Bubbles
During aspiration of the sample, bubbles can be drawn into the tip. Bubbles in the neon tip can reduce editing efficiency, and if large enough can ignite and kill all of the cells in the tip.
Potential Solution
To reduce the possibility of drawing bubbles into the tip, ensure that the sample is resuspended in a large enough volume. Our procedure has pipetting error built in to reduce this issue, but bubbles still do occur. To minimize bubble formation, make sure that the tip is fully extended before aspirating the sample slowly. If bubbles are pulled into the tip, depress the plunger and empty the sample back into the tube and try again. It is also possible to isolate bubbles on the side of the tube ensuring that they will not be able to enter the tip.
Problem 2
Electroporation Errors due to Incorrect Pipette Docking
After docking the pipette within the electroporation stand and pressing “Start” you may occasionally get an error saying: “Pipette Station Error.” This is likely due to a faulty connection between the pipette and the pipette stand itself due to the way the pipette was inserted.
Potential Solution
To resolve the issue, simply remove the pipette from the stand and re-insert it, making sure that it is vertical and snug with the back of the stand where the current comes from. In our hands, this gets rid of the error and the sample can be successfully electroporated.
Problem 3
Sequencing Results Are Unable to Analyze via ICE
After clean PCR amplification and submission for sequencing, the sequencing quality may occasionally be so low such that it cannot be analyzed by ICE and thus no knockout score can be obtained.
Potential Solution
This problem is likely due to either nonspecific priming by the sequencing primer of choice, or the guide sequence may not be far enough from the sequencing start site. If possible, use another primer within the genomic region that is capable of sequencing, or use the reverse primer. In either case, we recommend at least 150 bp before the guide sequence and at least 300 pb after the guide sequence to ensure quality analysis of editing efficiency.
Problem 4
Sequencing Results Indicate Little to No Editing Efficiency
After clean PCR, submission for sequencing, and sequencing results of are high quality, ICE analysis may indicate that there is no or very low editing efficiency.
Potential Solution
There are two possible solutions to this problem. The first is simply that the guide is poor and thus another guide should be tested. The second is that the electroporated cells have not replicated enough. To ensure that this is not the case, double check the culture conditions and make sure that enough time has passed since electroporation such that the population of edited cells will have divided at least once.
Resource Availability
Lead Contact
Further information and requests for resources, reagents or materials should be directed to, and will be fulfilled by the Lead Contact, Timothy O’Sullivan (tosullivan@mednet.ucla.edu).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
This study did not generate datasets/code. The data that support the findings of this study are available from the corresponding author upon request.
Acknowledgments
A.D.H. was supported by the Ruth L. Kirschstein National Research Service Award AI007323. T.E.O. was supported by the NIH (P30DK063491 and AI145997).
Author Contributions
L.R., A.D.H., and T.E.O. designed the study and wrote the manuscript.
Declaration of Interests
T.E.O. is a scientific advisor for NKMax America, Inc.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.xpro.2020.100113.
References
- Riggan L., Hildreth A.D., Rolot M., Wong Y., Satyadi W., Sun R., Huerta C., O’Sullivan T.E. CRISPR-Cas9 ribonucleoprotein-mediated genomic editing in mature primary innate immune cells. Cell Rep. 2020;31:107651. doi: 10.1016/j.celrep.2020.107651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T., Yu H., Hughes N.W., Liu B., Kendirli A., Klein K., Chen W.W., Lander E.S., Sabatini D.M. Gene essentiality profiling reveals gene networks and synthetic lethal interactions with oncogenic Ras. Cell. 2017;168:890–903.e15. doi: 10.1016/j.cell.2017.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
. Ensuring quick and precise aspiration, electroporation, and ejection of cells will maximize viability.
Data Availability Statement
This study did not generate datasets/code. The data that support the findings of this study are available from the corresponding author upon request.