

Abstract
Previous studies suggested that high consumption of saturated fatty acid (SFA) is a risk factor for liver cancer. However, it remains unclear how dietary SFA affects liver tumorigenesis. This study aimed to investigate the impact of a SFA-rich diet on hepatic tumorigenesis using hepatitis C virus core gene transgenic (HCVcpTg) mice that spontaneously developed hepatic steatosis and tumors with aging. Male HCVcpTg mice were treated for 15 months with a purified control diet or SFA-rich diet prepared by replacing soybean oil in the control diet with hydrogenated coconut oil, and phenotypic changes were assessed. In this special diet, almost all dietary fatty acids were SFA. Long-term feeding of SFA-rich diet to HCVcpTg mice increased hepatic steatosis, liver dysfunction, and the prevalence of liver tumors, likely due to stimulation of de novo lipogenesis, activation of the pro-inflammatory and pro-oncogenic transcription factor nuclear factor-kappa B (NF-κB), enhanced c-Jun N-terminal kinase/activator protein 1 (JNK/AP-1) signaling and induction of the oncogenes cyclin D1 and p62/sequestosome 1. The SFA-rich diet did not affect liver fibrosis or autophagy. Collectively, long-term SFA-rich diet consumption promoted hepatic tumorigenesis mainly through activation of lipogenesis, NF-κB, and JNK/AP-1 signaling. We therefore propose that HCV-infected patients should avoid excessive intake of SFA-rich foods to prevent liver cancer.
Keywords: Lipogenesis, NF-κB, Cyclin D1, p62, JNK, HCC
1. Introduction
Hepatocellular carcinoma (HCC), the most common primary tumor in the liver, is the sixth most common cancer in the world responsible for cancer-related death. HCC is frequently caused by persistent infection of hepatitis B virus, hepatitis C virus (HCV), and schistosomes, long-term exposure of alcohol and aflatoxins, and various genetic metabolic disorders [1,2]. Among them, HCC derived from non-alcoholic fatty liver disease (NAFLD) and non-alcoholic steatohepatitis (NASH) is increasing worldwide as a result of increased obesity, metabolic syndrome, and type 2 diabetes [2,3]. Since NAFLD/NASH is closely associated with dietary habits, clarifying the impact of diets on liver pathologies might provide new strategies to prevent NAFLD/NASH and related HCC.
Saturated fatty acid (SFA) is a type of fatty acid (FA) without double carbon bonds that is often solid at room temperature. SFA is mainly found in animal products, including dairy foods (butter, cream, and cheese), fatty cuts of meats, lards, and processed meats (salami, sausage, and bacon), but plant-derived palm oil and coconut oil are also regarded as a major source of SFA. SFAs are commonly found in many manufactured foods, such as snacks, cakes, biscuits, and deep fried take away foods. In the average adult western diet, 11% of energy is derived from SFA [2]. The 2015–2020 Dietary Guidelines for Americans recommended limitation of saturated fat intake to less than 10% of total energy. Excessive intake of red meat and dairy products can lead to dietary SFA overload, which may cause metabolic syndrome, obesity, type 2 diabetes, cardiovascular diseases, and NAFLD/NASH [2,4].
Since metabolic syndrome is often associated with carcinogenesis in various organs by promoting micro-inflammation and metabolic alterations, it is reasonable to conclude that higher consumption of SFA is a risk factor for breast and rectal cancer as revealed in several epidemiological studies [5,6]. Notably, palmitic acid (PA, C16:0), the most abundant and conventional SFA, can enhance oxidative and endoplasmic reticulum stress and injure several types of cells, including liver cells [7,8]. It is therefore plausible that SFA disrupts the microenvironment and nutrient metabolism in liver which may affect liver tumorigenesis. In fact, previous studies have shown that male liver cancer mortality is positively correlated with increased animal fat intake [9]. A prospective study in the United States showed a positive correlation between SFA and HCC risk, and another epidemiological study from Japan indicated inverse correlations between n-3 polyunsaturated fatty acid intake, fish consumption, and prevalence of HCC [9,10]. Although these findings have suggested the causality of SFA on hepatocarcinogenesis, the precise mechanism on how SFA-rich diet affects hepatic tumorigenesis has not been elucidated.
Additionally, the amount of dietary SFA has been estimated based on self-assessment of consumed foods from the individuals enrolled in several epidemiological studies. This estimate might not reflect the actual SFA intake or impact of SFA because most of SFA-rich foods are also rich in other hepatotoxic lipids, such as trans fatty acid [11,12].
Moriya et al. generated HCV core gene transgenic (HCVcpTg) mice that exhibited hepatic steatosis at the age of 3 months with subsequent emergence of liver tumors after the age of 16 months in the absence of obvious hepatic inflammation and fibrosis, thus mimicking the mode of hepatocarcinogenesis developed from hepatic steatosis [13]. In order to clarify the singular effect of dietary SFA overload as possible, original SFA-rich diet was made by replacing soybean oil in the control diet with hydrogenated coconut oil [14].Using these mice and diets, we aimed to investigate the effect of SFA-rich diet on hepatic tumorigenesis and its mechanistic insight.
2. Methods
2.1. Mice and treatment
All animal experiments were conducted by use of methods outlined in the “Guidelines for the Care and Use of Laboratory Animals” approved by Shinshu University School of Medicine. Male 8 to 12 week-old HCVcpTg mice on a C57BL/6 J genetic background weighing 25–30 g were selected, randomized into two groups, and maintained under the same controlled clean environment (25°C, 12-h light/dark cycle) [13]. One group (n=17) was fed a purified control diet, which was identical to the control group in the previous study [15]. The other group (n=10) was fed a SFA-rich diet, which was made by replacing soybean oil in the control diet with hydrogenated coconut oil. The calories in the SFA-rich diet were close to that of the control diet, and the detailed nutritional composition in these diets is shown in Supplementary Table 1. After treatment for 15 months, the mice were killed by CO2 asphyxiation and blood and liver tissues collected. Blood was centrifuged at 3000 rpm for 15 min twice to obtain sera, and the samples were stored at −80°C until use. Livers were removed, rinsed with physiological saline, weighed, and observed to detect tumors. After determining the number and diameter of liver tumors, livers were cut in 3–4 mm thickness and surfaces were carefully inspected to count and dissect small liver tumors. Liver tumors and non-tumorous tissues were divided into two parts, and one part was immediately fixed in 10% neutral formalin. The remaining liver tissues were immediately frozen in dry ice and stored at −80°C.
2.2. Biochemical analysis
Serum aspartate aminotransferase (AST), alanine aminotransferase (ALT), triglyceride (TG), total cholesterol (T-Chol), non-esterified fatty acid (NEFA), and glucose were measured with commercially available enzyme assay kits (Wako Pure Chemical Industries, Ltd., Osaka, Japan). Serum insulin was determined by a LBIS mouse insulin ELISA kit (AKRIN-011 T, FUJIFILM Wako Shibayagi, Gunma, Japan). Total liver lipids were extracted according to the hexane:isopropanol method and quantified using the same kits (Wako Pure Chemical Industries) [16].
2.3. Histological analysis
Formalin-fixed liver tissues were embedded in paraffin, cut into 3-μm sections, and stained with hematoxylin and eosin or Azan-Mallory staining using standard methods.
2.4. Quantification of mRNA levels
Total RNA was isolated from frozen liver tissues using RNeasy Mini Kit (Qiagen, Tokyo, Japan) and reverse-transcribed to cDNA using oligo-dT and random primers with PrimeScript RT Reagent Kit (Perfect Real Time, Takara Bio Inc., Shiga, Japan). The mRNA levels were determined by real-time quantitative polymerase chain reactions (qPCR) using a SYBR Premix Ex Taq II (Takara Bio) on an Applied Biosystems™ 7500 Fast Dx Real-Time PCR Instrument (Thermo Fisher Scientific, Waltham, MA). The primer pairs used for qPCR are shown in Supplementary Table 2. The mRNA levels were normalized to 18S ribosomal RNA (18S rRNA) levels and expressed as fold changes relative to those of HCVcpTg mice fed the control diet.
2.5. Immunoblot analysis
Frozen liver tissues (approximately 20 mg) were homogenized with protein lysis buffer (Sucrose 0.25 M, Tris chloride 25 mM, KCl 25 mM, MgCl2 5 mM, 0.5% (w/v) Triton X, pH 7.4), containing protease and phosphatase inhibitor (100-fold dilution, Thermo Fisher Scientific) [11] [16]. Nuclear fractions were isolated from liver homogenates with NE-PER Nuclear and Cytoplasmic Extraction Reagents (Thermo Fisher Scientific). Protein concentrations were measured colorimetrically with the BCA Protein Assay kit (Pierce, Rockford, IL, USA). For immunoblot analysis using whole liver homogenates, 30–90 μg of protein was loaded in each lane and separated by 7.5–15% sodium dodecyl sulfate-polyacrylamide gel electrophoresis. For the analysis using nuclear fractions, 45 μg of protein was separated in a 10% gel. After electrophoresis, the proteins were transferred to a polyvinylidene fluoride membranes (G9903119, Amersham, Freiburg, Germany) or nitrocellulose filter membranes (G9935264, Amersham). The membranes were blocked for 1 h with 1–5% non-fat dry milk or 1–10% bovine serum albumin solution dissolved in Tris-buffered saline and incubated overnight with the appropriate antibodies listed in Supplementary Table 3. After four washes with Tris-buffered saline with Tween 20, the membranes were incubated with alkaline phosphatase-conjugated secondary antibodies (#93785, Jackson ImmunoResearch Laboratories, West Grove, PA, 1:4000 dilution) and treatment with 1-step NBT/BCIP substrate (Pierce), or horseradish peroxidase-conjugated secondary antibodies (#115–035–003, Jackson ImmunoResearch Laboratories 1:4000 dilution) and detection of the chemiluminescent signal of target proteins using myECL Imager System (Thermo Fisher Scientific). The position of the proteins was confirmed by co-electrophoresing molecular weight marker (3 μL/well, PM2500, Smobio, Hsinchu, Taiwan). The band of β-actin (ACTB) or histone H1 was used as a loading control. The band intensities of target proteins were quantified using NIH ImageJ software (National Institutes of Health, Bethesda, MD, USA), normalized to those of ACTB or histone H1, and subsequently expressed as fold changes relative to those of HCVcpTg mice fed the control diet.
2.6. Statistical analysis
Results are expressed as the mean±standard error of the mean (S.E. M). The two-tailed Student’s t test and chi-square test were conducted for quantitative and qualitative data, respectively, using SPSS statistics version 22 (IBM, Armonk, NY). A P value of less than .05 was considered statistically significant.
3. Results
3.1. A SFA-rich diet aggravates hepatic steatosis and liver tumor prevalence in HCVcpTg mice
HCVcpTg mice were treated with a SFA-rich diet or purified control diet for 15 months. The prevalence of liver tumors and tumors with more than 5 mm in diameter, and the number of solitary tumors were increased in the SFA-rich diet-fed group (Fig. 1A–C). Liver-to-body weight ratios and serum AST, ALT, and glucose, were significantly higher in the SFA-rich diet group (Fig. 1D and E, Supplementary Table 4). Serum and hepatic levels of T-Chol and NEFA were not changed by SFA diet feeding (Supplementary Table 4). In the SFA-rich diet-fed mice, more abundant macrovesicular lipid droplets were detected throughout the section (Fig. 1F), as evidenced by significantly increased liver TG contents (Fig. 1G). These results show that SFA-rich diet enhances hepatic steatosis, hepatocyte injury, and hepatic tumorigenesis.
Fig. 1.
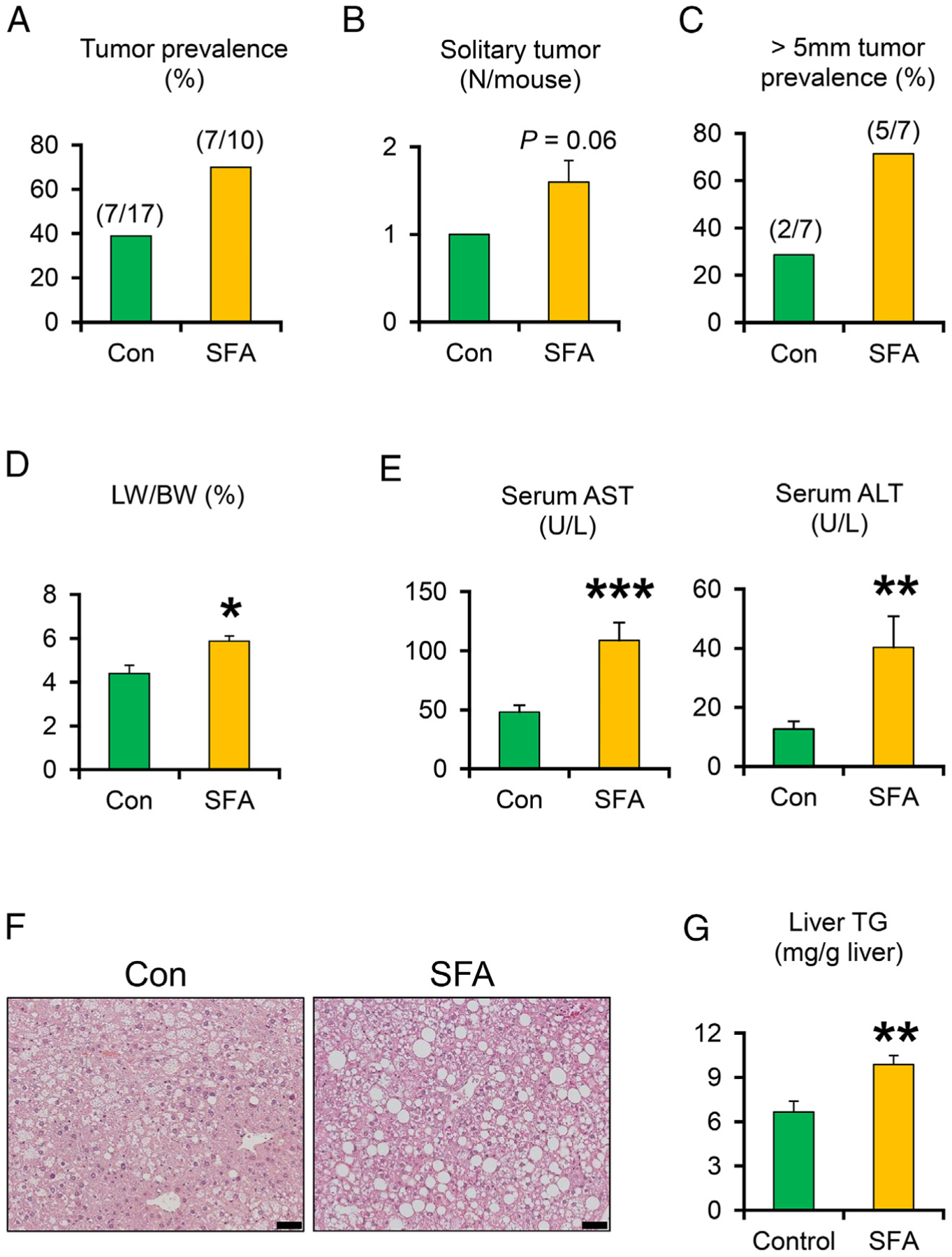
A SFA-rich diet promotes the development of hepatic steatosis and liver tumor prevalence in HCVcpTg mice after a 15-month treatment. (A) Liver tumor prevalence. (B-C) The number of solitary tumors in each mouse and prevalence rates of liver tumors larger than 5 mm in diameter. (D) Liver weight (LW) expressed as a percentage of body weight (BW). (E) Serum AST and ALT activities. (F) Photomicrographs of hematoxylin and eosin-stained liver sections. Bar=50 μm. (G) Liver content of triglycerides (TG). Data are expressed as mean±S.E.M. *P<.05, **P<.01, and ***P<.001 between control diet-fed and SFA-rich diet-fed HCVcpTg mice.
3.2. A SFA-rich diet induces the expression of lipogenic enzymes in HCVcpTg mice
In order to study the mechanism on how SFA-rich diet potentiates hepatosteatosis, hepatic expression of genes related to FA and TG metabolism was measured. The levels of mRNAs encoding cluster of differentiation 36 (Cd36) and liver-type FA-binding protein 1 (L-FABP, Fabp1), which play important roles in FA uptake from circulation and intracellular FA transport in hepatocytes, respectively, were significantly increased by long-term SFA-rich diet feeding (Fig. 2A). Furthermore, the levels of mRNAs involved in de novo FA synthesis, such as acetyl-CoA carboxylase α (ACCα, Acaca), FA synthase (FAS, Fasn), and stearoyl-CoA desaturase 1 (SCD1, Scd1) were significantly increased in the SFA-rich diet-treated mice. A similar tendency was found in the mRNA levels encoding diacylglycerol O-acyltransferase 1 (Dgat1), involved in TG synthesis (Fig. 2A). The mRNA levels of genes encoding microsomal TG transfer protein (Mttp), apolipoprotein B (Apob), and long-chain acyl-CoA dehydrogenase (Acadl) were increased (Fig. 2B), which could be the result of compensatory response to hepatic TG accumulation. Although the protein levels of CD36 and L-FABP were similar between the groups, significant increases in lipogenic enzymes ACCα, ACCβ, FAS, and SCD1 were noted in SFA-rich diet feeding group (Fig. 2C).
Fig. 2. A SFA-rich diet aggravates the hepatic expression of lipogenic enzymes in HCVcpTg mice.
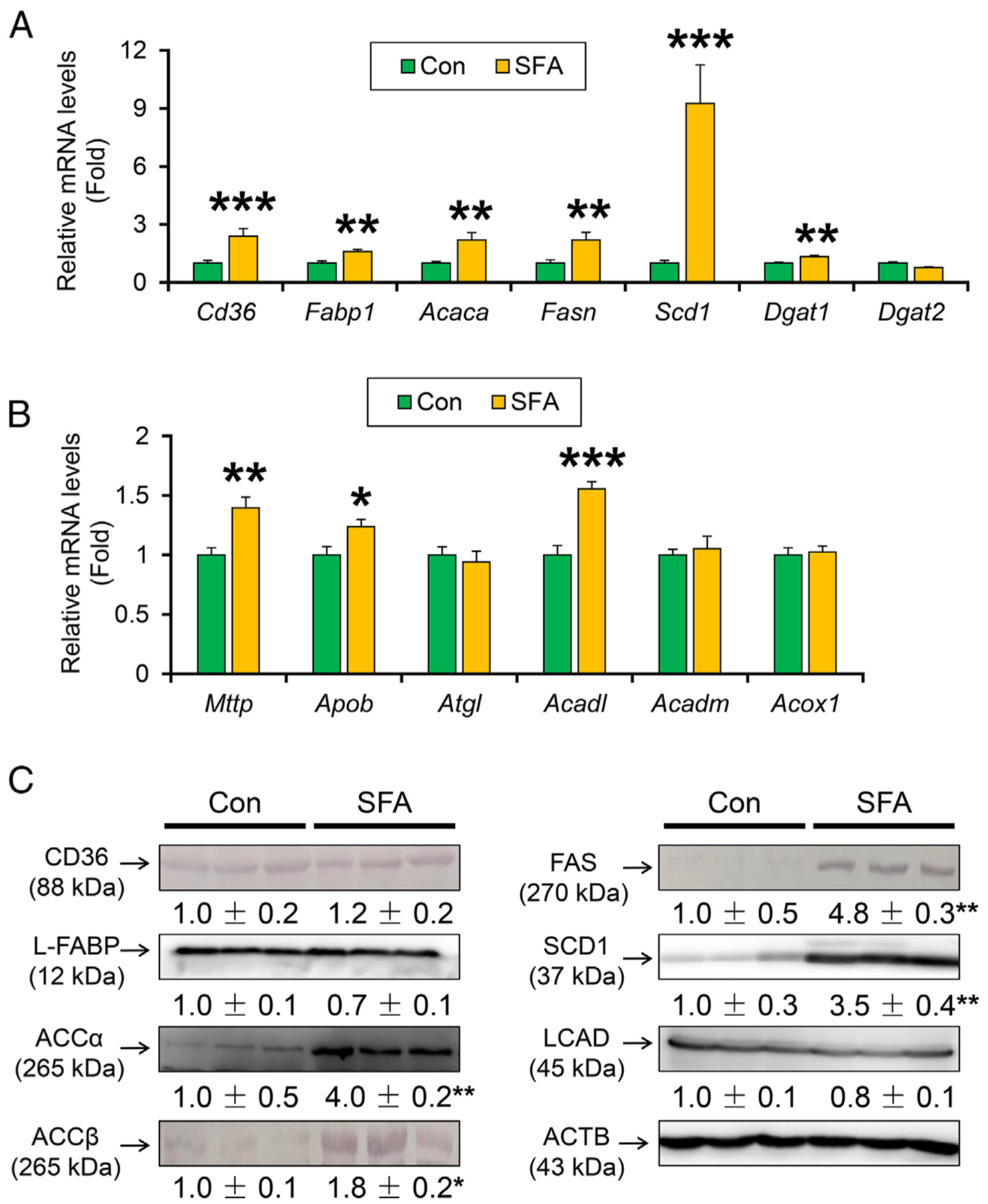
(A) Hepatic mRNA levels of genes related to FA uptake (Cd36, Fabp1) and FA de novo synthesis (Acaca, Fasn and Scd1) and TG synthesis (Dgat1 and Dgat2). (B) Hepatic mRNA levels of genes related to TG export (Mttp and Apob) and hydrolysis (Atgl) and FA catabolism (Acadl, Acadm, and Acox1).(C) Immunoblot analysis of CD36, L-FABP, ACCα, ACCβ, FAS, SCD1 and LCAD. Data are expressed as mean±S.E.M. *P<.05, **P<.01, and ***P<.001 between control diet-fed and SFA-rich diet-fed HCVcpTg mice. Con, control diet-fed HCVcpTg mice; SFA, SFA-rich diet-fed HCVcpTg mice.
Peroxisome proliferator-activated receptor (PPAR) α is a nuclear receptor mainly regulating lipid metabolism, and its activation is related to liver tumorigenesis in mice [17,18]. However, the expression of PPARα targets, such as CD36, L-FABP, LCAD, and nuclear PPARα levels were not changed (Fig. 2C and Supplementary Fig. 1A). Overall, an SFA-rich diet induces the expression of lipogenic enzymes in the liver, which is associated with more severe TG accumulation.
3.3. A SFA-rich diet activates NF-κB signaling through stimulation of TLR4 and inflammasomes
Since micro-inflammation is closely associated with tumorigenesis, the expression of key pro-inflammatory mediators was determined. The expression of mRNAs encoding macrophage marker cluster of differentiation 68 (Cd68) and pro-inflammatory factors released from M1 macrophages, such as interleukin 1β (Il1b), colony-stimulating factor 1 (Csf1), and C-C motif chemokine ligand 2 (Ccl2), were significantly increased in SFA-rich diet-fed HCVcpTg mice compared with the purified control diet (Fig. 3A). The mRNA levels of suppressor of cytokine signaling 3 (Socs3) were similar between the groups, and those of arginase 1(Arg1) were reduced by SFA-rich diet feeding (Fig. 3A). The expression of these M1 macrophage-related factors is reportedly induced by activation of nuclear factor-kappa B (NF-κB) [4]. Immunoblot analysis revealed that phosphorylation of NF-κB p65 component was significantly enhanced in SFA-rich diet-treated HCVcpTg mice (Fig. 3B). Additionally, the mRNA levels of genes associated with toll-like receptor (TLR) signaling (Tlr2, Tlr4, Cd14, and Myd88) and inflammasomes (Nlrp3, Casp1, and Aim2), both can activate NF-κB, were significantly increased in SFA-rich diet-fed HCVcpTg mice (Fig. 3C and D). Immunoblot analysis confirmed increased hepatic expression of TLR4 and caspase 1 precursor and its cleaved form, indicative of inflammasome activation following SFA diet administration. These results demonstrate that long-term SFA-rich diet leads to persistent activation of NF-κB through stimulating TLR4 and inflammasome signaling, which can accelerate hepatic tumor development.
Fig. 3. A SFA-rich diet activates TLR/inflammasome and NF-κB signaling in HCVcpTg mice.
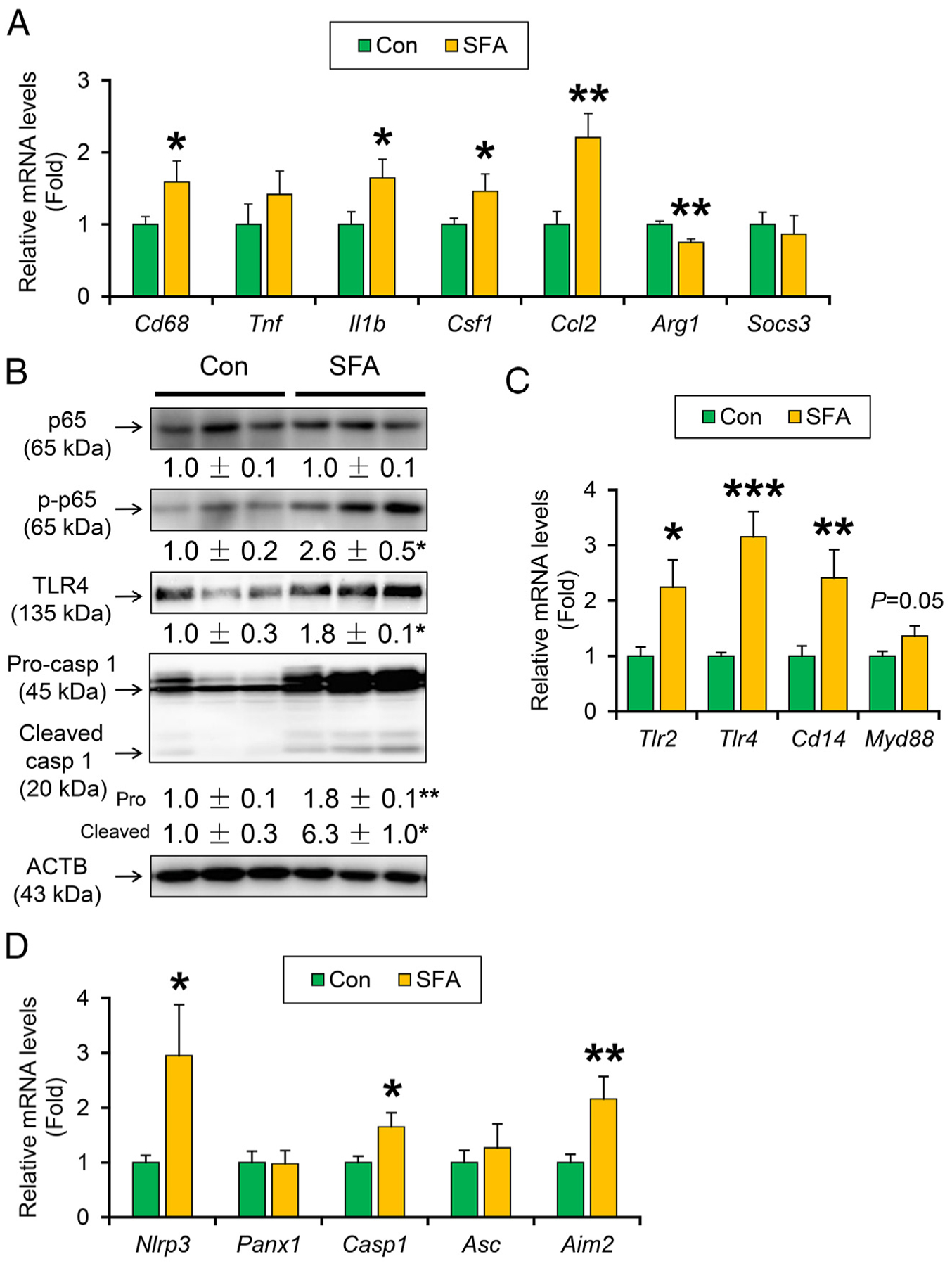
(A) Hepatic mRNA levels of genes encoding Cd68, M1 cytokines (Tnf, Il1b, Csf1, and Ccl2), M2 cytokine (Arg1), and STAT3 signaling suppressor (Socs3). (B) Immunoblot analysis of NF-κB p65, phosphorylated p65, TLR4 and caspase1. (C and D) Hepatic mRNA levels of genes related to TLRs (Tlr2, Tlr4, Cd14, and Myd88) (C) and inflammasomes (Nlrp3, Asc, Panx1, Casp1, and Aim2) (D). Data are expressed as mean±S.E.M. *P<.05, **P<.01, and ***P<.001 between control diet-fed and SFA-rich diet-fed HCVcpTg mice. Con, control diet-fed HCVcpTg mice; SFA, SFA-rich diet-fed HCVcpTg mice.
3.4. A SFA-rich diet activates JNK/AP-1 signaling
Among the key cyclins and cyclin-dependent kinases and its inhibitors related to hepatic tumorigenesis, the mRNA levels of genes encoding cyclin B1 (Ccnb1) and D1 (Ccnd1) were significantly increased in SFA-rich diet-treated HCVcpTg mice (Fig. 4A). Myc mRNA levels were similar between the groups (Fig. 4A). Immunoblot analysis revealed increased expression of cyclin D1 by SFA-rich diet (Fig. 4B). Cyclin D1 is often overexpressed in HCC and its expression is regulated by several intracellular signaling pathways, such as signal transducer and activator of transcription 3 (STAT3), mitogen-activated protein kinase, and activator protein 1 (AP-1). Although the SFA-rich diet gave no impact on the expression of total and phosphorylated STAT3 and extracellular regulated protein kinase (ERK) (Fig. 4C), the diet increased hepatic expression of total and phosphorylated c-Jun N-terminal kinase ν (JNK1/2) and the degree of phosphorylation in p56 component of JNK1/2 (Fig. 4D). JNK activation was indicated by increased mRNA levels of AP-1 components, such as Fos and Jun, and Jnk1 (Fig. 4E). The other pathways inducing cyclin D1, such as Wnt/β-catenin and hedgehog pathway, were not changed by SFA-rich diet because of unaltered mRNA levels of Ctnnb1 target genes, including Hnf1a and Gsk3b, and nuclear Gli1 levels (Supplementary Fig. 1A and C). Therefore, SFA-rich diet induces cyclin D1, a key driver of liver tumorigenesis, mainly due to activating JNK/AP-1 axis.
Fig. 4. A SFA-rich diet activates JNK/AP-1 signaling and induces cyclin D1 in HCVcpTg mice.
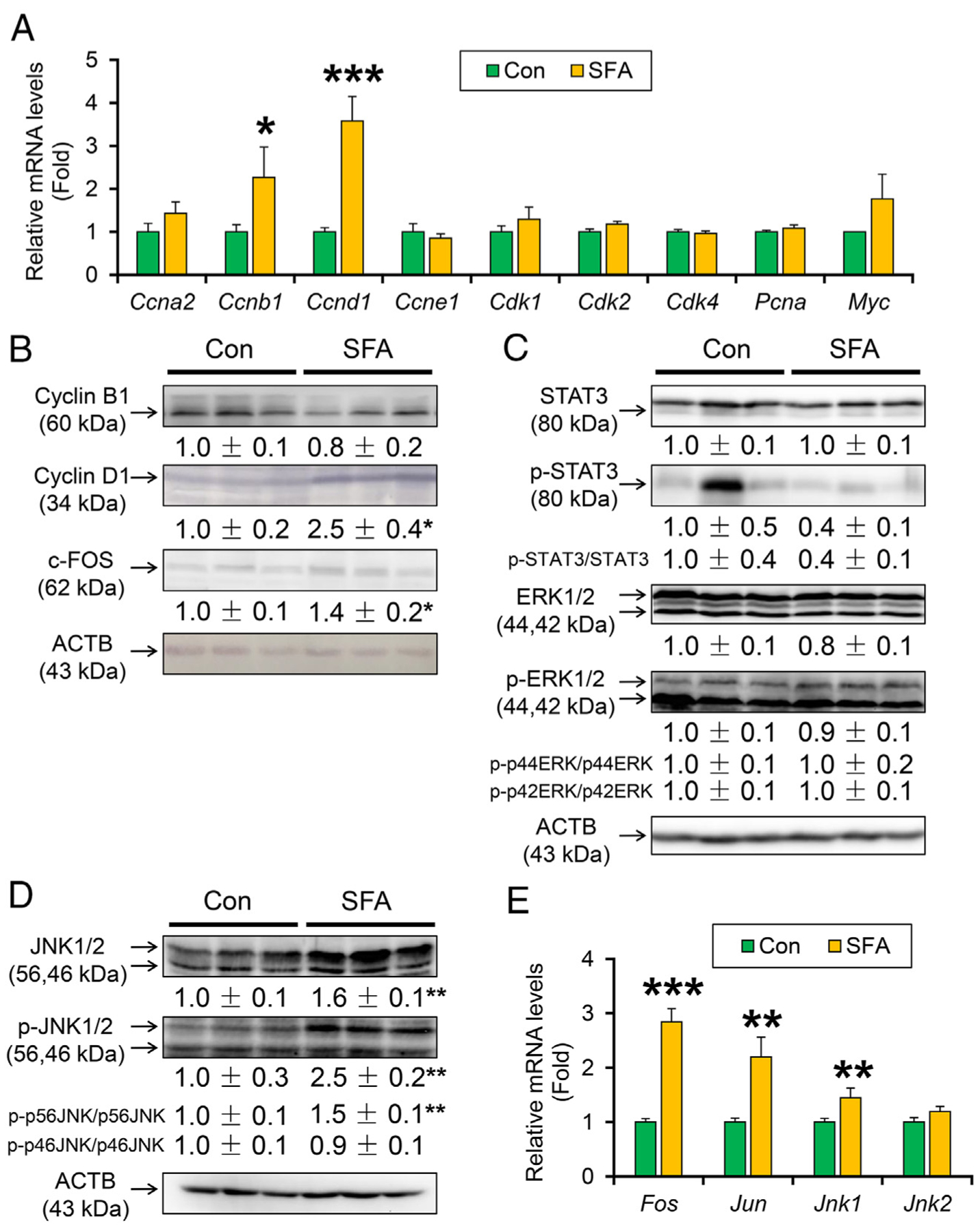
(A) Hepatic mRNA levels of cell cycle regulators. (B) Immunoblot analysis of cyclin B1, cyclin D1 and c-Fos. (C-D) Immunoblot analysis of STAT3, ERK1/2 and JNK1/2. (E) Hepatic mRNA levels of AP-1 (Fos and Jun) and Jnk1/2. Data are expressed as mean±S.E.M. *P<.05, **P<.01, and ***P<.001 between control diet-fed and SFA-rich diet-fed HCVcpTg mice. Con, control diet-fed HCVcpTg mice; SFA, SFA-rich diet-fed HCVcpTg mice.
3.5. A SFA-rich diet induces p62 at the transcriptional level
Accumulation of autophagy adaptor protein p62/sequestosome 1 (encoded by Sqstm1, thereafter designated as p62) and activation of nuclear factor erythroid 2-related factor 2 (NRF2, encoded by Nfe2l2) is documented in pre-cancerous liver tissues and HCC [19]. Since p62/NRF2 was induced by oxidative stress, lipotoxicity, and JNK activation, the expression of p62 and NRF2-regulated genes was assessed. Expression of Sqstm1, Nfe2l2 mRNAs and the downstream gene mRNAs, glutamate cysteine ligase catalytic subunit (Gclc) and NAD(P)H quinone dehydrogenase 1 (Nqo1), were significantly increased in the SFA-rich diet-treated HCVcpTg mice (Fig. 5A). The mRNA levels of genes involved in lipotoxicity, including death receptor 5 (encoded by Tnfrsf10b) and Bcl2-associated X protein (Bax) were also significantly increased in these mice (Fig. 5A). These mRNAs increases were corroborated by the corresponding protein analysis (Fig. 5B). While p62 is also accumulated by reduced autophagy flux [20], immunoblot analysis exhibited no remarkable suppression of key autophagy factors, including beclin 1, autophagy-related 5 and autophagy-related 12 complex (ATG5–12 complex), and microtubule-associated protein 1 light chain 3 (LC3) by dietary SFA overload (Fig. 5C).
Fig. 5. A SFA-rich diet induces p62/NRF2 signaling.
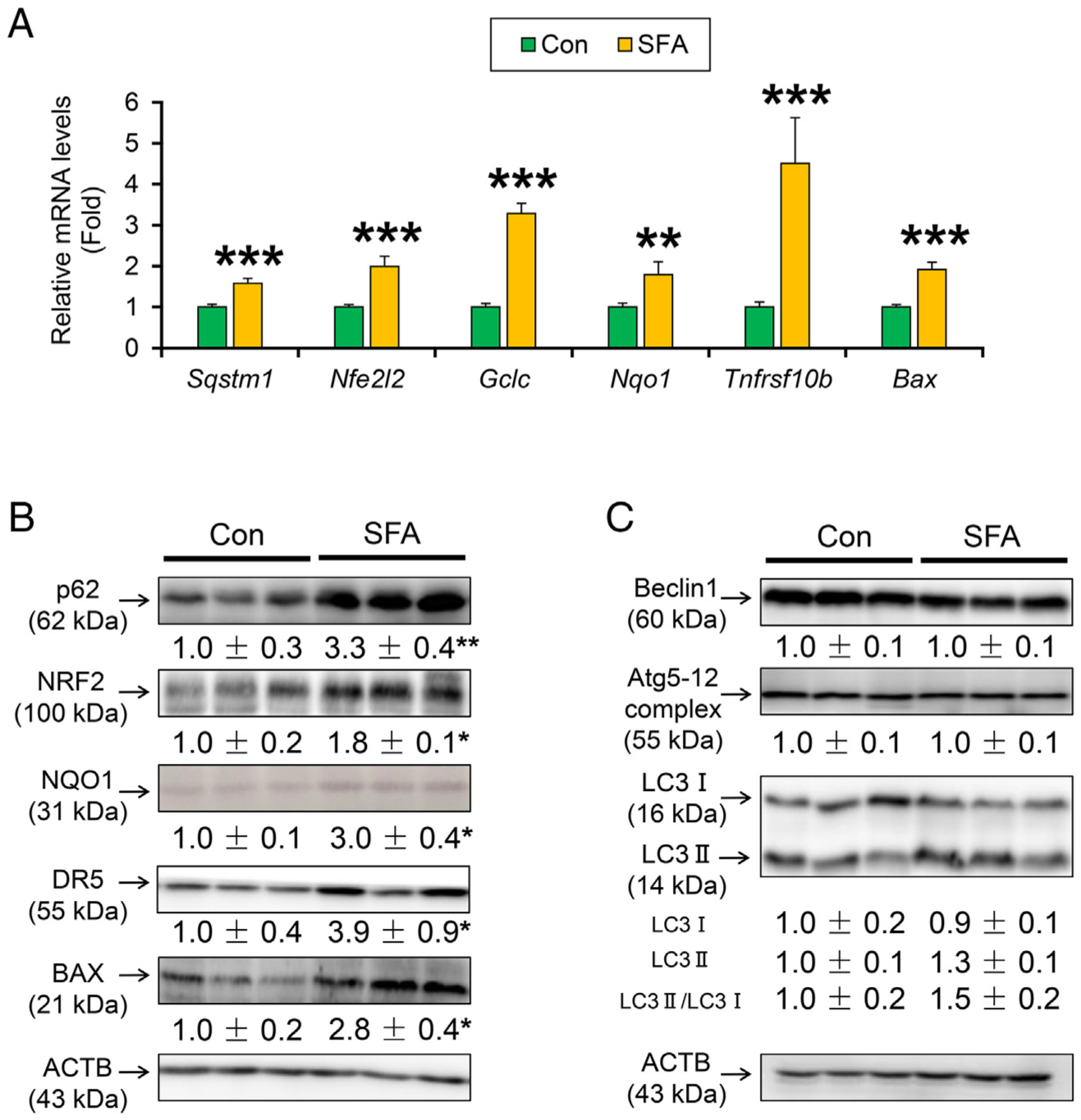
(A) Hepatic mRNA levels of Sqstm1, Nfe2l2 and its downstream genes (Gclc and Nqo1) and lipotoxicity genes (Tnfrsf10b, Bax). (B and C) Immunoblot analysis of p62, NRF2, NQO1, DR5, BAX (B), and autophagy pathway (Beclin1, Atg5–12 and LC3) (C).Data are expressed as mean±S.E.M. *P<.05, **P<.01, and ***P<.001 between control diet-fed and SFA-rich diet-fed HCVcpTg mice. Con, control diet-fed HCVcpTg mice; SFA, SFA-rich diet-fed HCVcpTg mice.
3.6. Impact of SFA-rich diet on liver fibrogenesis
Lastly, the liver fibrosis-cancer axis was investigated. There were no remarkable differences in the mRNA levels of genes encoding transforming growth factor β1 (Tgfb1), connective tissue growth factor (Ctgf), and collagen 1a1 (Col1a1). Although the mRNA levels of genes encoding α-smooth muscle actin (Acta2) and osteopontin (Spp1) were elevated, these protein levels were similar between the groups (Fig. 6A and B). Histological analysis uncovered the absence of pericellular fibrosis, a hallmark of steatohepatitis, in SFA-rich diet-treated mouse livers (Fig. 6C). These results indicate that liver tumor promotion caused by long-term SFA-rich diet intake is independent of the progression of hepatic fibrosis.
Fig. 6. A SFA rich diet gives no significant effect on fibrogenesis.
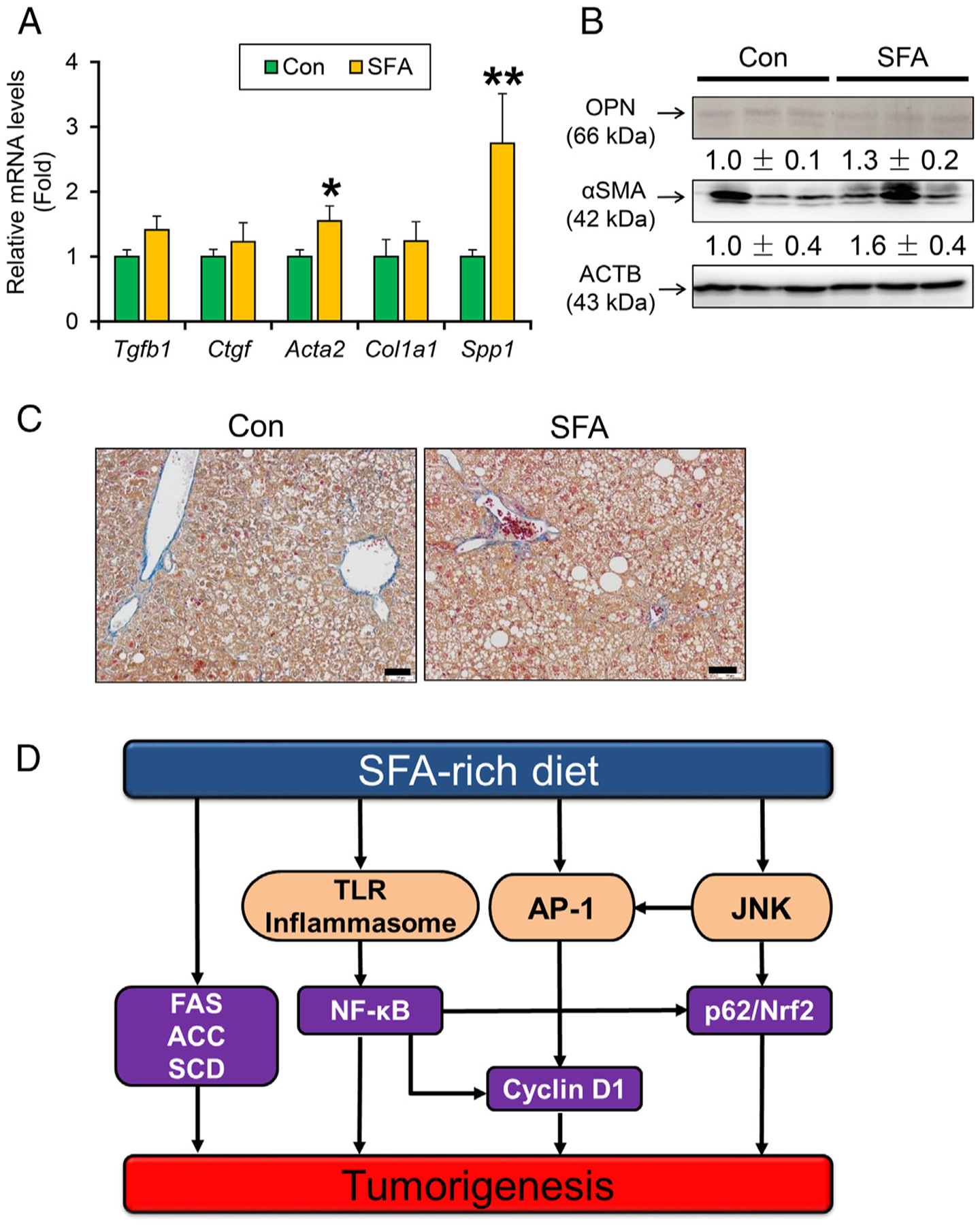
(A) Hepatic mRNA levels of genes related to fibrogenesis. (B) Immunoblot analysis of OPN and αSMA. (C) Representative photomicrographs of liver section stained with Azan-Mallory method. Bar=50 μm. Data are expressed as mean±S.E.M. *P<.05 and **P<.01 between control diet-fed and SFA-rich diet-fed HCVcpTg mice. Con, control diet-fed HCVcpTg mice; SFA, SFA-rich diet-fed HCVcpTg mice.(D) A proposed mechanism on how a SFA-rich diet enhances steatogenesis and liver tumorigenesis in HCV core gene transgenic mice.
4. Discussion
Although some studies have shown a link between excessive SFA intake, metabolic syndrome and HCC, it is unclear whether persistence of SFA-rich diet intake actually promotes HCC, one of the most common forms of human cancer [2]. The current study demonstrated that a SFA-rich diet promoted hepatic steatogenesis and tumorigenesis in HCVcpTg mice. Mechanistically, the SFA-rich diet markedly induced lipogenic enzymes, activated NF-κB and JNK/AP-1 axis, and up-regulated cyclin D1 and p62/NRF2 in the livers. These changes likely functioned as synergistic promoters of hepatic tumorigenesis. This is the first study revealing clues by which high dietary SFA promotes hepatic tumorigenesis (Fig. 6D).
Up-regulation of lipogenic enzymes was reported in various human cancers, indicating that tumor cells require lipids as a source of energy and cell membranes for rapid growth [21,22]. Among the lipogenic enzymes, FAS is the most studied protein implicated in tumorigenesis. FAS plays a key role in cell growth and malignant transformation through modulating lipid raft formation and activating epidermal growth factor signaling and various kinases [22,23]. In human HCC cell lines, FAS inactivation leads to a decline in cell proliferation through inhibiting AKT activation [24]. Therefore, significant up-regulation of lipogenic enzymes by the SFA-rich diet is responsible for not only steatogenesis, but also liver oncogenesis.
Another remarkable alteration in hepatic FA metabolism was overexpression of SCD1 by the SFA diet. SCD1 catalyzes the conversion of SFA-CoA into Δ9-monounsaturated fatty acid (MUFA)-CoA, e.g., palmitoyl-CoA and stearoyl-CoA into palmitoleoyl-CoA and oleoyl-CoA, respectively. Higher MUFA levels have also been found in transformed cells and cancerous tissue, and SCD1 overexpression and resultant MUFA overproduction is essential for the undisturbed proliferation, migration, and metastasis of cancer cells [25–27]. Indeed, stable knockdown of SCD1 in human fibroblasts decreased the rate of cell proliferation and induced apoptosis [28]. These findings corroborate that SCD1 is a crucial driver of neoplastic transformation and tumor progression. Since long-term SFA-rich diet intake induced the expression of metabolic tumor drivers, such as FAS, ACC, and SCD1, in the livers, avoiding SFA-rich foods might lead to prevention of liver tumor development.
In this study, excessive SFA intake stimulated TLR4 and inflammasome signaling, leading to persistent promotion of micro-inflammation, NF-κB activation and hepatic tumorigenesis. It was reported that PA can activate TLR4 and upregulate the expression of nucleotide binding and oligomerization domain-like receptor family pyrin domain-containing 3, a key component of the inflammasome [29,30]. Additionally, SFA-rich diet may modulate gut microbiota with an overproduction of lipopolysaccharide, enhancing TLR regulated signaling [29]. However, high dietary SFA-induced inflammation is not sufficient to promote liver fibrogenesis in this experimental model. It should be noted that high SFA consumption induces steatosis-derived liver tumorigenesis independent of liver fibrosis. Indeed, approximately 30% of NAFLD-associated HCC patients had none to mild liver fibrosis in non-tumorous tissues [31]. The proposed mechanism for promotion of hepatic tumorigenesis by long-term SFA-rich diet might partially apply to NAFLD-derived HCC in humans.
The current study uncovered that excessive SFA intake induced the expression of cyclin D1, a key oncogene in HCC development [32,33]. Among several inducers of cyclin D1, JNK/AP-1 signaling was activated by high SFA diet, as well as NF-κB. Several studies revealed that SFA and PA activated JNK, which subsequently enhanced the expression of AP-1 (c-Jun/c-Fos) and promoted hepatocyte survival and liver tumorigenesis in mice [19,34–36]. This finding is partially supported by results of a previous study that O-GlcNAc transferase plays an oncogenic role in human NAFLD-associated HCC through enhancing PA production and activating JNK/c-Jun/AP-1 and NF-κB cascades [37].
Excessive SFA intake also enhances p62 mRNA levels; p62 is an important driver of HCC development. Chronic p62 overexpression activates NRF2, allows pre-cancerous aberrant hepatocytes to resist oxidative stress and cell death, accumulate oncogenic mutations, and acquire genetic instability, leading to HCC initiation and progression [19]. p62 is also induced by NF-κB and NRF2 and activates these factors, forming self-amplifying loops [38]. More importantly, SFA enhanced the p62 mRNA levels in HCC cells through JNK activation [20]. This observation indicates a crucial role of JNK in the promotion of hepatic tumorigenesis by SFA-rich diet through up-regulating AP-1 and p62. Further investigations using JNK-disrupted mice might clarify the contribution of JNK to SFA-induced NAFLD-associated liver tumorigenesis.
It was reported that p62 was mainly regulated by autophagy flux, and that SFA suppressed autophagy [20,39]. However, the expression of autophagy-related proteins was not changed in the current study. Recent studies found that p62 expression did not always inversely correlate with autophagic activity, and that PA did not give impact on autophagy in HepG2 cells [39,40]. Thus, the p62 accumulation observed in SFA-rich diet-fed mouse livers was mainly due to JNK and NF-κB activation, rather than reduced autophagic degradation.
It was not demonstrated that SFA can directly influence the oncogenic properties of HCV core protein. However, recent studies proposed the significance of post-transcriptional protein modification by SFA, such as myristic acid and PA (myristoylation and palmitoylation, respectively) on various physiological and pathological processes [41–43]. For example, myristoylation of fibroblast growth factor (FGF) receptor substrate 2, a scaffold protein essential for FGF signaling, is essential for FGF10-induced tumorigenesis [44]. Additionally, palmitoylation of the nuclear transcription regulator C-terminal domain small phosphatase 1 directs it to the plasma membrane, promoting angiogenesis and tumor growth [45]. The impact of these SFA on the function of HCV core protein deserves further investigation.
In conclusion, long-term SFA-rich diet consumption promoted hepatic tumorigenesis mainly through activation of lipogenesis, NF-κB, and JNK/AP-1 signaling and ensuring overexpression of cyclin D1 and p62. This study suggests that excessive intake of SFA-rich foods should be avoided in HCV-infected patients to prevent liver cancer. Furthermore, these findings might apply to non-viral NAFLD-associated HCC since the experimental model used in the current study reproduces steatosis-derived liver tumorigenesis in the absence of significant fibrosis. Further studies will be needed using the other models of hepatocarcinogenisis to verify tumor-promoting effects of SFA-rich diet.
Supplementary Material
Acknowledgment
We thank Dr. Takero Nakajima and Dr. Ruan Guo (Shinshu University School of Medicine) for their invaluable help, advice, instruction, and encouragement.
Financial support: The authors have declared no financial support.
Abbreviations
- αSMA
α-smooth muscle actin
- ACC
acetyl-CoA carboxylase
- ACTB
β-actin
- ALT
alanine aminotransferase
- AP-1
activator protein 1
- AST
aspartate aminotransferase
- Bax
BCL2-associated X protein
- DR5
tumor necrosis factor receptor superfamily member 10b
- ERK
extracellular regulated protein kinase
- FA
fatty acid
- FAS
FA synthase
- FGF
fibroblast growth factor
- HCC
hepatocellular carcinoma
- HCV
hepatitis C virus
- HCVcpTg
HCV core gene transgenic
- JNK
c-Jun N-terminal kinase
- L-FABP
liver-type FA-binding protein
- MUFA
monounsaturated fatty acid
- NAFLD
non-alcoholic fatty liver disease
- NASH
non-alcoholic steatohepatitis
- NEFA
non-esterified fatty acid
- NF-κB
nuclear factor-kappa B
- NQO1
NAD(P)H quinone dehydrogenase 1
- NRF2
nuclear factor erythroid 2-related factor 2
- OPN
osteopontin
- PA
palmitic acid
- PPAR
peroxisome proliferator-activated receptor
- qPCR
quantitative polymerase chain reaction
- SCD1
stearoyl-CoA desaturase 1
- SEM
standard error of the mean
- SFA
saturated fatty acid
- STAT3
signal transducer and activator of transcription 3
- T-Chol
total cholesterol
- TG
triglyceride
- TLR
toll-like receptor
Footnotes
Potential conflict of interest: The authors declare no conflict of interest.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jnutbio.2020.108460.
References
- [1].Kimura T, Tanaka N, Fujimori N, Sugiura A, Yamazaki T, Joshita S, et al. Mild drinking habit is a risk factor for hepatocarcinogenesis in non-alcoholic fatty liver disease with advanced fibrosis. World J Gastroenterol. 2018;24:1440–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Duarte-Salles T, Fedirko V, Stepien M, Aleksandrova K, Bamia C, Lagiou P, et al. Dietary fat, fat subtypes and hepatocellular carcinoma in a large European cohort. Int J Cancer. 2015;137:2715–28. [DOI] [PubMed] [Google Scholar]
- [3].Tanaka N, Kimura T, Fujimori N, Nagaya T, Komatsu M, Tanaka E. Current status, problems, and perspectives of non-alcoholic fatty liver disease research. World J Gastroenterol. 2019;25:163–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Zacek P, Bukowski M, Mehus A, Johnson L, Zeng H, Raatz S, et al. Dietary saturated fatty acid type impacts obesity-induced metabolic dysfunction and plasma lipidomic signatures in mice. J Nutr Biochem. 2019;64:32–44. [DOI] [PubMed] [Google Scholar]
- [5].Pais R, Silaghi H, Silaghi AC, Rusu ML, Dumitrascu DL. Metabolic syndrome and risk of subsequent colorectal cancer. World J Gastroenterol. 2009;15:5141–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Xia H, Ma S, Wang S, Sun G. Meta-analysis of saturated fatty acid intake and breast Cancer risk. Medicine (Baltimore). 2015;94:e2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Wang D, Wei Y, Pagliassotti MJ. Saturated fatty acids promote endoplasmic reticulum stress and liver injury in rats with hepatic steatosis. Endocrinology. 2006;147:943–51. [DOI] [PubMed] [Google Scholar]
- [8].Wei Y, Wang D, Topczewski F, Pagliassotti MJ. Saturated fatty acids induce endoplasmic reticulum stress and apoptosis independently of ceramide in liver cells. Am J Physiol Endocrinol Metab. 2006;291:E275–81. [DOI] [PubMed] [Google Scholar]
- [9].Freedman ND, Cross AJ, McGlynn KA, Abnet CC, Park Y, Hollenbeck AR, et al. Association of meat and fat intake with liver disease and hepatocellular carcinoma in the NIH-AARP cohort. J Natl Cancer Inst. 2010;102:1354–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Sawada N, Inoue M, Iwasaki M, Sasazuki S, Shimazu T, Yamaji T, et al. Japan public health center-based prospective study G: consumption of n-3 fatty acids and fish reduces risk of hepatocellular carcinoma. Gastroenterology. 2012;142:1468–75. [DOI] [PubMed] [Google Scholar]
- [11].Hu X, Wang X, Jia F, Tanaka N, Kimura T, Nakajima T, et al. A trans-fatty acid-rich diet promotes liver tumorigenesis in HCV core gene transgenic mice. Carcinogenesis. 2020;41:159–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Hu X, Tanaka N, Guo R, Lu Y, Nakajima T, Gonzalez FJ, et al. PPARalpha protects against trans-fatty-acid-containing diet-induced steatohepatitis. J Nutr Biochem. 2017;39:77–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Moriya K, Fujie H, Shintani Y, Yotsuyanagi H, Tsutsumi T, Ishibashi K, et al. The core protein of hepatitis C virus induces hepatocellular carcinoma in transgenic mice. Nat Med. 1998;4:1065–7. [DOI] [PubMed] [Google Scholar]
- [14].Nakajima T, Yang Y, Lu Y, Kamijo Y, Yamada Y, Nakamura K, et al. Decreased fatty acid beta-oxidation is the Main cause of fatty liver induced by polyunsaturated fatty acid deficiency in mice. Tohoku J Exp Med. 2017;242:229–39. [DOI] [PubMed] [Google Scholar]
- [15].Wang X, Tanaka N, Hu X, Kimura T, Lu Y, Jia F, et al. A high-cholesterol diet promotes steatohepatitis and liver tumorigenesis in HCV core gene transgenic mice. Arch Toxicol. 2019;93:1713–25. [DOI] [PubMed] [Google Scholar]
- [16].Jia F, Diao P, Wang X, Hu X, Kimura T, Nakamuta M, et al. Dietary restriction suppresses steatosis-associated hepatic tumorigenesis in HCV core gene transgenic mice. Liver Cancer. 2020;9:529–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Tanaka N, Aoyama T, Kimura S, Gonzalez FJ. Targeting nuclear receptors for the treatment of fatty liver disease. Pharmacol Ther. 2017;179:142–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Tanaka N, Moriya K, Kiyosawa K, Koike K, Gonzalez FJ, Aoyama T. PPARalpha activation is essential for HCV core protein-induced hepatic steatosis and hepatocellular carcinoma in mice. J Clin Invest. 2008;118:683–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Umemura A, He F, Taniguchi K, Nakagawa H, Yamachika S, Font-Burgada J, et al. p62, Upregulated during preneoplasia, induce shepatocellular carcinogenesis by maintaining survival of stressed HCC-initiating cells. Cancer Cell. 2016;29:935–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Suzuki A, Kakisaka K, Suzuki Y, Wang T. Takikawa Y. c-Jun N-terminal kinase-mediated Rubicon expression enhances hepatocyte lipoapoptosis and promotes hepatocyte ballooning. World J Gastroenterol. 2016;22:6509–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Hao Q, Li T, Zhang X, Gao P, Qiao P, Li S, et al. Expression and roles of fatty acid synthase in hepatocellular carcinoma. Oncol Rep. 2014;32:2471–6. [DOI] [PubMed] [Google Scholar]
- [22].Mounier C, Bouraoui L, Rassart E. Lipogenesis in cancer progression (review). Int J Oncol. 2014;45:485–92. [DOI] [PubMed] [Google Scholar]
- [23].Menendez JA. Fine-tuning the lipogenic/lipolytic balance to optimize the metabolic requirements of cancer cell growth: molecular mechanisms and therapeutic perspectives. Biochim Biophys Acta. 1801;2010:381–91. [DOI] [PubMed] [Google Scholar]
- [24].Li L, Pilo GM, Li X, Cigliano A, Latte G, Che L, et al. Inactivation of fatty acid synthase impairs hepatocarcinogenesis driven by AKT in mice and humans. J Hepatol. 2016; 64:333–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Moriya K, Todoroki T, Tsutsumi T, Fujie H, Shintani Y, Miyoshi H, et al. Increase in the concentration of carbon 18 monounsaturated fatty acids in the liver with hepatitis C: analysis in transgenic mice and humans. Biochem Biophys Res Commun. 2001;281:1207–12. [DOI] [PubMed] [Google Scholar]
- [26].Miyoshi H, Moriya K, Tsutsumi T, Shinzawa S, Fujie H, Shintani Y, et al. Pathogenesis of lipid metabolism disorder in hepatitis C: polyunsaturated fatty acids counteract lipid alterations induced by the core protein. J Hepatol. 2011;54:432–8. [DOI] [PubMed] [Google Scholar]
- [27].Moriya K, Miyoshi H, Tsutsumi T, Shinzawa S, Fujie H, Shintani Y, et al. Tacrolimus ameliorates metabolic disturbance and oxidative stress caused by hepatitis C virus core protein: analysis using mouse model and cultured cells. Am J Pathol. 2009; 175:1515–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Tracz-Gaszewska Z, Dobrzyn P. Stearoyl-CoA desaturase 1 as a therapeutic target for the treatment of Cancer. Cancers (Basel). 2019;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Rocha DM, Caldas AP, Oliveira LL, Bressan J, Hermsdorff HH. Saturated fatty acids trigger TLR4-mediated inflammatory response. Atherosclerosis. 2016;244:211–5. [DOI] [PubMed] [Google Scholar]
- [30].Ganz M, Csak T, Nath B, Szabo G. Lipopolysaccharide induces and activates the Nalp3 inflammasome in the liver. World J Gastroenterol. 2011;17:4772–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Kimura T, Kobayashi A, Tanaka N, Sano K, Komatsu M, Fujimori N, et al. Clinicopathological characteristics of non-B non-C hepatocellular carcinoma without past hepatitis B virus infection. Hepatol Res. 2017;47:405–18. [DOI] [PubMed] [Google Scholar]
- [32].Chen J, Li X, Cheng Q, Ning D, Ma J, Zhang ZP, et al. Effects of cyclin D1 gene silencing on cell proliferation, cell cycle, and apoptosis of hepatocellular carcinoma cells. J Cell Biochem. 2018;119:2368–80. [DOI] [PubMed] [Google Scholar]
- [33].Joo M, Kang YK, Kim MR, Lee HK, Jang JJ. Cyclin D1 overexpression in hepatocellular carcinoma. Liver. 2001;21:89–95. [DOI] [PubMed] [Google Scholar]
- [34].Tsutsumi T, Suzuki T, Moriya K, Yotsuyanagi H, Shintani Y, Fujie H, et al. Alteration of intrahepatic cytokine expression and AP-1 activation in transgenic mice expressing hepatitis C virus core protein. Virology. 2002;304:415–24. [DOI] [PubMed] [Google Scholar]
- [35].Trierweiler C, Hockenjos B, Zatloukal K, Thimme R, Blum HE, Wagner EF, et al. The transcription factor c-JUN/AP-1 promotes HBV-related liver tumorigenesis in mice. Cell Death Differ. 2016;23:576–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Guo L, Guo Y, Xiao S, Shi X. Protein kinase p-JNK is correlated with the activation of AP-1 and its associated Jun family proteins in hepatocellular carcinoma. Life Sci. 2005;77:1869–78. [DOI] [PubMed] [Google Scholar]
- [37].Xu W, Zhang X, Wu JL, Fu L, Liu K, Liu D, et al. O-GlcNAc transferase promotes fatty liver-associated liver cancer through inducing palmitic acid and activating endoplasmic reticulum stress. J Hepatol. 2017;67:310–20. [DOI] [PubMed] [Google Scholar]
- [38].Katsuragi Y, Ichimura Y, Komatsu M. Regulation of the Keap1–Nrf2 pathway by p62/SQSTM1. Current Opinion in Toxicology. 2016;1:54–61. [Google Scholar]
- [39].Mei S, Ni HM, Manley S, Bockus A, Kassel KM, Luyendyk JP, et al. Differential roles of unsaturated and saturated fatty acids on autophagy and apoptosis in hepatocytes. J Pharmacol Exp Ther. 2011;339:487–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Lee YJ, Jang BK. The role of autophagy in hepatocellular carcinoma. Int J Mol Sci. 2015;16:26629–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Martinez L, Torres S, Baulies A, Alarcon-Vila C, Elena M, Fabrias G, et al. Myristic acid potentiates palmitic acid-induced lipotoxicity and steatohepatitis associated with lipodystrophy by sustaning de novo ceramide synthesis. Oncotarget. 2015;6: 41479–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Yuan M, Song ZH, Ying MD, Zhu H, He QJ, Yang B, et al. N-myristoylation: from cell biology to translational medicine. Acta Pharmacol Sin. 2020. [in press]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Fraser NJ, Howie J, Wypijewski KJ, Fuller W. Therapeutic targeting of protein S-acylation for the treatment of disease. Biochem Soc Trans. 2020;48:281–90. [DOI] [PubMed] [Google Scholar]
- [44].Li Q, Alsaidan OA, Ma Y, Kim S, Liu J, Albers T, et al. Pharmacologically targeting the myristoylation of the scaffold protein FRS2α inhibits FGF/FGFR-mediated oncogenic signaling and tumor progression. J Biol Chem. 2018;293:6434–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Liao P, Wang W, Li Y, Wang R, Jin J, Pang W, et al. Palmitoylated SCP1 is targeted to the plasma membrane and negatively regulates angiogenesis. eLife. 2017;6: e22058 10.7554/Elife22058. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.