

Abstract
New antifungal drugs are urgently needed to address the emergence and transcontinental spread of fungal infectious diseases, such as pandrug-resistant Candida auris. Leveraging the microbiomes of marine animals and cutting-edge metabolomics and genomic tools, we identified encouraging lead antifungal molecules with in vivo efficacy. The most promising lead, turbinmicin, displays potent in vitro and mouse-model efficacy toward multiple-drug-resistant fungal pathogens, exhibits a wide safety index, and functions through a fungal-specific mode of action, targeting Sec14 of the vesicular trafficking pathway. The efficacy, safety, and mode of action distinct from other antifungal drugs make turbinmicin a highly promising antifungal drug lead to help address devastating global fungal pathogens such as C. auris.
Infectious fungal diseases are among the deadliest threats to global human health. Worldwide, nearly 2 million people die each year from fungal infections, and the death toll continues to rise with increasing at-risk immunocompromised populations (1, 2). Poor outcomes are exacerbated further with the emergence of pathogens that are resistant to first-line antifungal drugs (3–5). Most recently, the pandrug-resistant “killer fungus, ”C. auris, has emerged and is spreading in health care facilities worldwide, prompting an urgent-threat alert from the Centers for Disease Control and Prevention (CDC) (6, 7).
To address the global threat of multidrug-resistant (MDR) pathogens, new antifungal agents are urgently needed. Today, only three antifungal drug classes are available for clinical use. The development of new antifungals has been hampered, in part, by the evolutionary history fungi and animals share, limiting treatment options to drugs because of limited efficacy and/or toxic side effects. Most antifungal agents, as with other antimicrobial leads, originate from a natural product source, including two of the three available antifungal classes (8, 9). However, conventional screening strategies have been seriously challenged by the high frequency of rediscovering known molecules. More recently, however, natural product-based screening has enjoyed a renaissance driven by development of screening approaches and sources of natural products (10–13).
To identify antifungal candidates, we implemented a discovery platform that leverages liquid chromatography-mass spectrometry (LC-MS)-based metabolomics, genomics, and antimicrobial activity screening of metabolomic arrays from bacterial isolates from the microbiome of marine animals (10, 11). Here, we describe the discovery and early development of a promising antifungal, turbinmicin, from a sea squirt microbiome constituent, Micromonospora sp. The compound exhibits in vitro and in vivo broad-spectrum activity against emerging MDR human fungal pathogens, including C. auris. Turbinmicin’s safety profile and highly selective mechanism of action (MOA) against C. auris and Aspergillus fumigatus support development of the compound for clinical use while also unveiling an exploitable fungal target.
Chemical diversity is critical to discovery programs (11, 14). To generate a diversity library, we did LC-MS profiling on 1482 actino-bacteria from marine invertebrates collected in the Florida Keys between 2012 and 2016. We then applied strain prioritization by metabolomics using HCAPCA (hierarchical cluster analysis principal components analysis), an LC-MS-based metabolomics tool that we recently published (supplementary materials) (15). Data resulting from HCAPCA enabled us to prioritize 174 chemically diverse strains. As shown in the PCA scores plot (Fig. 1C), strain WMMC-415 separated from the group, indicating chemical variance. The PCA loadings plot (Fig. 1D) showed a number of compounds that contributed to the variance, including turbinmicin. After fermentations, a two-step orthogonal chromatographic approach was employed to array molecules into 174 96-well plates (supplementary materials) (12, 16, 17). We assessed the in vitro activity of these metabolomic arrays against Candida albicans by high-throughput screening. One hit from a Micromonospora sp. was prioritized on the basis of potency, MS, and nuclear magnetic resonance (see supplementary materials). This led to the discovery of the antifungal agent that we named turbinmicin (Fig. 1). Turbinmicin belongs to a small group of highly oxidized type II polyketides. Although representatives such as actinomadurone, lysolipin, and xantholipin display an array of biological activities, none have been thoroughly evaluated for MOA, but changes in structure affect the spectrum of activity (18–23).
Fig. 1. Turbinmicin discovery from marine microbiome using MDR drug discovery platform.

(A) Turbinmicin-producing strain was isolated from the ascidian Ecteinascidia turbinata. (B) Turbinmicin-producing bacterium Micromonospora sp. WMMC-415. (C and D) WMMC-415 was prioritized on the basis of its chemical diversity after HCAPCA processing of a 174-strain library. PCA scores plot (C) revealed WMMC-415’s chemical diversity, and the PCA loadings plot (D) showed a number of diverse metabolites produced by WMMC-415, including turbinmicin; for the purposes of clarity, data in (C) and (D) are from a 30-member subpool of the 174-strain library. (E) Structure of turbinmicin. (F) X-ray ORTEP drawing of turbinmicin shown with 50% probability ellipsoids depicting its absolute configuration.
We used high-purity turbinmicin for expanded, clinically relevant in vitro antifungal screens and in vivo evaluation for both efficacy and safety through determination of a maximum tolerated dose (MTD) in a mouse model. To assess the spectrum of activity for turbinmicin, we analyzed its activity against a collection of 39 clinical isolates using microbroth methods from the Clinical Laboratory Standards Institute (CLSI M27 and M38). Clinical isolates in this group included MDR pathogens representing each of the defined resistance mechanisms to available antifungal classes for common fungal pathogens (C. albicans, C. auris, Candida glabrata, Candida tropicalis, A. fumigatus, Fusarium spp., Scedosporium spp., and Rhizopus spp.) (Fig. 2A and table S2) (24, 25). As examples, the collection includes panresistant C. auris [strain B11211, turbinmicin minimum inhibitory concentration (MIC) 0.25 μg/mL, fluconazole MIC >256 μg/mL, amphotericin B MIC 2 μg/mL, and micafungin MIC 4 μg/mL], echinocandin- and triazole-resistant C. glabrata, and triazole-resistant A. fumigatus. The turbinmicin MICs ranged from 0.03 to 0.5 μg/mL across most genera, with the exception of the zygomycetes, which required concentrations of 4 to 8 μg/mL for growth inhibition. Turbinmicin exhibited similar activity against isolates resistant to available antifungal classes, indicating a lack of cross-resistance and, potentially, a distinct MOA. We selected the panresistant isolate of C. auris for further pharmacodynamic characterization of the activity of various turbinmicin concentrations over time. Fungicidal activity was observed at concentrations exceeding the MIC, with the highest concentrations exhibiting a reduction of more than 2-log in organism burden at 4 hours (Fig. 2B).
Fig. 2. Turbinmicin displays potent in vitro and in vivo efficacy against multiple MDR fungal pathogens and mammalian safety.
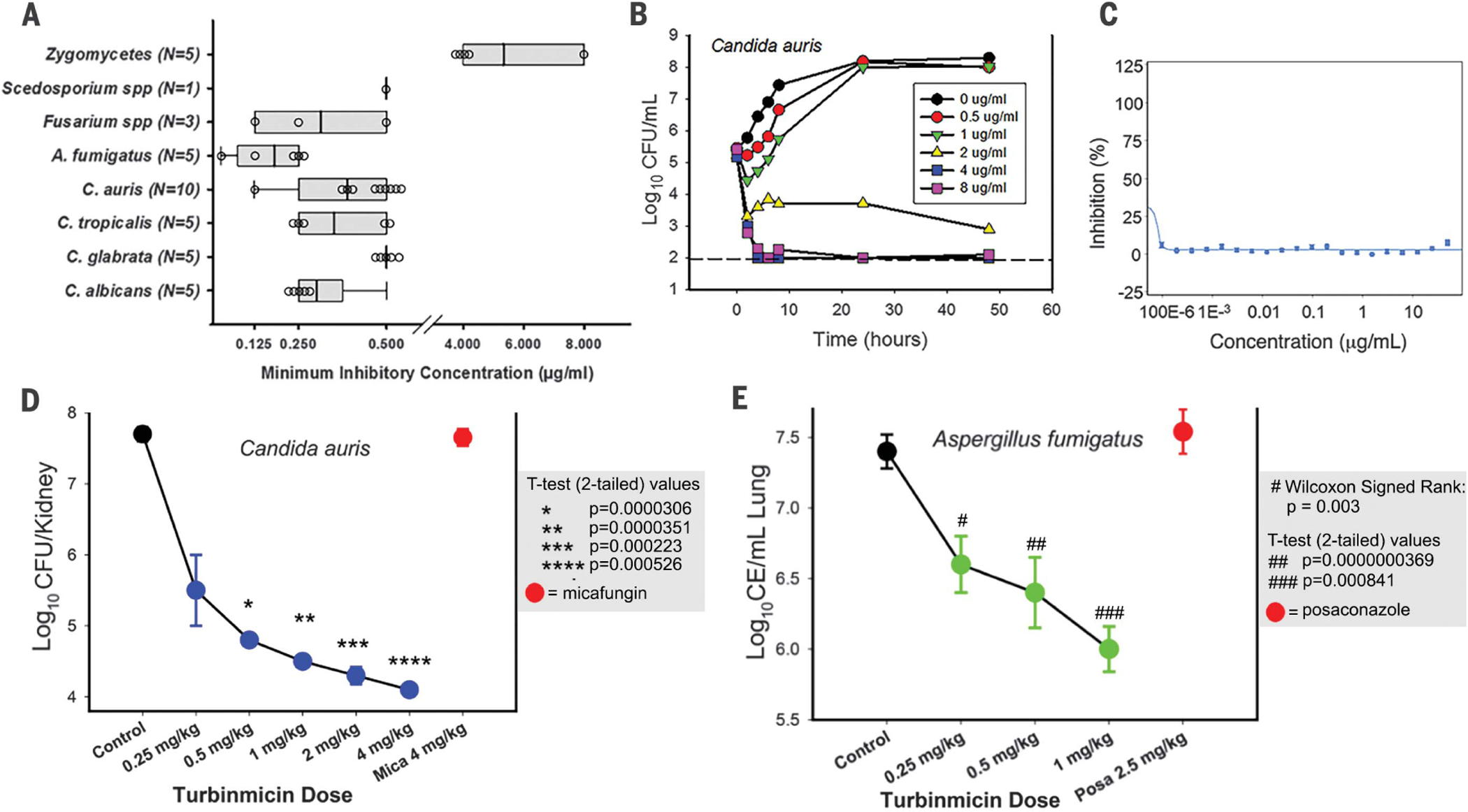
(A) In vitro activity of turbinmicin against 39 fungal isolates. (B) Time-kill curves for turbinmicin against C. auris B11211. Kill curves are generated from data collected at 0, 2, 4, 6, 8, 24, and 48 hours after subjection to turbinmicin at concentrations spanning 1 to 16 times the MIC. CFU, colony-forming units. (C) Toxicity of turbinmicin on erythrocytes was performed by hemolysis assay. No hemolytic activity was detected for turbinmicin at all test concentrations. (D) Multidose in vivo experiments with turbinmicin against C. auris B11211 by using a neutropenic, mouse, disseminated candidiasis model (3 mice per dose). The multidose experiments invoked the administration of turbinmicin at doses of 0.25, 0.5, 1, 2, or 4 mg/kg at 6-hour intervals (over a 24-hour period) by intraperitoneal (IP). Mica, micafungin. (E) In vivo multidose experiments with turbinmicin against A. fumigatus F11628 by using a pneumonia model (3 mice per dose). Multidose experiments invoked the administration at doses of 0.25, 0.5, or 1 mg/kg at 6-hour intervals (over a 4-day period) by IP. CE, conidial equivalents; Posa, posaconazole. Statistics handling for both (D) and (E) used paired t test for normally distributed data and Wilcoxon signed-rank for non-normal data; P values for each data point are indicated in each panel (shaded box).
Preliminary safety was established by using a human red blood cell (RBC) hemolysis assay as well as MTD determinations by using a mouse model. Turbinmicin concentrations exceeding the MIC by 1000-fold did not exhibit RBC toxicity, suggesting a wide therapeutic window for this compound (Fig. 2C). To identify the MTD, mice were administered single turbinmicin doses beginning at 1 mg/kg and increasing twofold. Mice showed no evidence of toxicity at dose levels as high as 256 mg/kg. We next explored in vivo efficacy using the U.S. Food and Drug Administration (FDA) standard fungal model for invasive candidiasis. This neutropenic mouse model involves Candida injection into the bloodstream and assessment of treatment response by viable fungal burden in the kidney (26, 27). Mice injected with a panresistant strain of C. auris (strain B11211) received five doses of turbinmicin, increasing by twofold and administered every 6 hours (0.25 to 4 mg/kg every 6 hours) over a 24-hour treatment period. We observed dose-dependent efficacy over the concentration range and a 3.6 log10 reduction in organism burden at the highest dose level compared with vehicle-treated control mice (Fig. 2D). There were no signs of toxicity observed over the dose range for any of the mice. Standard-of-care therapy with a humanized regimen of the echinocandin micafungin produced an outcome similar to that of untreated controls, as expected for the MDR organism (28, 29). Notably, the antifungal activity displayed by turbinmicin in these models has correlated well with efficacy in these infection models; in fact, decreases in fungal burden (<1 log10) much lower than those observed in this work have been linked to efficacy in humans (28, 30).
To further assess the clinical utility of turbinmicin, we also evaluated its efficacy toward a filamentous fungal pathogen using triazole-resistant A. fumigatus (strain F11628, CYP51 G138C mutation, posaconazole MIC 8 μg/mL and turbinmicin MIC 0.03 μg/mL) (table S2). This pathogen is particularly difficult to treat with currently available antifungals and leads to a high mortality rate (>50%). For the in vivo model, we selected a neutropenic and corticosteroid immuno-suppressed mouse model of invasive pulmonary aspergillosis that assesses treatment response by quantitative polymerase chain reaction (PCR) measurement of lung fungal burden (31). A. fumigatus-infected mice were administered turbinmicin every 6 hours over a 4-day treatment period. We empirically chose a lower dose range (0.25 to 1 mg/kg) of turbinmicin for these studies given the lower MIC observed for A. fumigatus when compared with C. auris. Turbinmicin similarly produced dose-dependent reductions in fungal burden with a 1.5 log10 drop in Aspergillus in the lungs of mice treated with 1 mg/kg every 6 hours (Fig. 2E). All mice appeared healthy after the 16 turbinmicin administrations over the 4-day treatment period. Treatment with a clinically recommended triazole (posaconazole), using doses approximating exposures in humans, was ineffective, consistent with the known azole resistance (32, 33).
MOA inquiries for turbinmicin began with our application of Saccharomyces cerevisiae DNA-barcoded knockout and knockdown libraries. For essential genes, we used the Decreased Abundance by mRNA Perturbation (DAmP) knockdown library (~1000 knockdowns), in which all essential genes were effectively knocked down through a modification of the 5′ untranslated region that destabilizes corresponding mRNA transcripts (34). In parallel, we evaluated the effect of turbinmicin in the diagnostic DNA-barcoded knockout library (310 knockouts) of nonessential gene mutants (35). After exposure to turbinmicin (1 to 5 μg/mL), genomic DNAs were extracted and their bar-codes amplified by using multiplexed PCR; amplified sequences were determined by Illumina sequencing (36–38). Chemical-genetic profiles at each concentration revealed DAmP mutants that were most sensitive to turbinmicin. SEC14 was the most notable, as reflected by the most pronounced negative chemical-genetic interactions at three of five tested concentrations (Fig. 3A). Sec14p, encoded by SEC14, is a phosphatidylinositol-phosphatidylcholine transfer protein required for correct trans-Golgi network dynamics. Sec14p is a validated fungal target, and no approved antifungal agents target Sec14p (39–41).
Fig. 3. MOA hypothesis of turbinmicin.
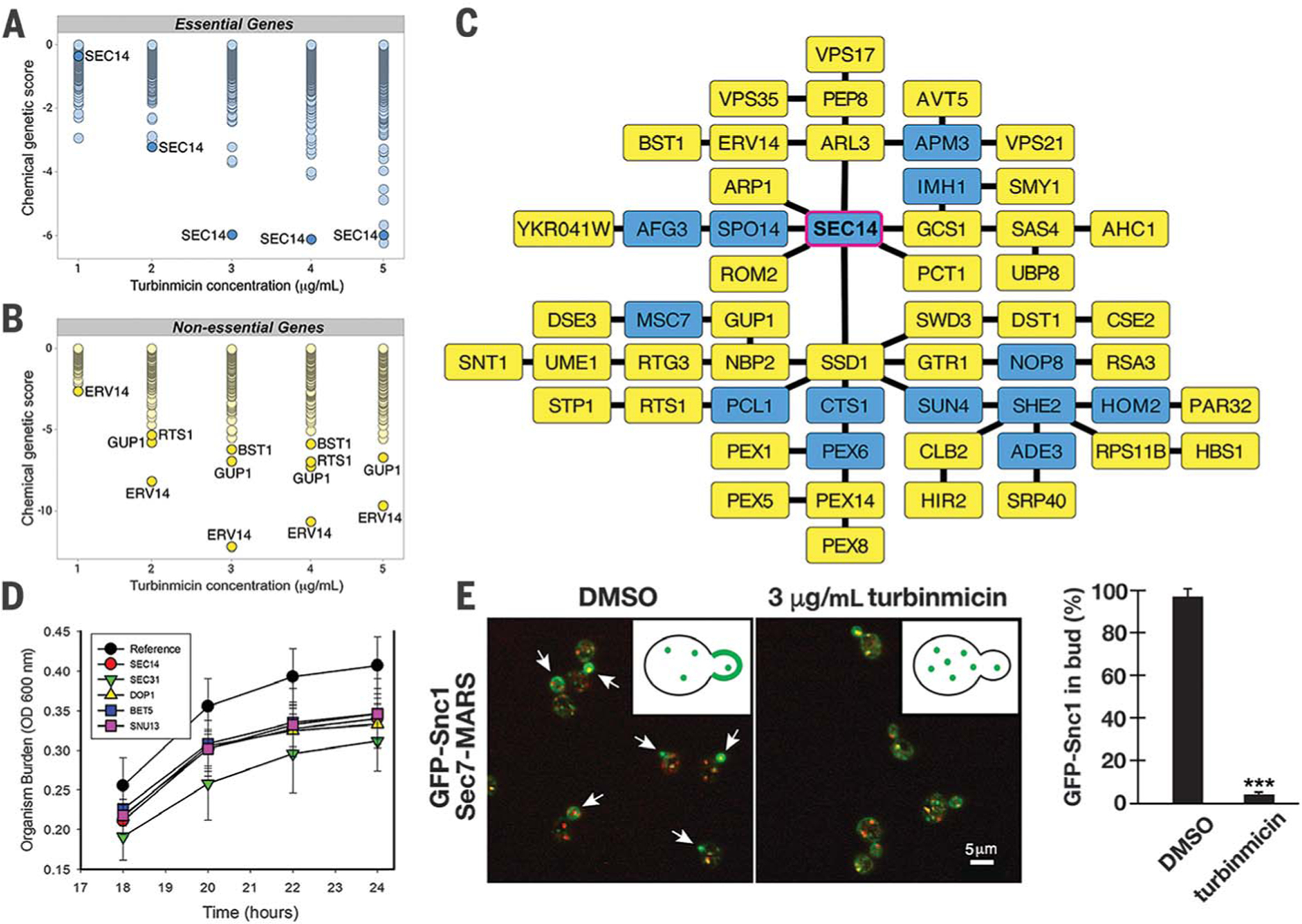
Dose-wise chemical-genetic profiles of turbinmicin against the S. cerevisiae DAmP essential gene pool (A) and the diagnostic nonessential gene pool (B). Mean negative chemical-genetic interactions are represented in green [n = 6 except for 2 μg/mL turbinmicin in (C), in which n = 5]. (C) Resulting TransposeNET genetic interaction network built by using sensitive, nonessential gene mutants. Input genes are represented by yellow nodes, and “hidden” nodes are represented in blue. SEC14 was a hidden node and highlighted with a magenta border. (D) Time-kill curve for turbinmicin against wild type and C. albicans mutants. Mutant C. albicans SEC14 H1 5C10, DOP1 H1 27E12, BET5 H1 5C5, SUN13 H1 4B5, SEC31 H1(PGA63) 29A6, and SEC31 H1(PGA63) 51H11 were used in this study. OD, optical density. (E) Representative images of GFP-Snc1 localization (green) relative to Golgi membranes (red) in the presence and absence of 3 μg/mL turbinmicin (five biological replicates each and at least 100 cells examined per condition; ***P < 0.001 based on a t test; error bars are SEM). Spectroscopy with pure turbinmicin (3 μg/mL) revealed fluorescence properties distinct and noninterfering with respect to those of the GFP and MARS (mRFPmars). Arrows highlight the distribution of GFP-Snc1 in buds in the absence of turbinmicin (also shown in the graphic). Scale bar, 5 μm. DMSO, dimethyl sulfoxide.
The relevance of Sec14p as one of if not the dominant target of turbinmicin was supported by the nonessential knockout library. ERV14, GUP1, and BST1 deletion mutants showed concentration-dependent hypersensitivities to turbinmicin (Fig. 3B), and all encode proteins involved with vesicle-mediated trafficking. Using the most sensitive nonessential deletion mutants (CG score ≤ −1.5), we then built a genetic interaction network using TransposeNET, an algorithm that imputes network nodes using sparse data from diagnostic gene sets (42). The resulting network revealed SEC14 as a central “hidden” node within this network (Fig. 3C), unifying the DAmP and diagnostic nonessential chemical-genetic data and further supporting Sec14p as the putative target of turbinmicin. Time-kill studies using C. albicans mutants from a haploinsufficiency library provided further support for Sec14p as turbinmicin’s principal target (43). C. albicans deletion mutants in the orthologs of SEC14, DOP1, BET5, SUN13, and SEC31 (Fig. 3D) all displayed hypersensitivity relative to wild-type C. albicans, consistent with turbinmicin’s ability to impair vesicle-mediated transport (43).
To further test the hypothesis that turbinmicin impairs vesicle-mediated trafficking by inhibiting Sec14p, we directly examined membrane trafficking through the secretory and endocytic pathways of S. cerevisiae using the model cargo protein GFP (green fluorescent protein)-Snc1 (44). During exponential growth, GFP-Snc1 accumulates largely on the plasma membrane of nascent buds, with more modest localization to Golgi and endosomal membranes. However, in the presence of turbinmicin, GFP-Snc1 no longer concentrated within buds and instead remained entirely associated with the Golgi and endosomes (Fig. 3E and fig. S23). By contrast, cell polarity was not noticeably affected (fig. S23). Again, these data are consistent with previous studies showing that Sec14p inactivation localizes Snc1 to the Golgi and endosomes (44–46).
Finally, docking of turbinmicin into the phospholipid binding pocket of Sec14p produced a predominant binding mode with turbinmicin’s heptacyclic ring system overlapping the co-crystallized ligand positions of picolinamide (6F0E) and β-octylglucoside (1AUA) (39) and turbinmicin’s polyene tail extending into a hydrophobic cleft left vacant by the co-crystallized ligands (fig. S24). This observation was consistent with experimental findings in which cleavage of the turbinmicin side chain (through hydrolysis) reduced antifungal activity (see supplementary materials). These in silico studies, as with chemical genomics, haploinsufficiency, and membrane trafficking analyses, implicate Sec14p as the principal fungal liability exploited by turbinmicin and is foundational to its MOA.
The current and predicted impact of emerging antimicrobial resistance is a public health crisis underscored by recent reports from the World Health Organization and the CDC (7, 47). For the first time, the CDC has classified the fungal pathogen C. auris as a highest-priority urgent risk (7). A major developmental challenge for antifungal agents is the establishment of their safety in humans and ability to evade or forestall resistance mechanisms. For instance, the echinocandins, despite their efficacy and generally excellent safety profiles, are inherently prone to resistance mechanisms associated with cryptococcal organisms; this susceptibility represents a major limitation and underscores the need for improved approaches. Both amphotericin and the azoles are characterized and limited by toxicity issues. The azoles, by virtue of their affinity for specific CYP450 enzymes, are particularly limited by toxicities stemming from drug-drug interactions. Studies to map turbinmicin’s potential for toxicity are warranted, but the results of mouse models are thus far promising. Our data implicate Sec14p as the primary antifungal target of turbinmicin; potential off-target effects appear to be limited and devoid of downstream toxicities. Careful attention to monitoring for adverse effects with more prolonged treatments similar to those required for management of fungal disease in patients (typically 2 to 6 weeks), as well as studies in additional animal species, will be needed before further development toward human administration.
Our results illustrate that animal microbiomes harbor bacterial diversity readily translating to chemical diversity in the form of biosynthetic gene clusters (BGCs) that encode previously untested natural product scaffolds. Such scaffolds may use MOAs against microbial pathogens distinct from current drugs. Turbinmicin, with its highly functionalized polycyclic core and lipophilic side chain, appears to constitute one such scaffold. The BGC class associated with turbinmicin is rare (supplementary materials and figs. S21 and S22). The therapeutic potential of turbinmicin demonstrates the continued importance of microbial sources as beacons of discovery, especially given what we now know about evolutionarily selected microbiomes and ecosystems as well as the use of cutting-edge dereplication approaches. The promising in vitro and in vivo activity against urgent fungal pathogens, mammalian safety, and exploitation of Sec14p as a target argue for further preclinical development of turbinmicin as an antifungal lead.
Supplementary Material
ACKNOWLEDGMENTS
This work is dedicated to the spirit and legacy of Prof. Robert M. Williams (February 1953 to May 2020). We thank the Analytical Instrumentation Center and the Medicinal Chemistry Center within the School of Pharmacy, University of Wisconsin-Madison for instrumentation as well as the National Magnetic Resonance Facility at Madison (NMRFAM). We also thank D. DeMaria (Sea Samples, Summerland Key, Florida, USA) for the collection of marine invertebrate specimens.
Funding: This work was supported by funding from the University of Wisconsin-Madison School of Pharmacy and the Graduate School at the University of Wisconsin, NIH (grants U19 AI109673 and U19 AI142720 to T.S.B., C.R.C., and D.R.A.), NIGMS (grant R35 GM134865 to A.A. and grant R01 GM104192 to T.S.B.), and NIAID (grant R01 AI073289 to D.R.A.). This study made use of the National Magnetic Resonance Facility at Madison, which is supported by the NIH (grant P41GM103399) (NIGMS) (old number: P41RR002301). Equipment was purchased with funds from the University of Wisconsin-Madison, the NIH (P41GM103399, S10RR02781, S10RR08438, S10RR023438, S10RR025062, S10RR029220), the NSF (DMB-8415048, OIA-9977486, BIR-9214394), and the USDA.
Footnotes
Competing interests: F.Z., D.R.B., T.S.B., M.Z., and D.R.A. are inventors on patent application no. PCT/US19/68786 filed through the Wisconsin Alumni Research Foundation (WARF), which covers turbinmicin and related compositions and uses thereof as antifungal agents with activity against MDR fungal infections. The remaining authors declare no competing interests.
Data and materials availability: All data are available in the main text and/or supplementary materials. Materials are subject to standard Uniform Biological Materials Transfer Agreement. Crystallographic data for turbinmicin have been deposited with the Cambridge Crystallographic Data Centre (deposition number CCDC 1556369).
REFERENCES AND NOTES
- 1.Brown GD et al. , Sci. Transl. Med 4, 165rv13 (2012). [DOI] [PubMed] [Google Scholar]
- 2.Pappas PG et al. , Clin. Infect. Dis 50, 1101–1111 (2010). [DOI] [PubMed] [Google Scholar]
- 3.Fairlamb AH, Gow NA, Matthews KR, Waters AP, Nat. Microbiol 1, 16092 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van der Linden JW, Arendrup MC, Melchers WJ,Verweij PE, Emerg. Infect. Dis 22, 158–159 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Perlin DS, Clin. Infect. Dis 61, S612–S617 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Meis JF, Voss A, N. Engl. J. Med 380, 890–891 (2019). [DOI] [PubMed] [Google Scholar]
- 7.CDC, “Antibiotic resistance threats in the United States, 2019” (U.S. Department of Health and Human Services, 2019). [Google Scholar]
- 8.Clardy J, Fischbach MA, Currie CR, Curr. Biol 19, R437–R441 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Newman DJ, Cragg GM, J. Nat. Prod 83, 770–803 (2020). [DOI] [PubMed] [Google Scholar]
- 10.Chanana S et al. , Metabolites 7, 34 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hou Y et al. , Anal. Chem 84, 4277–4283 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Earl DC et al. , Nat. Commun 9, 39 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chevrette MG et al. , Nat. Commun 10, 516 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hernandez A, Nguyen LT, Dhakal R, Murphy BT, Nat. Prod. Rep (2020). [DOI] [PMC free article] [PubMed]
- 15.Chanana S, Thomas CS, Zhang F, Rajski SR, Bugni TS, Metabolites 10, 297 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang F et al. , Org. Lett 20, 5529–5532 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Adnani N, Michel CR, Bugni TS, J. Nat. Prod 75, 802–806 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bunyapaiboonsri T et al. , Tetrahedron 72, 775–778 (2016). [Google Scholar]
- 19.Bunyapaiboonsri T et al. , Tetrahedron Lett 58, 3223–3225 (2017). [Google Scholar]
- 20.Annang F et al. , J. Nat. Prod 81, 1687–1691 (2018). [DOI] [PubMed] [Google Scholar]
- 21.Zhang W et al. , Chem. Biol 19, 422–432 (2012). [DOI] [PubMed] [Google Scholar]
- 22.Lopez P et al. , Gene 461, 5–14 (2010). [DOI] [PubMed] [Google Scholar]
- 23.Chu M et al. , J. Nat. Prod 60, 525–528 (1997). [DOI] [PubMed] [Google Scholar]
- 24.Clinical Laboratory Standards Institute, “Reference method for broth microdilution antifungal susceptibility testing of yeasts” (Approved standard M27-A3, Clinical Laboratory Standards Institute, ed. 3, 2007). [Google Scholar]
- 25.Clinical Laboratory Standards Institute, “Reference method for broth dilution antifungal susceptibility testing of filamentous fungi” (Approved standard M38, Clinical Laboratory Standards Institute, 2017). [Google Scholar]
- 26.Zhao M et al. , Antimicrob. Agents Chemother 62, e02542–e17 (2018).29378706 [Google Scholar]
- 27.Lepak AJ, Andes DR, “Animal models to evaluate anti-infective pharmacodynamics” in Antibiotic Pharmacodynamics, Rotschafer J, Andes D, Rodvold K, Eds. (Humana Press, 2016), pp. 59–87. [Google Scholar]
- 28.Andes D et al. , Antimicrob. Agents Chemother 55, 2113–2121 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lepak AJ, Zhao M, Berkow EL, Lockhart SR, Andes DR, Antimicrob. Agents Chemother 61, e00791–17 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lepak AJ, Marchillo K, Vanhecker J, Andes DR, Antimicrob. Agents Chemother 57, 6284–6289 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhao M et al. , Antimicrob. Agents Chemother 63, e02372–18–8 (2019).30670426 [Google Scholar]
- 32.Andes D, Pascual A, Marchetti O, Antimicrob. Agents Chemother 53, 24–34 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lepak AJ, Marchillo K, Vanhecker J, Andes DR, Antimicrob. Agents Chemother 57, 579–585 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yan Z et al. , Nat. Methods 5, 719–725 (2008). [DOI] [PubMed] [Google Scholar]
- 35.Piotrowski JS et al. , Nat. Chem. Biol 13, 982–993 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Costanzo M et al. , Science 353, aaf1420 (2016).27708008 [Google Scholar]
- 37.Nelson J et al. , Bioinformatics 34, 1251–1252 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Simpkins SW et al. , PLOS Comput. Biol 14, e1006532 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pries V et al. , Cell Chem. Biol 25, 279–290.e7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Filipuzzi I et al. , PLOS Genet 12, e1006374 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nile AH et al. , Nat. Chem. Biol 10, 76–84 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Khurana V et al. , Cell Syst 4, 157–170.e14 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xu D et al. , PLOS Pathog 3, e92 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.D’Arcangelo JG, Stahmer KR, Miller EA, Biochim. Biophys. Acta 1833, 2464–2472 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Curwin AJ, Fairn GD, McMaster CR, J. Biol. Chem 284, 7364–7375 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McDonold CM, Fromme JC, Dev. Cell 30, 759–767 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.World Health Organization (WHO). “Antimicrobial resistance-global report on surveillance” (WHO, 2014). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.