

Abstract
Combinations of metabolic blockers (including fenofibrate) with chemotherapeutic drugs interfere with the drug‐resistance of prostate cancer cells. However, their effect on cancer stem cells‐dependent microevolution of prostate cancer malignancy remains unaddressed. Here, we hypothesize that the combined docetaxel/fenofibrate treatment prompts the selective expansion of cancer stem cells that affects the microevolution of their progenies. Accordingly, we adapted a combined in vitro/in vivo approach to identify biological and therapeutic consequences of this process. Minute subpopulations of docetaxel‐resistant CD133high and/or CD44high cancer stem cell‐like (SCL) cells were found in prostate cancer DU145 and PC3 cell populations. When pretreated with docetaxel, they readily differentiated into docetaxel‐resistant CD44negative “bulk” cells, thus accounting for the microevolution of drug‐resistant cell lineages. Combined docetaxel/fenofibrate treatment induced the generation of poly(morpho)nuclear giant cells and drug‐resistant CD44high SCL cells. However, the CD44negative offspring of docetaxel‐ and docetaxel/fenofibrate‐treated SCLs remained relatively sensitive to the combined treatment, while retaining enhanced resistance to docetaxel. Long‐term propagation of drug‐resistant SCL‐derived lineages in the absence of docetaxel/fenofibrate resulted in their reverse microevolution toward the drug‐sensitivity and invasive phenotype. Consequently, prostate tumors were able to recover from the combined docetaxel/fenofibrate stress after the initial arrest of their expansion in vivo. In conclusion, we have confirmed the potential of fenofibrate for the metronomic treatment of drug‐resistant prostate tumors. However, docetaxel/fenofibrate‐induced selective expansion of hyper‐resistant CD44high SCL prostate cells and their “bulk” progenies prompts the microevolution of prostate tumor drug‐resistance. This process can limit the implementation of metabolic chemotherapy in prostate cancer treatment.
Keywords: cancer microevolution; cancer stem cells; CD44, prostate cancer; drug‐resistance; fenofibrate
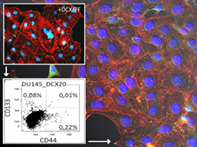
CD44+ stem cell‐related microevolution of prostate cancer drug‐resistance under the combined chemotherapeutic/metabolic stress. Combined (DCX)/fenofibrate (FF) treatment induces the formation of polymorphonuclear cells (upper insert) and DCX/FF‐resistant CD44+/CD133+ SCL cells (lower insert) that give rise to DCX‐ (but not DCX/FF‐) “super‐resistant” cell populations (right).
Abbreviations
- CSC
cancer stem cells
- DCX
docetaxel
- EMT
epithelial‐mesenchymal transition
- FACS
fluorescence‐activated cell sorter
- FF
fenofibrate
- SCID
Severe Combined ImmunoDeficiency
- SCL
stem cell‐like cells
Significance statement.
Fenofibrate has recently been shown to interfere with the drug resistance of prostate tumor cells. The data of this study confirmed that fenofibrate increases sensitivity of drug‐resistant prostate tumors to chemotherapeutic stress. However, the data also revealed the resistance of CD44+ prostate cancer stem cells to the combined docetaxel/fenofibrate treatment, accompanied by docetaxel/fenofibrate‐induced microevolution of hyper‐resistant CD44− progenies. Thus, the balance between the sensitivity of CD44low prostate cancer cells to chemotherapeutics and the adaptation of CD44high stem‐like cells to metabolic stress determines the response of prostate tumors to metabolic chemotherapy.
1. INTRODUCTION
Tumor homeostasis and development is governed by minute (<1%) populations of multipotent cells (cancer stem cells [CSCs]). 1 , 2 CSCs can evolve from transformed tissue‐specific stem cells. Alternatively, they originate from the “bulk” cancer cells in the process of “retro‐differentiation,” which can be triggered by genetic/epigenetic aberrations and/or by aberrant intratumoral communication systems. 3 Because of their self‐renewal capacity and multipotency, CSCs reside at the top of hierarchy of tumor cell phenotypes and give rise to intermediate progenitors and terminally differentiated progeny. 4 , 5 Thus, they participate in the phenotypic plasticity and diversity of cancer cell populations. 4 , 6 Furthermore, the activity of DNA repair systems and membrane efflux pumps, accompanied by relative dormancy of CSCs, accounts for their high drug‐resistance. 7 , 8 Consequently, CSC abundance increases during tumor treatment. These features account for the adaptability of tumors to the extrinsic stress. 4 , 9 As its consequence, premature cessation of chemotherapy commonly results in an expansion of drug‐resistant CSCs and their progenies in prostate tumors. 10 These cells subsequently rebuild the prostate tumor and other tumors in the more malignant form. 11 , 12 , 13 , 14
Prostate cancer is one of the most frequently diagnosed cancers and the second leading cause of cancer‐related deaths in Euro‐American populations. 15 Its latency period is relatively long; however the risk of prostate tumor formation increases with the age of the patient. Conventional strategies of prostate cancer treatment (surgical intervention, chemotherapy, and androgen ablation) usually provide a temporary symptom relief but hardly interfere with the cancer progression. Usually, they also prompt the microevolution of tumor drug resistance, enforcing the intensification of chemotherapy against continuously more aggressive tumor within the weakening organism of the patient. 13 Harmful effects of this vicious cycle are of particular importance for the palliative/elderly patients, who are oversensitive to the adverse effects of chemotherapeutics. 16 These effects enforce premature treatment cessations that facilitate tumor recurrence and its metastatic cascade. “Clonal evolution” and activation of the dormant, drug‐resistant, phenotypically plastic, and multipotent CSCs 3 , 17 , 18 participates in this process and ultimately leads to abrupt tumor relapses.
CSCs are routinely distinguished on the basis of stem surface and intracellular stem cell markers: CD44, CD133, Oct3/4, Sox2, Klf4, Nanog, etc. 13 Their presence is commonly observed in the biopsies from prostate tumors. 13 , 17 , 19 Like other CSCs, prostate CSCs possess self‐renewal capacity and genetic/epigenetic/phenotypic plasticity that prompt them to drive tumor microevolution and metastatic dissemination in response to the signals from niche. 14 , 19 , 20 , 21 The activation of CSCs in prostate tumors under chemotherapeutic stress has particularly dreadful consequences for prostate cancer patients. A narrow window between patient's collapse and cancer relapse poses a crucial limitation for prostate cancer chemotherapy. Accordingly, new regimens are required that would reduce the effective doses of chemotherapeutics, while preventing the CSC‐dependent microevolution of prostate tumor drug resistance.
Metronomic therapies based on the combined application of cytostatic drugs and metabolic blockers can potentially interfere with the progression of drug‐resistant prostate tumors, concomitantly exerting less intense systemic adverse effects than conventional therapies. We have recently shown that fenofibrate (FF)‐based metronomic regimens interfere with the drug resistance and malignancy of prostate cancer cells, while reducing adverse effects of chemotherapeutics. 22 FF is commonly used to improve high:low‐density lipoprotein ratio in hyperlipidemia. 23 The peroxisome‐proliferator activated receptor‐α and reactive oxygen species‐dependent signaling systems “canonically” mediate its biological activity. 24 They also account for its anticancer properties. 25 , 26 Interference of FF with cancer cell expansion and systemic dissipation 27 is accompanied by its effects on cancer “stroma,” including vascular cells, 24 , 28 and cellular energy metabolism. 22 , 29 , 30 Thus, FF interferes with drug‐resistance systems and sensitizes drug‐resistant prostate cancer cells to chemotherapy. Along with its systemic tolerability, these facts justify FF application in the palliative treatment of malignant prostate tumors. However, its effect on CSC‐related microevolution of drug‐resistant tumors remains unknown.
CD133+/CD44+ cells are commonly present within prostate tumors in vivo. 13 , 17 , 19 Even though a full “stem cell” signature of CSCs in vitro is still the matter of debate, 18 , 31 CD133+ and/or CD44+ cancer stem cell‐like (SCL) cells have been observed in prostate cancer cell DU145 and PC3 lines in vitro. 32 , 33 Thus, they imitate the hierarchical structure of prostate tumors, providing a tool for the identification of processes that underlie their clonal expansion under chemotherapeutic/metabolic stress in vivo. Here, we hypothesized that the combined docetaxel (DCX)/FF treatment can prompt the clonal evolution of CSC/SCL cells. Accordingly, we adapted a combined in vitro/in vivo approach to identify biological and therapeutic consequences of this activity. We estimated the effect of the combined DCX/FF treatment on the phenotypic microevolution of prostate cancer cell populations. In particular, we focused (a) on the pattern of clonal expansion of CD44+ DU145/PC3 SCL cells under DCX and/or FF stress, (b) on the phenotype of their CD44− “bulk” progeny, and (c) on the interference of FF with the chemoresistance pattern and invasiveness of CD44− “bulk” cells.
2. MATERIALS AND METHODS
2.1. Cell cultures, preselection, and fluorescence‐activated cell sorting
Human prostate carcinoma DU145 (ATCC; HTB81), PC3 cells (ATCC; CRL1435), and their SCL‐derived offspring (see below) were routinely cultivated in Dulbecco' modified Eagle's Medium/F12 HAM medium supplemented with 10% FBS and antibiotics (Sigma, St. Louis, Missouri). 24 , 34 For endpoint experiments, media supplemented with docetaxel (DCX; 0.125‐50 nM, Sigma) and FF (5‐25 μM; F6020, Sigma) were added to cancer cell cultures at the concentrations given in the text. For the isolation of CD44+/CD133+ subpopulations, the cells were cultivated in the presence of DCX (10 nM) and/or FF (25 μM) for 48 hours, dissociated with 1 mM EDTA in Ca++/Mg++‐free PBS, suspended in culture medium, centrifuged/washed in Ca++/Mg++‐free PBS and incubated in mouse phycoerythrin‐conjugated anti‐CD133 IgG/mouse fluorescein isothiocyanate‐conjugated anti‐CD44 IgG (1:100) solution for 30 minutes. After washing, CD44high and/or CD133high cells were sorted out with fluorescence‐activated cell sorter (FACS) Aria (BD Systems, Heidelberg, Germany), using 488/512 nm excitation and a 525BP(FL1)/575LP(FL2) emission filter sets, or with ImageStreamX system (Amnis). For each analysis, 107 cells were initially identified according to a particle diameter exceeding 8 μm.
Microevolution of multi‐drug resistance in DU145/PC3 cell populations was prompted by the intermittent cell exposition to DCX administered at the increasing concentrations (0.5‐50 nM). 22 Shortly, the cells were cultivated for 3 days in DCX (0.5 nM)‐containing medium, followed by 72 hours of incubation in the pure medium containing 30% of DU145‐conditioned medium, before the next round of DCX treatment. After 3 cycles of treatment/recovery, the procedure was repeated in the presence of 1, 2, 5, 10, 20, and 50 nM DCX. The established cell lineages were cultivated in standard medium and repeatedly subjected to pulse 20/50 nM DCX treatment to sustain drug‐resistance in short‐term (ST) populations. Concomitantly, established lineages were maintained in standard DCX‐free medium for at least 30 passages (1:8) to obtain drug‐sensitive long‐term (LT) variants. SCL‐derived, drug‐resistant DU145/PC3 cells between 5th and 15th passage after the lineage establishment were used in the experiments. Stability of their acquired phenotype was assessed after freezing/thawing and following drug withdrawal throughout this period. Untreated parental cells were cultivated alongside.
2.2. Immunofluorescence
For the immunofluorescence studies of Oct‐4, Nanog, and CD44, the cells were fixed with methanol:acetone (7:3, −20°C) for 15 minutes. α‐Tubulin and vinculin were visualized in the cells that had been fixed with formaldehyde (3.7%; 20 minutes in RT) and permeabilized with Triton X‐100 (0.1%; 10 minutes in RT). Primary antibodies: mouse anti‐Oct3/4 IgG, mouse anti‐CD44 IgG, rabbit anti‐Nanog IgG, rabbit anti‐vinculin IgG, and mouse anti‐α‐tubulin IgG (all from Sigma) were applied for 1 hour, immediately after the incubation in the presence of 3% BSA. Then, the cells were labeled with Alexa 488‐conjugated goat anti‐mouse IgG and/or Alexa 488‐conjugated goat anti‐rabbit IgG (No. A11001 and A11008, Invitrogen, Carlsbad, California). When indicated, the cells were counterstained with TRITC‐conjugated phalloidin (No. 77418, Sigma) and Hoechst 33258 (No. B2883, Sigma). 34 Image acquisition was performed with a Leica DMI6000B microscope (DMI7000 version; Leica Microsystems, Wetzlar, Germany) equipped with the differential interference contrast. Images were registered with ×40, NA‐1.4 oil immersion objective using Leica DFC360FX CCD camera under the control of the Leica Application Suite X software. 35
2.3. Cell motility and invasion assays
The cells were seeded into 12‐well plates at a density of 5 × 103 cells/cm2 (short‐term incubation variant) or 5 × 102 cells/cm2 (long‐term incubation variant). Cell movement was recorded for 6 hours at 300 seconds time intervals (using a dry ×10, NA‐0.75 objective) with a time‐lapse Leica DMI6000B video‐microscopy system equipped with the temperature chamber (37C° ± 0.2°C/5% CO2), IMC contrast optics and a DFC360FX CCD camera. Single cell trajectories were constructed from the sequences of cell centroid positions to estimate the total lengths of single cell trajectories (distance [μm]), total lengths of single cell displacements (displacement [μm]), speed of cell movement (speed: distance/recording time [μm/h]) and speed of cell displacement (displacement/recording time [μm/h]). Single cell parameters were further compiled to calculate the averaged values of the parameters at the population level (from >3 independent experiments; number of cells >50). 36 For transmigration assays, cells (2 × 103) were seeded on the upper side of transwell inserts in 24‐well plates, allowed to transmigrate in chemodynamic conditions for 48 hours, fixed with 3.7% formaldehyde and stained in a 0.2% solution of Coomassie brilliant blue R250 in methanol:acetic acid:water (46.5:7:46,5; [v/v/v]) for 1 hour. Areas of cell colonies were further estimated with Leica DMI6000B system.
2.4. Proliferation and apoptosis assays
Cells were seeded into 24‐well plates (Corning) at the density of 5 × 103 cells/cm2, cultivated in the standard medium for 24 hours, before the administration of the media containing DCX and/or FF. After 48 hours, the cells were harvested, resuspended in the original culture medium and counted with a Coulter Z2 Counter (Beckman Coulter Inc, Fullerton, California). For the quantification of apoptosis, cells were dissociated, resuspended in original medium and stained with AnnexinV/propidium iodide according to manufacturer's protocol (BD Pharmingen, San Diego, California). Flow cytometric detection of apoptotic cells was performed with a ImageStreamX system (Amnis). 22 At least 5 × 104 cells were analyzed for each condition.
2.5. Clonogenic potential of SCL progenies
SCL‐derived cells were seeded at the density of 5000 cells/cm2. After reaching the confluence (usually 48 hours), their progeny was trypsinized and seeded into 6‐well dishes (Corning) at the density of 500/cm2. Then, the cells were allowed to develop the clones for the next 72 hours in the absence/presence of DCX and/or FF, fixed with 3.7% formaldehyde, and stained in a 0.2% solution of Coomassie brilliant blue R250 in methanol:acetic acid:water (46.5:7:46,5; [v/v/v]) for 1 hour. After washing in distilled water, the numbers of cell colonies/well were assessed with Leica DMI6000B microscope in the bright‐field mode.
2.6. In vivo analyses
Severe combined immunodeficient (SCID) mice (age: ca. 5 weeks; Charles River Laboratories) were maintained in a temperature‐controlled, pathogen‐free room, in community cages on a standard laboratory diet with free access to drinking water and a 12 hour day/night regime. Before the experiments, the animals were subjected to the quarantine and acclimatized for at least 2 weeks. Cancer cells were mixed with BD Matrigel Basement Membrane Matrix High Concentration (1:1 in PBS; BD Biosciences), and 40 μL of the cold suspension (1.5 × 105 cells) was subcutaneously injected into abdominal flank of SCID mice. DCX (10 or 20 mg/kg) was administered intraperitoneally every 6 or 4 days, respectively, whereas FF (60 mg/kg) was administered intragastrically every day or 2 days for both DCX schemes. The mice were observed for 2 to 4 weeks for the appearance and development of tumors. Volumes of developing tumors were calculated according to the following formula: V = (Π/6)a × b × c, where a, b, and c are perpendicular diameters of the ellipsoid approximating the shape of the tumor. Afterward, the animals were sacrificed and the tumor biopsies subjected to sectioning and to immunohistochemical CD44 staining. Animals were handled according to the protocols and guidelines approved by the 2nd Local Ethics Committee for Experiments on Animals at the Jagiellonian University in Cracow (Dec. No. 290/2017).
2.7. Calcein efflux assay
Cells were seeded into 12‐well plates at a density of 5 × 103 cells/cm2, cultivated for 24 hours and immersed in culture medium supplemented with 0.25 μM calceinAM (Life Technologies, Carlsbad, California, C3099) for 30 minutes at 37°C. Then, the cells were rinsed and the sequences of fluorescence images of at least 16 randomly chosen confluent culture regions were collected in green channel (A4; GFP excitation ‐ BP470/40; emission ‐ BP525/50) 5 and 30 minutes after calcein AM administration. In each experiment, the stacks were obtained with the same excitation/exposure settings (excitation/camera gain/time of exposition). Efflux Index was estimated for each stack with LasX software (Leica) and calculated for each specimen. 22
2.8. Statistical analysis
All data were expressed as mean ± SEM from at least three independent experiments (N > 3). The statistical significance was tested with t‐Student test or one‐way ANOVA followed by post hoc Tukey's comparison for variables with non‐normal (tested with Levene's comparison) and normal distribution, respectively. Statistical significance was shown at P < .05.
3. RESULTS
3.1. CD133 +/−/CD44 +/− cancer SCL cells display enhanced drug resistance
CD133 and CD44 have previously been identified as the markers of prostate CSCs. 13 , 33 Flow‐cytometric analyses revealed very small (<0.05%) subpopulations of CD133+, CD133+/CD44+, and of CD44+ cells in DU145 populations (Figure 1A). Their abundance remained stable during the long term propagation of the cell line. Furthermore, these SCL cells displayed a relatively high resistance to DCX, as illustrated by their more abundant fractions in DCX‐exposed populations (>0.2%). In the presence of serum (FBS), naive and DCX‐treated CD44+ cells progressively acquired CD133−/CD44− phenotype in vitro, which indicates that they display the potential corresponding to CSCs in vivo. Due to the considerable plating efficiency of their direct progenies (Figure 1B), SCL cells finally gave rise to CD133−/CD44− lineages of proliferating “bulk” cells (nSCL_DU145 and dcxSCL_DU145, respectively; Figure 1C). These lineages (in particular, dcxSCL_DU145 cells) displayed slightly more abundant stress fibers and matured focal adhesions than their naive counterparts. Concomitantly, slightly lower proliferation and motility rates were seen in both SCL progenies in control conditions (Figure 1D). They were accompanied by their increased resistance to DCX, illustrated by relatively high motility and proliferation rates (Figure 1E), and a low apoptosis ratio of nSCL_DU145 and dcxSCL_DU145 cells cultivated under DCX stress (Figure 1F; cf. Figure S1). Corresponding potential was displayed by CD44+PC3 SCL and CD133+DU145 SCL cells, as illustrated by increased DCX‐resistance of DCX‐treated SCL progenies (Figures S2 and S3, respectively; see Supplementary Material). Thus, CD44+ SCLs and the selective expansion of their CD44− progenies may lead to the formation drug‐resistant cell lineages in vitro.
FIGURE 1.
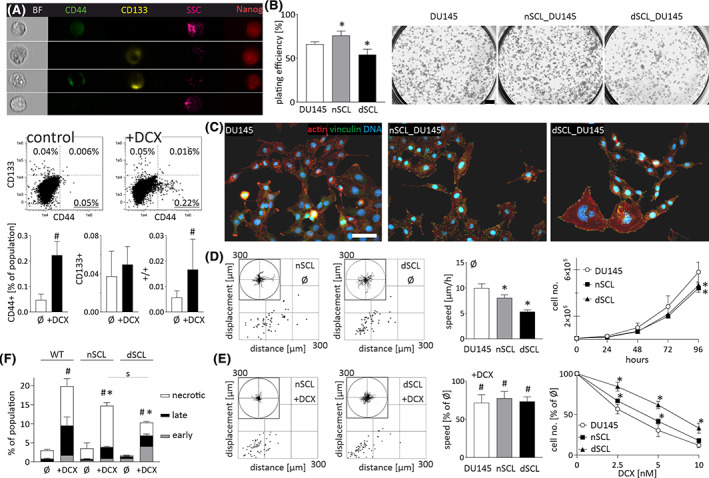
Docetaxel (DCX)‐resistance and differentiation potential of DU145 stem cell‐like (SCL) CD44+ cells. A, Abundance of CD133+/CD44+ SCL cells in DU145_DCX20 and DU145_DCX50 populations (calculated as % of total cell number) in the absence/presence of DCX (10 nM). The values in compensated dot‐plots represent relative SCL fractions (N = 50 000). B, Clonogenic activity of CD44+ SCL progenies (500/cm2) estimated with CBB R250 staining. Scale bar = 2 mm. C, nSCL_DU145 and dcxSCL_DU145 cells were cultivated for 2‐6 passages and their morphology/actin cytoskeleton architecture was estimated with fluorescence microscopy. Scale bar = 50 μm. D‐F, The effect of 10 nM DCX on the motility (D, E; left), proliferation (D, E; right) and apoptosis (F) of nSCL_DU145 and dcxSCL_DU145 cells estimated with time‐lapse videomicroscopy (after 6 hours), Coulter counter (after 48 hours), and FACS (after 72 hours), respectively. Cell trajectories are depicted in circular diagrams (axis scale in μm) drawn with the initial point of each trajectory placed at the origin of the plot (registered for 6 hours; N > 50). Dot‐plots and column charts show movement parameters at the single cell and population level, respectively (plotted as % of control). Apoptosis was assessed in 50 000 AnnexinV/PI‐stained cells. The statistical significance of the differences was tested with t‐Student test (A, B, F and proliferation in D, E; #P ≤ .05 vs untreated control; *P ≤ .05 vs wild‐type [WT] lineage; s P ≤ .05 vs indicated bars) or by one‐way ANOVA followed by post hoc Tukey's HSD (D, E motility; *P ≤ .05 vs WT lineage). All results are representative of a least three independent experiments (N ≥ 3). Note the drug resistance of CD44+ SCL cells and their ability to differentiate into DCX‐resistant “bulk” lineages
3.2. CD44 + cells underlie DCX‐induced microevolution of prostate cancer drug resistance
To further confirm the involvement of SCLs in the microevolution of drug resistance, we subjected DU145 cells to the long term DCX treatment in vivo and in vitro. In vivo analyses revealed a DCX‐induced temporary arrest of DU145 tumor growth, followed by its apparent recovery (Figure 2A). This complex response indicates a gradual adaptation of prostate cancer cells to DCX. To trace this adaptation process in vitro, we intermittently exposed DU145 cells to DCX administered at increasing concentrations. 22 Thus, the microevolution of drug‐resistant DU145 lineages was induced (DU145_DCX20 and DU_DCX50). They displayed considerably higher DCX‐resistance than dcxSCL_DU145 cells, as illustrated by negligible DCX effects on their motility and proliferation (Figure 2B). FACS analyses revealed relatively ample CD44+ cells in DU145_DCX20 and DU_DCX50 populations (Figure 2C). Again, their abundance remained stable during the long term propagation of the cell line. DCX (10 nM) treatment had a minute effect on the abundance of SCLs in these lineages and on their plating efficiency (Figure 2D). The lineages derived from DU145_DCX20 SCL cells in the absence/presence of DCX (nSCL_DU145_DCX20 and dcxSCL_DU145_DCX20 cells, respectively) displayed a phenotype corresponding to “maternal” cells, that is, a stable nonpolarized morphology (not shown), paralleled by slightly lower proliferation and motility rates (Figure 2E). Their response to 10 nM DCX was similar to that of maternal cells, whereas SCL progenies showed less pronounced apoptotic response to DCX treatment (Figure 2F). This was correlated with the high efficiency of efflux systems as illustrated by calcein efflux assays (Figure 2G). Corresponding reactions to DCX were observed in the progenies of CD44+ PC3_DCX20 cells, which retained very high DCX resistance (Figure S4), whereas the progenies of CD133+ DU145_DCX20 cells were less drug resistant than their CD44+‐derived counterparts (Figure S5). Thus, we confirmed the relevance of DCX‐induced clonal SCL expansion for the microevolution of prostate cancer drug‐resistance in vitro and in vivo.
FIGURE 2.
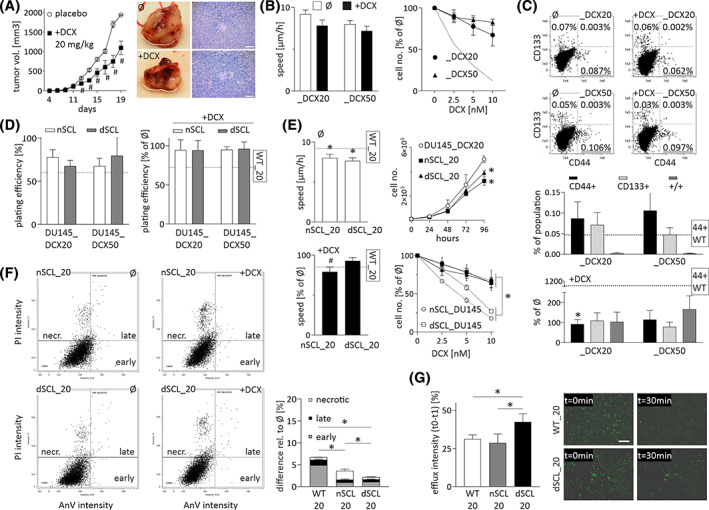
CD44+ stem cell‐like (SCL) cells participate in the microevolution of prostate cancer drug‐resistance in vitro and in vivo. A, DU145 cells were subcutaneously injected into abdominal flank of Severe Combined ImmunoDeficiency (SCID) mice and tumors growth was estimated for 2 to 4 weeks in the presence/absence of docetaxel (injected at 20 mg/kg b.w.). At least 10 animals were taken for each variant (N > 10). Scale bar = 250 μm. B, Effect of docetaxel (DCX) on the motility and proliferation of DU145_DCX20 and DU145_DCX50 cells estimated after 6 and 48 hours of incubation, respectively. Dotted line illustrates the data for wtDU145 cells. C, The abundance of CD133+/CD44+ SCL cells in DCX (10 nM)‐treated DU145_DCX20 and DU145_DCX50 populations (calculated as % of total cell number). D, Clonogenic potential of CD44+ SCL cell progenies (cf. Figure 1B; dotted line = wtDU145 cells). E, Effect of 10 nM DCX on motility (left) and proliferation (right) of nSCL_and dcxSCL‐derived lineages of DU145_DCX20 and DU145_DCX50 cells. F, Cellular apoptotic response to 10 nM DCX estimated with AnnexinV/PI assay. G, Calcein efflux intensity in nSCL_and dcxSCL_DU145 populations. Scale bar = 200 μm. The statistical significance of the differences was tested with t‐Student test (A, C, D, F, G and proliferation in B, E; #P ≤ .05 vs untreated control; *P ≤ .05 vs wild‐type [WT] lineage or selected bars) or by one‐way ANOVA followed by post hoc Tukey's HSD (A and motility in B, E; #P ≤ .05 vs untreated control; *P ≤ .05 vs WT lineage (or selected bars/points). All results are representative of a least three independent experiments (N ≥ 3). Note the microevolution of prostate cancer drug resistance under DCX stress in vivo and in vitro
3.3. Fenofibrate impairs drug resistance of CD44 + SCL cell offspring
We have previously shown the interference of FF with the drug resistance of prostate cancer cells. 22 Analyses of the cytostatic effects of combined DCX/FF treatment on the progenies of CD44+ SCL cells demonstrated the attenuating effect of FF on their DCX‐resistance (Figure 3A). This is illustrated by the inhibition of DU145, nSCL_DU145, and dcxSCL_DU145 cell motility immediately after DCX/FF administration. Corresponding effects were seen in the populations of DU145_DCX20, nSCL_DCX20, dcxSCL_DCX20 cells. These responses were followed by impairment of their clonal efficiency (Figure S6), the retardation of cell proliferation (Figure 3B, cf. Figure 2E for DCX activity) and accompanied by the decomposition of microtubular cytoskeleton, which apparently results from the impaired activity of efflux systems in the analyzed cells (Figure 3C). The cells that survived long term (48 hours) combined DCX/FF treatment displayed nonpolarized morphology, disrupted cytoskeleton, and relatively low motility (Figure 3D). Similar DCX/FF activity was observed in the populations derived from CD44+ DU145_DCX50 SCL cells (Figure S7), CD133+ DU145 SCL cells (Figure S8A), and from CD44+ PC3 SCL cells (Figure S8B). On the other hand, we also observed less pronounced reactions of nSCL and dcxSCL cells to the combined DCX/FF treatment (cf. Figure 3D; Figure S8), which were accompanied by their diminished apoptotic response (cf. Figure 3E). These observations show that DCX‐directed clonal expansion of SCL cells does not prompt the microevolution of DCX/FF resistance; however DCX‐resistant cells can partly adapt to the combined DCX/FF treatment.
FIGURE 3.
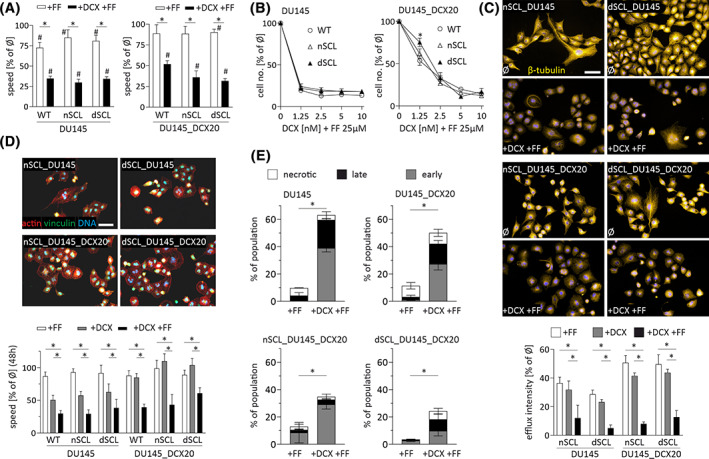
Stem cell‐like (SCL) cell derived DU145 lineages display sensitivity to the combined docetaxel/fenofibrate (DCX/FF) treatment. A, Motility of naïve DU145 and DU145_DCX20 cells, and their counterparts derived from naive and DCX‐treated SCL cells (nSCL_ and dcxSCL, respectively) was estimated with time‐lapse videomicroscopy immediately after the administration of DCX and/or FF (10 nM/25 μM). B, Cells were treated with DCX and/or FF as in A and counted with Coulter counter after 48 hours. C, The architecture of microtubular cytoskeleton was estimated with immunofluorescence after 48 hours of DCX/FF treatment along with calcein efflux assay (lower panel; cf. Figure 1G). D, Cells were treated with DCX/FF for 48 hours and their morphology/actin cytoskeleton architecture was visualized with immunofluorescence. Time‐lapse videomicroscopy was employed to assess long‐term (48 hours) effects of FF, DCX and DCX/FF on cell motility (lower panel). E, Cells were DCX/FF treated for 72 hours and their apoptotic response was estimated by annexinV/PI assay. Compensated dot‐plots comprise 30 000/50 000 events, classified based on their bright‐field ratios and/or nuclear contrast. Scale bars = 50 μm. D, The statistical significance of the differences was tested with t‐Student test (B, C, E; #P ≤ .05 vs untreated control; *P ≤ .05 vs wild‐type [WT] lineage or selected bars) or by one‐way ANOVA followed by post hoc Tukey's HSD (A, D; *P ≤ .05 vs WT lineage or selected bars/points). Note the sensitivity of SCL‐derived lineages to the combined DCX/FF‐treatment
3.4. SCL cells are resistant to the combined DCX/FF treatment
Further analyses were performed to address the mechanisms underlying cell adaptation to the combined DCX/FF stress. The role of SCL cells in this process is indicated by their increased numbers in DCX/FF‐treated DU145 populations (Figure 4A). Flow cytometric studies revealed considerably increased fractions of CD133+, CD133+/CD44+, and CD44+ cells in DCX/FF‐treated DU145 (0.82% vs 0.011% in DCX‐treated variant) and PC3 populations (Figure S9A). Furthermore, the appearance of poly(morpho)nuclear giant cells (PGCs; Figure 4B) and the expression profile of multipotency markers in DCX/FF‐treated populations of DU145 cells revealed the interrelations between DCX/FF‐activated PGCs and the generation of SCL cells. In particular, CD44+ expression was observed in PGCs (Figure 4C). Concomitantly, scattered Nanog+ and Oct+ cells were seen in DCX/FF‐treated DU145 specimens. Less prominent, although significant increase of SCL fraction was also observed in DCX/FF‐treated populations of DU145_DCX20 and DU145_DCX50 cells (Figure 4A). It was accompanied by a less prominent induction of PGC phenotype 22 and by a higher clonogenic potential of DU145_DCX20/50 SCL cells than of their wild‐type counterparts (Figure 4D). These observations confirm the activation of a self‐defense system(s) in prostate cancer cell populations in response to the DCX/FF stress. It leads to the formation of DCX/FF‐resistant CD44+ SCL cell population via stress‐induced activation of PGCs.
FIGURE 4.
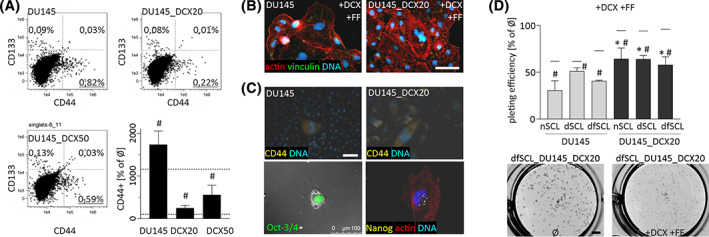
Docetaxel/fenofibrate (DCX/FF)‐treatment activates the generation of PGCs. A, FACS analyses of the abundance of CD133+ and/or CD44+ cells in the populations of wild‐type and DCX‐resistant DU145 lineages under DCX/FF stress. Compensated dot‐plots comprise 50 000 events, classified based on their bright‐field ratios and nuclear contrast. The values in the plots represent relative stem cell‐like (SCL) fractions. Dotted lines in bar plot illustrate SCL percentages in control (100%) and after DCX treatment. B, DU145 cells were cultivated in the presence of DCX/FF for 48 hours, stained against vinculin (green), F‐actin (red), and DNA (blue) to show polymorphonuclear cells. C, Cells were treated as in B, followed by their staining against CD44, Oct3/4, Nanog and CD44, and polymorphonuclear cells were vizualized with fluorescence microscopy. Scale bars = 50 μm. D, Clonogenic potential of CD44+ SCL cells derived from wild‐type and DCX‐resistant DU145 populations cultivated in the presence of DCX/FF. Horizontal lines above bars represent plating values upon DCX treatment. Scale bar = 2 mm. The statistical significance of the differences was tested with t‐Student test (A, D; #P ≤ .05 vs untreated control; *P ≤ .05 vs wild‐type [WT] lineage[s] or selected bars). All results are representative of a least three independent experiments (N ≥ 3). Note the increased fraction of PGCs and the abundance of CD44+ cells in DCX/FF‐treated cell populations
3.5. Progeny of DCX/FF‐resistant SCL cells display relatively high resistance to DCX
In an attempt to further assess the consequences of DCX/FF treatment for SCL cells' potential, we focused on the “bulk” progeny of DCX/FF‐treated SCL cells (dcx/ffSCL_DU145 and dcx/ffSCL_DCX20) in vitro and in vivo. They displayed remarkable DCX‐resistance, while remaining sensitive to the combined DCX/FF treatment. This is illustrated by the inhibition of their motility (Figure 5A), proliferation (Figure 5B), and by the induction of their apoptosis under DCX/FF stress. Concomitant disorganization of microtubular cytoskeleton (Figure 5C) could be ascribed to the FF‐induced inhibition of P‐gp, 22 as also determined for DU145wt and DU145_DCX20 cells (cf. Figure 3B). In vivo assays revealed the initial growth arrest of DCX/FF‐treated DU145_DCX20 tumors, accompanied by the presence of CD44+ cells, and followed by tumor growth recovery from DCX/FF stress (Figure 5D). Concomitantly, the attenuation of dcx/ffSCL_DU145 and dcx/ffSCL_DCX20 cell invasiveness under DCX/FF stress (Figure 5E) was paralleled by their epithelioid phenotype, low proliferation and motility in DCX/FFlow conditions (Figure 5F). Thus, these cells are predestined to form the local barriers that potentially reduce the intratumor bioavailability of DCX/FF. Together with the increased DCX‐resistance of dcx/ffSCL_DCX20 cells (Figure 5B; cf. Figure 2E), this may account for the discrepancy between cell DCX/FF‐sensitivity in vitro and tumor growth recovery in vivo. Along with the corresponding data on CD44+ PC3 and CD133+ DU145 cells (Figure S9), our observations confirm the role of CD44+/CD133+cells as a “reservoir” of drug resistance. Phenotypic reprogramming and restricted DCX/FF bioavailability may facilitate the adaptation of prostate tumors to the combined DCX/FF stress.
FIGURE 5.
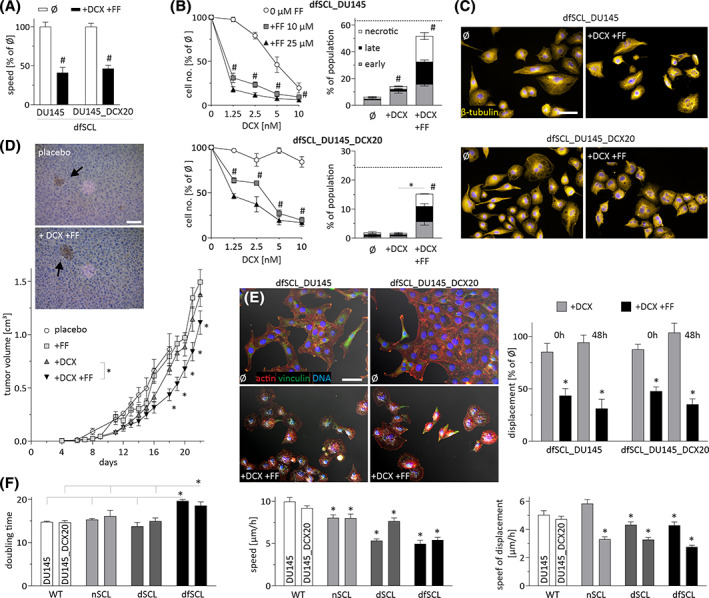
DU145 stem cell‐like (SCL) cells generate docetaxel/fenofibrate (DCX/FF)‐sensitive offspring. A,B, dcx/ffSCL_DU145 and dcx/ffSCL_DCX20 cells were cultivated in the presence of DCX and/or FF (10 nM/25 μM). Their motility (A), proliferation, and apoptotic response (B) was quantified after 6, 48 and 72 hours, respectively. C, Cells were cultivated as in A and the architecture of microtubular cytoskeleton was estimated after 48 hours of in DCX/FF treatment. Scale bar = 50 μm. D, DU145_DCX20 cells were subcutaneously injected into abdominal flank of Severe Combined ImmunoDeficiency (SCID) mice and the growth of tumors was estimated for 2 to 4 weeks in the presence/absence of DCX (10 mg/kg) and FF (60 mg/kg). At least 10 animals were taken for each variant (N > 10). E, The morphology and displacement of dfSCL_DU145 and _DCX20 cells quantified after 48 hours of DCX or DCX/FF treatment. Scale bar = 50 μm. F, Doubling times and motility rates of naïve DU145 cell lineages and their SCL counterparts. The statistical significance of the differences was tested with t‐Student test (D and proliferation in B, F; #P ≤ .05 vs untreated control; *P ≤ .05 vs wild‐type [WT] lineage(s) or selected bars, or by one‐way ANOVA followed by post hoc Tukey's HSD (A and motility in B, E, F; *P ≤ .05 vs WT lineage(s); #P ≤ .05 vs untreated control). All results are representative of a least three independent experiments (N ≥ 3). Note the increased DCX‐resistance and slightly reduced sensitivity of dfSCL_DU145 lineages to the combined DCX/FF treatment
3.6. DCX/FF‐induced phenotypic microevolution increases cell malignancy
Furthermore, we focused on the effect of limited DCX/FF bioavailability on the phenotype of drug‐resistant cells. Long term propagation (>30 passages, 1:8) of the progenies of naive and DCX/FF‐treated CD44+ cells in the absence of both agents enabled us to establish DU145 lineages, characterized by reduced resistance to DCX (Figure 6A). For instance, the proliferation of dfSCL_DCX20/LT cells in the presence of 5 nM DCX was ca. twofold slower than that of dfSCL_DCX20 cells. Attenuated growth and viability of dfSCL_DCX20/LT cells in the presence of DCX was accompanied by the decreased activity of drug‐efflux systems (Figure 6B) and increased invasive potential of LT cells. This is illustrated by their increased motility in control conditions (Figure 6C) and their increased capability of penetrating Matrigel‐coated microporous membranes (Figure 6D). Concomitantly, we observed increased fractions of Snail‐1high dfSCL_DCX20/LT cells in comparison to their drug‐resistant counterparts (Figure 6E). Together with their rear‐front polarized, invasive morphology (characteristic for post‐epithelial‐mesenchymal transition [EMT] cells; Figure 6F), they confirm that the limited DCX/FF bioavailability can prompt the reversed microevolution of drug‐resistant cell lineages toward the expansive phenotype. This notion was further substantiated by relatively weak effects of the combined DCX/FF treatment on the invasiveness of dfSCL_DCX20/LT cells (Figure 6G). Thus, the oscillations of intratumoral DCX/FF bioavailability may occasionally facilitate the microevolution of DCX/FF‐resistant prostate cancer cell lineages. However, this effect should not be overestimated as other DCX‐resistant and DCX‐sensitive DU145_SCL progenies retained sensitivity to the combined DCX/FF treatment (Figure 6G).
FIGURE 6.
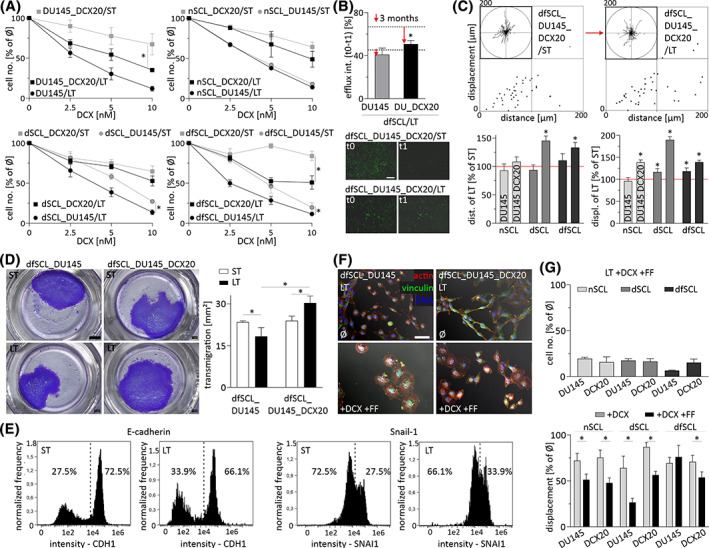
Reversed microevolution of drug‐resistant DU145 cells increases their invasive potential. A, Proliferation of SCL‐derived DU145 lineages (ST) and their (LT) counterparts estimated after 48 hour‐long docetaxel (DCX) treatment. B, Calcein efflux intensity in dfSCL_DU145/LT and dfSCL_DU145_DCX20/LT in comparison to their ST counterparts (indicated by dotted lines/arrows). Scale bar = 200 μm. C, Motility of dfSCL_DU145/LT and dfSCL_DU145_DCX20/LT measured as % of ST control. D, Transmigration of dfSCL_DU145/LT and dfSCL_DU145_DCX20/LT cells through microporous membranes. Scale bar = 2 mm. E, Relative fractions of Snail1‐/E‐cadherinhigh cells in dfSCL_DU145_DCX20/LT populations quantified with ImageStream. F, Morphology of DCX/fenofibrate (FF)‐treated dfSCL_DU145/LT and dfSCL_DU145_DCX20/LT cells. Scale bar = 50 μm. G, Effect of DCX and/or FF on the proliferation (upper panel) and displacement (lower panel) of LT DU145 and DU145_DCX20 lineages. All results are representative of a least three independent experiments (N ≥ 3). The statistical significance of the differences was tested with t‐Student test (A, B, D; #P ≤ .05 vs untreated control; *P ≤ .05 vs ST lineage or selected bars/points) or by one‐way ANOVA followed by post hoc Tukey's HSD (C, G; *P ≤ .05 vs wild‐type [WT] lineage or selected bars/points). Note the increased invasiveness of drug‐sensitive/Snail‐1high cells undergone reversed microevolution under the limited DCX/FF bioavailability
4. DISCUSSION
“Clonal evolution of cancer stem cells” is believed to determine stress‐induced cancer microevolution. 18 , 37 Drug‐resistance, self‐renewal capacity, and multipotency of CSCs underlie their survival under chemotherapeutic stress in vivo and the selective expansion of their stress‐resistant progenies. 5 , 7 Interference of FF with prostate cancer drug‐resistance 22 suggests its potential application in metronomic therapies of prostate tumors. Following these facts, we focused on the reactions of CD44+ SCL cells to the combined DCX/FF treatment and on their involvement in DCX/FF‐induced prostate cancer microevolution. We further traced the consequences of DCX/FF‐induced selective expansion of CD44+ SCL cells and their differentiation into hyper‐resistant “bulk” progenies. In particular, we addressed SCL cell involvement in the adaptation of cell populations to the combined DCX/FF treatment in vitro and in vivo. These studies confirmed that FF can be applied for the treatment of drug‐resistant prostate tumors. 22 However, they also revealed the transitions between CD44+ SCL and CD44− phenotype, which maintain phenotypic “steady‐state” of prostate cancer cell populations. FF amplifies the expansion of hyper‐resistant CD44+ SCL cells and their progenies, limiting the effectiveness of FF‐based treatment strategies.
Prostate cancer CD44+ SCL cells are commonly observed in vitro. In serum‐free conditions their differentiation is inhibited, which petrifies their “primitive” phenotype. 32 , 38 We observed the persistence of CD44+/Nanog+/Oct‐4+ DU145/PC3 cells during the long term propagation of prostate cancer cells, accompanied by the differentiation of CD44+ SCL cells toward CD44− phenotype(s). These observations suggest a continuous self‐renewal of CD44+ cells in the presence of serum. It can occur through symmetric divisions of CD44+ SCL cells and/or through asymmetric divisions of “retro‐differentiating” CD44− “bulk” cells. Apparently, bidirectional transitions between CD44+ and CD44− phenotype maintain the phenotypic “steady‐state” of heterogeneous prostate cancer cell populations. These transitions also determine the selective expansion of drug‐resistant prostate cancer cell sub‐populations under the chemotherapeutic stress. Their involvement in the microevolution of drug‐resistance and the role of CD44+ cells as a “reservoir” of drug resistance 13 , 14 is illustrated by their increased fractions observable in DCX‐treated DU145 and PC3 populations and increased drug resistance of their progenies. CD133+ cells may play a corresponding role. However, differences in the reactivity of CD44+ and CD133+ cell progenies to DCX/FF (generally lower resistance of CD133+‐derived cells) prompt questions whether they represent a discrete subpopulation or are interspersed with CD44+ population(s). 39
Analyses of DU145 and PC3 reactions to the combined DCX/FF treatment confirmed the potential of FF for the treatment of drug‐resistant prostate tumors. It is illustrated by cytostatic and pro‐apoptotic effects that we observed in DCX/FF‐treated “bulk” (CD44−) progenies of naive and DCX‐induced SCL cells. Notably, CD44+ SCL cells remained resistant to the combined chemotherapeutic/metabolic stress; however it prompted their differentiation toward “hyper‐resistant” phenotype(s). This finding reveals the adaptation/survival strategy, which potentially prompts the microevolution of prostate tumor drug resistance. Several lines of evidence indicate the involvement of poly(morpho)nuclear cells (PGCs) in this process. First, PGCs are known to act as cellular “spores” that survive the microenvironmental cataclysm and give rise to CSCs. 40 , 41 , 42 Then, increased fractions of polyploid PGCs cells in DCX/FF‐treated cell populations (8.9% in comparison to 0.8% in control) 22 correlated with the abundance of SCL cells. This correlation was accompanied by the presence of CD44 in DCX/FF‐induced PGCs. The origins of PGCs, in particular the involvement of big mononuclear (diploid) cells in their generation, and the budding of SCL cells from PGCs remains to be directly confirmed. Further transcriptomic, proteomic and lipidomic analyses of PGCs and SCL cells should also help to define PGC/SCL phenotype in the absence/presence of chemotherapeutic/metabolic stress. They are necessary to elucidate whether chemotherapy‐induced development of drug resistance follows strictly a CSC program. 18 , 31 However, our findings collectively reveal DCX/FF‐induced acquisition of PGC phenotype by the “bulk” cells and indicate the potential role of PGCs in SCL cell generation. DCX/FF‐induced activation of this sequence apparently facilitates chemotherapeutic/metabolic stress‐induced microevolution of prostate cancer drug resistance.
Biological significance of these events was confirmed by our in vivo data. Inhibitory effect of DCX on the growth of wild‐type DU145 tumors, followed by their recovery after the initial DCX‐induced arrest, can be explained by the microevolution of DCX‐resistance in DU145 populations. Somewhat surprising recovery of wild‐type and drug‐resistant DU145 tumors observed in the presence of DCX/FF can be explained by the evolution of cellular hyper‐resistance to DCX. Concomitant dormancy, immobility and epithelioid phenotype of super‐resistant cells facilitates the formation of DCX/FFlow tumor compartments. When subsequently colonized by “hyper‐resistant” CD44+/CD133+ cells, these compartments may further promote the growth of tumors under chemotherapeutic/metabolic stress. Additionally, DCX/FF may selectively promote/impair the expansion of discrete cell lineages within heterogeneous mass of tumor cells. In our hands, lineage‐specificity of DCX/FF resistance was illustrated by relatively low clonal capacity of the direct CD44+ PC3 progenies under DCX/FF stress, accompanied by their considerable invasive potential after prolonged expansion. Reversed microevolution of drug‐sensitive SCL/“bulk” cells induced by low DCX/FF bioavailability may further participate in this process, as indicated by the signs of EMT in DU145 populations after long term cultivation in the absence of DCX/FF. Thus, the synergy of cytostatic, anti‐invasive, and pro‐apoptotic DCX/FF effects in vitro confirm the potential of the metabolic chemotherapy for palliative treatment of drug‐resistant prostate tumors. However, the adaptation of prostate tumors to metabolic stress may counteract this effect, determining tumor recovery from the combined DCX/FF stress in vivo. 22 , 30
Collectively, our data indicate that bidirectional transitions between metastable CD44− “bulk” and CD44+/CD133+ SCL phenotype maintain the phenotypic “steady‐state” of heterogeneous prostate cancer cell populations and their microevolution toward microenvironmentally favorable phenotypes. Accordingly, chemotherapeutic stress biases phenotypic “steady‐state” of prostate cancer cell populations toward the generation of increasingly drug‐resistant CD44+ cells by their increasingly drug‐resistant CD44− progenies. Combined DCX/FF stress amplifies this effect by inducing the adaptation responses of CD44− bulk cells. Apart from growth retardation and autophagy, 43 , 44 they include the formation of PGCs, 40 which apparently generate CD44+ SCL cells that initiate the expansion of super‐resistant CD44− cell lineages. Concomitant reconstitution of intratumoral barriers (potentially limiting DCX/FF bioavailability in intratumoral niches) prompts the reverse microevolution of resident cells toward more invasive post‐EMT phenotype. Thus, also the transitions between mesenchymal and epithelioid phenotype can participate in a progressive prostate tumor adaptation to the combined chemotherapeutic/metabolic stress in vivo. 45 , 46 The mechanisms underlying PGC generation, mesenchymal‐epithelial transition/EMT‐related adaptation processes, interrelations between CD44+ and CD133+ phenotypes and their relation to the “true” CSC phenotype require further research. However, we provide the evidence for the cooperation of CD44+/CD133+ cells and their clonally expanding progenies in the prostate tumor adaptation to the chemotherapeutic/metabolic stress, hampering the effectiveness of prostate cancer treatment.
5. CONCLUSION
Our study confirms the potential of FF for the treatment of drug‐resistant prostate tumors. This agent apparently reduces effective doses of chemotherapeutics via the interference with the drug‐resistance systems in prostate cancer cells, thus being the potential tool to improve the living standard of prostate cancer patients. However, we also reveal the limitations of metronomic prostate cancer treatment strategies. Interference of FF with the transitions between CD44+ SCL and CD44− phenotype affects phenotypic “steady‐state” of prostate cancer cell populations and biases their microevolution toward drug‐resistant phenotype. This activity can petrify/augment tumor drug‐resistance and prompt cancer relapse via allowing super‐resistant cells to repopulate old “niches” and reconstitute the tumor in its “hyper‐resistant” form. Thus, the adaptation responses of prostate cancer cells to FF‐induced metabolic stress can progressively reduce the efficiency of combined DCX/FF application in the palliative therapy of prostate cancer. This stresses the need for the comprehensive look on the functions of CSCs in prostate cancer drug resistance and for a more thorough elucidation of side effects and restrictions of metronomic anticancer strategies as the leading challenges of the current oncology.
ACKNOWLEDGEMENT
The open‐access publication of this article was funded by the Priority Research Area BioS under the program “Excellence Initiative –Research University” at the Jagiellonian University in Krakow.
CONFLICT OF INTEREST
The authors declared no potential conflicts of interests.
AUTHOR CONTRIBUTIONS
T.W.: conception and design, provision of study material or patients, collection and/or assembly of data, data analysis and interpretation, manuscript writing, final approval of manuscript; M.L.: provision of study material or patients; J.C., M.S.: collection and/or assembly of data, data analysis and interpretation; E.K., G.D.: collection and/or assembly of data; K.P.: data analysis and interpretation; D.R.: conception and design, collection and/or assembly of data; E.Z.‐S., M.S., Z.M., M.E.: data analysis and interpretation, administrative support; J.C.: conception and design, financial support, administrative support, data analysis and interpretation, manuscript writing, final approval of manuscript.
Supporting information
Appendix S1: Supporting information
Wróbel T, Luty M, Catapano J, et al. CD44 + cells determine fenofibrate‐induced microevolution of drug‐resistance in prostate cancer cell populations. Stem Cells. 2020;38:1544–1556. 10.1002/stem.3281
Funding information Narodowe Centrum Nauki, Grant/Award Number: 2015/17/B/NZ3/01040
DATA AVAILABILITY STATEMENT
The data that supports the findings of this study are available in the supplementary material of this article or available from the corresponding author upon reasonable request.
REFERENCES
- 1. Sell S. On the stem cell origin of cancer. Am J Pathol. 2010;176:2584‐2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Batlle E, Clevers H. Cancer stem cells revisited. Nat Med. 2017;23:1124‐1134. [DOI] [PubMed] [Google Scholar]
- 3. Rybak AP, Bristow RG, Kapoor A. Prostate cancer stem cells: deciphering the origins and pathways involved in prostate tumorigenesis and aggression. Oncotarget. 2015;6:1900‐1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shibata M, Shen MM. The roots of cancer: stem cells and the basis for tumor heterogeneity. Bioessays. 2013;35:253‐260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rich JN. Cancer stem cells: understanding tumor hierarchy and heterogeneity. Medicine (Baltimore). 2016;95:S2‐S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Heppner GH. Tumor heterogeneity. Cancer Res. 1984;44:2259‐2265. [PubMed] [Google Scholar]
- 7. Najafi M, Mortezaee K, Majidpoor J. Cancer stem cell (CSC) resistance drivers. Life Sci. 2019;234:116781. [DOI] [PubMed] [Google Scholar]
- 8. Prieto‐Vila M, Takahashi RU, Usuba W, et al. Drug resistance driven by cancer stem cells and their niche. Int J Mol Sci. 2017;8(12):2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Alison MR, Lim SM, Nicholson LJ. Cancer stem cells: problems for therapy? J Pathol. 2011;223:147‐161. [DOI] [PubMed] [Google Scholar]
- 10. Maitland NJ, Collins A. A tumour stem cell hypothesis for the origins of prostate cancer. BJU Int. 2005;96:1219‐1223. [DOI] [PubMed] [Google Scholar]
- 11. McIntosh K, Balch C, Tiwari AK. Tackling multidrug resistance mediated by efflux transporters in tumor‐initiating cells. Expert Opin Drug Metab Toxicol. 2016;12:633‐644. [DOI] [PubMed] [Google Scholar]
- 12. Zhao J. Cancer stem cells and chemoresistance: the smartest survives the raid. Pharmacol Ther. 2016;160:145‐158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Harris KS, Kerr BA. Prostate cancer stem cell markers drive progression, therapeutic resistance, and bone metastasis. Stem Cells Int. 2017;2017:8629234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Skvortsov S, Skvortsova II, Tang DG, Dubrovska A. Concise review: prostate cancer stem cells: current understanding. Stem Cells. 2018;36:1457‐1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66:7‐30. [DOI] [PubMed] [Google Scholar]
- 16. Hurria A, Togawa K, Mohile SG, et al. Predicting chemotherapy toxicity in older adults with cancer: a prospective multicenter study. J Clin Oncol. 2011;29:3457‐3465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Maitland NJ, Collins AT. Prostate cancer stem cells: a new target for therapy. J Clin Oncol. 2008;26:2862‐2870. [DOI] [PubMed] [Google Scholar]
- 18. van Niekerk G, Davids LM, Hattingh SM, Engelbrecht AM. Cancer stem cells: a product of clonal evolution? Int J Cancer. 2017;140:993‐999. [DOI] [PubMed] [Google Scholar]
- 19. Li JJ, Shen MM. Prostate stem cells and cancer stem cells. Cold Spring Harb Perspect Med. 2019;9:a030395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lang SH, Frame FM, Collins AT. Prostate cancer stem cells. J Pathol. 2009;217:299‐306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nie D. Cancer stem cell and niche. Front Biosci (Schol Ed). 2010;2:184‐193. [DOI] [PubMed] [Google Scholar]
- 22. Luty M, Piwowarczyk K, Labedz‐Maslowska A, et al. Fenofibrate augments the sensitivity of drug‐resistant prostate cancer cells to docetaxel. Cancers (Basel). 2019;11:77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. McKeage K, Keating GM. Fenofibrate: a review of its use in dyslipidaemia. Drugs. 2011;71:1917‐1946. [DOI] [PubMed] [Google Scholar]
- 24. Piwowarczyk K, Wybieralska E, Baran J, et al. Fenofibrate enhances barrier function of endothelial continuum within the metastatic niche of prostate cancer cells. Expert Opin Ther Targets. 2015;19:163‐176. [DOI] [PubMed] [Google Scholar]
- 25. Wybieralska E, Szpak K, Gorecki A, et al. Fenofibrate attenuates contact‐stimulated cell motility and gap junctional coupling in DU‐145 human prostate cancer cell populations. Oncol Rep. 2011;26:447‐453. [DOI] [PubMed] [Google Scholar]
- 26. Lian X, Wang G, Zhou H, Zheng Z, Fu Y, Cai L. Anticancer properties of fenofibrate: a repurposing use. J Cancer. 2018;9:1527‐1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Panigrahy D, Kaipainen A, Huang S, et al. PPARalpha agonist fenofibrate suppresses tumor growth through direct and indirect angiogenesis inhibition. Proc Natl Acad Sci USA. 2008;105:985‐990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Piwowarczyk K, Kwiecien E, Sosniak J, et al. Fenofibrate interferes with the diapedesis of lung adenocarcinoma cells through the interference with Cx43/EGF‐dependent intercellular signaling. Cancers (Basel). 2018;10:363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wilk A, Wyczechowska D, Zapata A, et al. Molecular mechanisms of fenofibrate‐induced metabolic catastrophe and glioblastoma cell death. Mol Cell Biol. 2015;35:182‐198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hirpara J, Eu JQ, Tan JKM, et al. Metabolic reprogramming of oncogene‐addicted cancer cells to OXPHOS as a mechanism of drug resistance. Redox Biol. 2019;25:101076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Valent P, Bonnet D, De Maria R, et al. Cancer stem cell definitions and terminology: the devil is in the details. Nat Rev Cancer. 2012;12:767‐775. [DOI] [PubMed] [Google Scholar]
- 32. Rybak AP, He L, Kapoor A, Cutz JC, Tang D. Characterization of sphere‐propagating cells with stem‐like properties from DU145 prostate cancer cells. Biochim Biophys Acta. 2011;1813:683‐694. [DOI] [PubMed] [Google Scholar]
- 33. Wang L, Huang X, Zheng X, et al. Enrichment of prostate cancer stem‐like cells from human prostate cancer cell lines by culture in serum‐free medium and chemoradiotherapy. Int J Biol Sci. 2013;9:472‐479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Piwowarczyk K, Paw M, Ryszawy D, et al. Connexin43high prostate cancer cells induce endothelial connexin43 up‐regulation through the activation of intercellular ERK1/2‐dependent signaling axis. Eur J Cell Biol. 2017;96:337‐346. [DOI] [PubMed] [Google Scholar]
- 35. Baran B, Bechyne I, Siedlar M, et al. Blood monocytes stimulate migration of human pancreatic carcinoma cells in vitro: the role of tumour necrosis factor ‐ alpha. Eur J Cell Biol. 2009;88:743‐752. [DOI] [PubMed] [Google Scholar]
- 36. Sroka J, Antosik A, Czyz J, et al. Overexpression of thioredoxin reductase 1 inhibits migration of HEK‐293 cells. Biol Cell. 2007;99:677‐687. [DOI] [PubMed] [Google Scholar]
- 37. Marusyk A, Polyak K. Cancer. Cancer cell phenotypes, in fifty shades of grey. Science. 2013;339:528‐529. [DOI] [PubMed] [Google Scholar]
- 38. Weiswald LB, Bellet D, Dangles‐Marie V. Spherical cancer models in tumor biology. Neoplasia. 2015;17:1‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Eun K, Ham SW, Kim H. Cancer stem cell heterogeneity: origin and new perspectives on CSC targeting. BMB Rep. 2017;50:117‐125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhang S, Mercado‐Uribe I, Xing Z, Sun B, Kuang J, Liu J. Generation of cancer stem‐like cells through the formation of polyploid giant cancer cells. Oncogene. 2014;33:116‐128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Liu J. The dualistic origin of human tumors. Semin Cancer Biol. 2018;53:1‐16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bharadwaj D, Mandal M. Senescence in polyploid giant cancer cells: a road that leads to chemoresistance. Cytokine Growth Factor Rev. 2020;52:68‐75. [DOI] [PubMed] [Google Scholar]
- 43. Cristofani R, Montagnani MM, Cicardi ME, et al. Dual role of autophagy on docetaxel‐sensitivity in prostate cancer cells. Cell Death Dis. 2018;9:889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Smith AG, Macleod KF. Autophagy, cancer stem cells and drug resistance. J Pathol. 2019;247:708‐718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Brabletz T. EMT and MET in metastasis: where are the cancer stem cells? Cancer Cell. 2012;22:699‐701. [DOI] [PubMed] [Google Scholar]
- 46. Ryszawy D, Sarna M, Rak M, et al. Functional links between Snail‐1 and Cx43 account for the recruitment of Cx43‐positive cells into the invasive front of prostate cancer. Carcinogenesis. 2014;35:1920‐1930. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1: Supporting information
Data Availability Statement
The data that supports the findings of this study are available in the supplementary material of this article or available from the corresponding author upon reasonable request.