

Summary
Lysosomes are dynamic organelles that serve as regulatory hubs in cellular homeostasis. Changes in lysosome morphology, composition, and turnover are typically linked to disease. These characteristics make enrichment protocols based on biophysical parameters challenging. However, organelle enrichment methods are essential to facilitate their biomolecular analysis. We describe the synthesis and use of superparamagnetic iron oxide nanoparticles (SPIONs) for high-yield purification of lysosomes compatible with “omics” analysis. NANOLYSE (Nanoparticles for Lysosome Isolation) provides a reliable strategy in fingerprinting the biomolecular composition of lysosomes.
For complete details on the use and execution of this protocol, please refer to Tharkeshwar et al. (2017).
Graphical Abstract
Highlights
-
•
Lysosome isolation from adherent cells using superparamagnetic iron oxide nanoparticles
-
•
Enriched lysosomes with high intactness, compatible for functional, biochemical studies
-
•
NANOLYSE is highly reproducible and compatible for proteome and lipidome profiling
-
•
NANOLYSE is an unbiased method to identify lysosomal dyshomeostasis in a disease context
Lysosomes are dynamic organelles that serve as regulatory hubs in cellular homeostasis. Changes in lysosome morphology, composition, and turnover are typically linked to disease. These characteristics make enrichment protocols based on biophysical parameters challenging. However, organelle enrichment methods are essential to facilitate their biomolecular analysis. We describe the synthesis and use of superparamagnetic iron oxide nanoparticles (SPIONs) for high-yield purification of lysosomes compatible with “omics” analysis. NANOLYSE (Nanoparticles for Lysosome Isolation) provides a reliable strategy in fingerprinting the biomolecular composition of lysosomes.
Before You Begin
Cleaning and Preparation of Glassware
Timing: 4–5 h
Note: This part describes the cleaning and preparation of the glassware used to synthesize surface-functionalized superparamagnetic iron oxide nanoparticles (SPIONs).
CAUTION: The below described cleaning procedure involves the use of highly corrosive acidic solutions, which, when mixed, also produces a lot of heat. Not handling the solutions properly may result in adverse consequences. It is, therefore, mandatory and extremely important to wear Personal Protective Equipment (PPE) at all times during this procedure and to handle it with utmost care and attention!
Requirements: Three-neck round bottom flask, side necks angled (500 mL)
Graham Type Reflux Condenser
Glass centrifuge tube with conical bottom and cap (50 mL)
-
1.
Prepare 2 liters of piranha solution by mixing 1 part of 30% hydrogen peroxide (H2O2) and 4 parts of concentrated sulfuric acid (H2SO4) in a 5 L flat bottomed glass beaker.
CAUTION: Wear Personal Protective Equipment (PPE) before starting this process. Piranha solution must be prepared by adding sulfuric acid (H2SO4) to hydrogen peroxide (H2O2). Piranha solution is very dangerous, highly corrosive, and mixing the solutions as such is extremely exothermic. Therefore, prepare the piranha solution with utmost care and attention!
-
2.
Using a long stainless steel tong, carefully immerse all glassware (one-by-one) in the piranha solution.
CAUTION: Before immersing, make sure that the glassware is reasonably clean and free of any organic residues from previous wash steps. Piranha solution cleans by dissolving organic contaminants, and the presence of large amounts of such contaminants will cause violent bubbling, release of gas, and may result in an explosion.
CRITICAL: Cleaning usually requires about 30–60 min. However, the temperature of the solution can, at times, be above 100°C. It is, therefore important to let the solution cool down before removing the glassware.
Pause Point: 1–3 h (cooling period)
-
3.
Using a long stainless steel forceps, slowly remove the glassware from the piranha solution and carefully wash it under running tap water.
CAUTION: Always treat the mixture as an “active” piranha solution as it may still contain traces of hydrogen peroxide. Make sure the drain is flushed with sufficient amounts of tap water and collect the used piranha solution in appropriate bins.
-
4.
Rinse glassware with acetone before placing it on a rack to air dry.
-
5.
Air dry the glassware for 30 min (Figure 1).
Figure 1.

Glassware Used in the Synthesis of SPIONs
Preparation of Chemicals
Note: All chemicals and reagents have to be at room temperature (∼20°C) before using them.
-
6.
Carefully weigh Iron (III) acetylacetonate and 1,2-hexadecanediol, as indicated in “Reagents used for SPION synthesis.”
-
7.
Place a magnetic stirrer in the 3-neck flask and carefully transfer the weighed iron (III) acetylacetonate followed by 1,2-hexadecanediol into the flask.
-
8.
Using a pipette, measure and add exact volumes (as mentioned in “Reagents used for SPION synthesis”) of oleic acid followed by oleylamine, and finally benzyl ether into the 3-neck flask.
Preparation and Plating of Cells
Note: The lysosome purification protocol described here works on adherent cells. As an example, the use of HeLa cells is described in this protocol. The cells are cultured in DMEM/F-12 medium supplemented with 10% fetal bovine serum (FBS) and grown at 37°C under 5% CO2 in an incubator.
CAUTION: All steps described below must be carried out in a biosafety cabinet and wearing appropriate PPE.
-
9.
Trypsinize and plate 1 × 106 HeLa cells on 100 mm diameter sterile culture dishes.
Alternatives: Other culture dishes and adherent cell types, including neurons, may also be used. However, this would require specific modifications and optimizations in the protocol (please refer to the troubleshooting section for more detailed information).
-
10.
Culture the cells until they reach 90% confluency.
Preparation of the Cell Homogenizer
Note: A ball-bearing cell homogenizer is used to break and homogenize cells during the magnetic isolation procedure. The device contains a tungsten carbide ball in a stainless steel block and uses two disposable syringes to pass cells repeatedly through a bore in the block. Since the diameter of the ball is not much smaller than the diameter of the bore, the cells are forced through a narrow passage that breaks them up. Using this device and with a small number of such passages (e.g., 10 passages for HeLa cells (see also the section on the magnetic isolation of lysosomes)), a quantitative and high reproducibility of cell breakage can be achieved.
CAUTION: The cell homogenizer comes along with tungsten carbide balls of different sizes and thus can be used on various cell types. However, depending on the size of the cell type in the study, it requires some optimization to choose the right size of the tungsten carbide ball. For instance, HeLa and Mouse Embryonic Fibroblast (MEF) cell types require the use of a tungsten carbide ball that provides a 10-μm clearance space to efficiently break and homogenize cells through sheer force.
Alternatives: From our experience, a ball-bearing cell cracker improves or facilitates the reproducibility of the homogenization process. We, therefore, recommend using it for homogenizing the cells. Alternatively, a dounce homogenizer can be used, but as stated above, also in this case, cell-type-specific optimizations are required to warrant consistency and reproducibility in the homogenization process.
-
11.
Prior to using the cell homogenizer, dismantle the device on a clean surface and wash it thoroughly with milliQ water.
-
12.
Use a wash bottle with a pointed nozzle to clean bores and small openings in the cell homogenizer.
-
13.
Wipe the device with clean tissue paper and let it air dry.
-
14.
Choose the right size of the tungsten carbide ball, place it in the bore of the stainless steel block, and secure the openings with the screws.
CAUTION: The tungsten carbide balls that are provided have a precise diameter with μm range differences. Keep them separated in individual tubes so that they are not mixed as it is very difficult to distinguish them again.
-
15.
At least 30 min before use, place the cell homogenizer on a surface of packed ice, and apply pressure until it is partially embedded in the ice. This will ensure that the cell homogenizer remains ice-cold throughout the procedure.
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, Peptides, and Recombinant Proteins | ||
| Iron (III) acetylacetonate (≥99.9%) | Sigma-Aldrich | 517003 |
| 1,2-Hexadecanediol (technical grade, 90%) | Sigma-Aldrich | 213748 |
| Oleic acid (technical grade, 90%) | Sigma-Aldrich | 364525 |
| Benzyl 4-bromobutyl ether (Benyl ether) (98%) | Sigma-Aldrich | 108014 |
| meso-2,3-Dimercaptosuccinic acid (DMSA) (~98%) | Sigma-Aldrich | D7881 |
| Dimethyl sulfoxide (DMSO), anhydrous (>99.9%) | Sigma-Aldrich | 276855 |
| Hexane (≥99% (GC)) | Honeywell | 32293 |
| Oleylamine (>70%) | Acros Organics | AC129540010 |
| Toluene (99.8%, Extra Dry) | Acros Organics; Fisher Scientific | AC610951000; 10477172 |
| Ethanol (Absolute) | Honeywell | 24194 |
| Glycine (≥99% (HPLC)) | Sigma | G7126 |
| Sodium chloride (≥99%) | Sigma | S9625 |
| Sucrose (≥99.5% (GC)) | Sigma | S0389 |
| Tris-HCL | Sigma | T5941 |
| Ethylene glycol-bis(2-aminoethylether)- N,N,N′,N′-tetraacetic acid (EGTA) (≥97.0%) | Sigma | E3889 |
| Protease Inhibitor Cocktail (EDTA-free) | Roche | 11836170001 |
| Experimental Models: Cell Lines | ||
| HeLA | ATCC | CCL-2TM |
| Software and Algorithms | ||
| ImageJ software | Schneider et al., 2012 | ImageJ software |
| Other | ||
| Thermal Gravimetric Analysis (TGA) | TA instruments, New Castle, DE, USA | TGA Q5000 IR |
| 100 μL, Platinum-HT pans | TA instruments, New Castle, DE, USA | 957571.901 (PS2226) |
| Transmission Electron Microscope (TEM) | Philips | Philips CM30 (300KV) |
| SuperMACS™ II Separator | Miltenyi Biotec | 130-044-104 |
| LS Column | Miltenyi Biotec | 130-042-401 |
| Heating Mantle | Horst (Merck) | Z602140 |
| Mantle Thermocouples | Horst | 070213 |
| Graham Condenser | Homemade (Alternative: DWK Life Sciences) | Home made (Alternative: K439000-2440) |
| Three-Neck Round Bottom Flasks, Side Necks Angled (500 ml) | Homemade (Alternative: Aldrich) | Homemade (Alternative: CLS4965250) |
| Glass centrifuge tube with conical bottom and cap (50 ml) | Homemade (Alternative: Witeg, Germany) | Homemade (Alternative: 4945303) |
| Cell homogenizer | Isobiotec | H8 KIT (S8 2 -10; S8 12-18) |
| DMEM/F-12, With L-glutamine, HEPES | Gibco | 11039047 |
| DPBS, no calcium, no magnesium | Gibco | 14190169 |
| Zetasizer Nano (DLS) | Malvern Panalytical Ltd | ZEN 3600 |
| Spectrosil UV Quartz cell | Starna | 23-Q-10 |
| Multi Reax orbital shaker | Heidolph Instruments | 545-10000-00 |
| FBS | Gibco | 10270106 |
Materials and Equipment
Reagents used for SPION synthesis
| Reagent | Final Concentration | Mass/Stock Concentration | Add | Storage |
|---|---|---|---|---|
| Iron (III) acetylacetonate | 2 mM | 353.18 g/mol | 0.70636 g | Amber glass bottles, Desiccator cabinet, room temperature (~20°C) |
| 1,2-hexadecanediol | 10 mM | 258.16 g/mol | 2.5845 g | Desiccator cabinet, room temperature (~20°C) |
| Oleic acid | 6 mM | 282.46 g/mol | 1.981 mL | 4°C |
| Oleylamine | 6 mM | 267.5 g/mol | 1.910 mL | Room temperature (~20°C) |
| Benzyl ether | n/a | n/a | 20 mL | Room temperature (~20°C) |
Composition of acidic buffer
| Reagent | Final Concentration | Stock Concentration | Volume |
|---|---|---|---|
| Glycine | 0.2 M | 2 M | 50 mL |
| Sodium chloride | 0.15 M | 2 M | 37.5 mL |
| ddH2O | Adjust to pH 3.0 with 5% HCL, make up the volume to 500 mL | n/a | n/a |
| Total | n/a | n/a | 500 mL |
| Store at 4°C | |||
Composition of homogenizing buffer (HB)
| Reagent | Final Concentration | Stock Concentration | Add to mL |
|---|---|---|---|
| Sucrose | 250 mM | 2 M | 6.25 mL |
| Tris-HCl | 5 mM | 1 M | 0.25 mL |
| EGTA (pH 7.4) | 1 mM | 0.5 M | 0.1 mL |
| Protease inhibitor (EDTA free) | 1 tablet per 50 mL | n/a | add before use |
| ddH2O | Adjust pH to 7.4 with 1 M NaOH, make up the volume to 50 mL | n/a | n/a |
| Total | n/a | n/a | 50 mL |
| Store at 4°C | |||
Note: The homogenization buffer (HB) is used in several steps, we therefore recommend preparing at least 50 mL of HB. However, depending on the number of cell lines used (e.g., control vs diseased or control vs diseased vs treatment) the volume of HB needed might vary. Be sure to calculate the volume of HB beforehand and prepare accordingly. As an alternative 10 x, stock solutions of HB can be prepared and frozen if smaller volumes of lysis buffer are to be used.
Step-By-Step Method Details
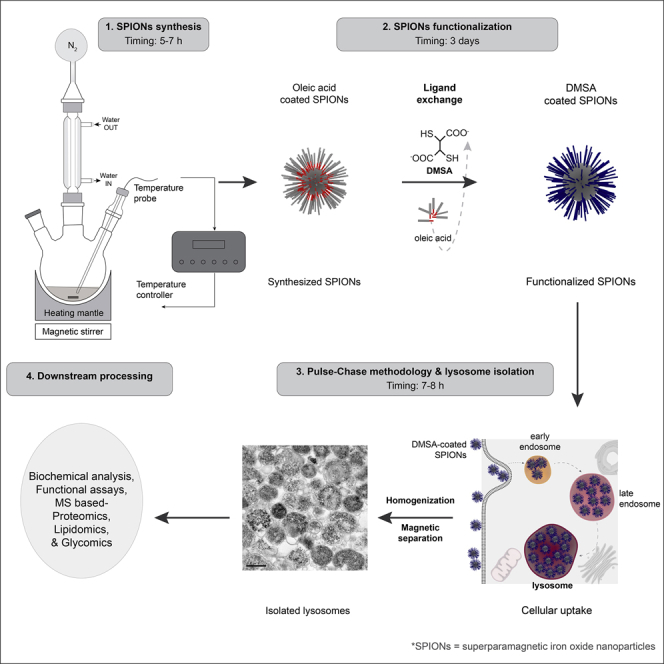
Synthesis of Superparamagnetic Iron Oxide Nanoparticles (SPIONs)
A thermal decomposition method is used for the synthesis of SPIONs.
Note: This protocol is modified from(Sun and Zeng, 2002). See Figures 1 and 2 for more information on the synthesis setup.
Figure 2.

Setup of the Thermal Decomposition Method Used in SPION Synthesis
(Note: For presentation purposes, we have placed the SPION synthesis setup on an open bench. However, it is crucial to place the setup in a secure environment such as hood/cabinet before starting the SPION synthesis).
CAUTION: All steps mentioned below must be carried out under a fume hood. The protocol involves high-temperature reactions. It is, therefore important to wear PPE at all times.
-
1.
Place the 3-neck flask containing the reagents on the heating mantle.
-
2.
Connect the reflux condenser to the opening in the center of the 3-neck flask.
-
3.
Clamp the entire setup (using a proper sized clamp) in the middle of the reflux condenser and secure it to a metal stand providing enough support to the setup.
-
4.
Connect the inlet and outlet openings of the reflux condenser to the water flow and turn on the water flow.
-
5.
Place a glass stopper on one of the two remaining openings of the 3-neck flask.
-
6.
Insert the temperature probe through the remaining opening of the 3-neck flask and seal the opening.
-
7.
Turn on the magnetic stirrer (35 × g) and using a tube, slowly pass nitrogen gas (∼1 bar) into the 3-neck flask from the top.
-
8.
Slowly remove the glass stopper connected to the 3-neck flask. Leave it open for 5–10 min and let the nitrogen gas flow out through this opening.
-
9.
Replace the nitrogen gas delivering tube with a balloon filled with nitrogen gas, as shown in Figure 2.
-
10.
Place the glass stopper back and seal all joints using metal holders.
-
11.
Turn on the heating and set the temperature to 200°C.
CAUTION: In most cases, the heating mantle does not have a temperature/power control and, therefore, use an external device to control the power input (Figure 2). Do not plug the heating mantle directly into a wall plug.
-
12.
From the moment the temperature reaches 200°C, let it continue for 2 h.
Note: The presence of benzyl ether aids in reaching such high temperatures. During this step, the metal precursors decompose (or disintegrate) into small molecules forming nuclei that eventually act as seeds for the formation of SPIONs.
-
13.
After 2 h, increase the temperature to 300°C.
CAUTION: In most cases, the heating mantle does not have a temperature/power control and therefore use an external device to control the power input. Do not plug the heating mantle directly on to a wall plug.
-
14.
Once the temperature reaches 300°C, keep it at that temperature for 1 h.
-
15.
Turn off the magnetic stirrer and heating. Allow the black colored slurry that is formed to cool down to room temperature (∼20°C) (this takes about 1 h).
-
16.
Carefully release the pressure developed inside the 3-neck flask by slowly removing the glass stopper and disassemble the setup.
-
17.
Carefully remove the magnetic stir bar and slowly add 300 mL of absolute ethanol to the slurry to precipitate the newly formed SPIONs.
-
18.
Transfer the contents from the 3-neck flask to 50 mL glass tubes.
-
19.
Place the tube on a magnetic separation stand to separate the SPIONs from unreacted chemicals (Figure 3A).
Figure 3.

Magnetic Separation and Purification of Synthesized SPIONs
Note: For optimal magnetic separation, we recommend using the Magnetight™ Separation Stand from Novagen (Cat. No. 69964-3). Alternately, LifeSep magnetic separation stand (#Z740157) or similar can be used.
-
20.
Leave the tube undisturbed for 5–10 min.
-
21.
While the SPIONs remain attracted to the magnet, use a glass pipette and carefully remove large particles and unreacted chemicals that remain suspended in ethanol.
CAUTION: Collect the ethanol in a separate waste container.
Note: When repeating this step several times, instant clearing of the solvent can be observed.
-
22.
Remove the tubes from the magnetic separation stand (Figure 3B) and re-suspend the SPIONs in 40–50 mL absolute ethanol per tube.
-
23.
Repeat steps 19–21, 5–6 times until the ethanol appears transparent following the attraction of the SPIONs to the magnet.
-
24.
Remove the ethanol completely and re-suspend SPIONs in 20 mL of hexane (Figure 3C).
-
25.
Centrifuge the tube at 4,100 × g for 10 min to remove aggregates.
-
26.
Carefully collect the supernatant containing the SPIONs and transfer it into a glass vial with a screw cap. Store at 4°C.
Note: The SPIONs suspended in hexane have a shelf life of about 2–3 months when stored at 4°C. However, when SPIONs have been stored for such long periods, we highly recommend repeating the quality control measures (such as concentration, size measurement) described below on a small aliquot of the stored SPIONs and ensure that the SPIONs are not prone to any aggregation/precipitation.
Measuring SPION Concentration
Thermal Gravimetric Analysis (TGA) is used for measuring the concentration of the synthesized SPIONs. The instrument measures the mass of SPIONs over time as the temperature changes under a nitrogen atmosphere. In addition to measuring the mass concentration of SPIONs, the TGA data also provides information on the physical and chemical properties of SPIONs.
Note: 100 μL Platinum-HT pans (#957571.901, TA instruments, New Castle, DE, USA) provided by the manufacturer are needed for measuring SPION concentrations. Platinum-HT pans have to be cleaned in piranha solution first, followed by flaming of the pans until it is red hot before use. These steps ensure that the pans are free of any contaminating or dust particles.
CAUTION: The protocol involves high-temperature reactions. It is, therefore important to wear PPE at all times.
-
27.
Place two clean, empty Platinum-HT pans on the carousel of the auto-sampler to measure their weights.
-
28.
Press TARE to bring the base weights to 0.
-
29.
Place 100 μL of SPIONs suspended in hexane on each pan and wait for 3–5 min for the hexane to evaporate.
-
30.
Heat the pans to 80°C to remove solvent traces.
-
31.
Increase the temperature to 850°C with a heating rate of 20°C per min.
-
32.
Record the mass of the pans every minute until the temperature reaches 850°C.
-
33.
Use the Universal Analysis 2000, v 4.5A software provided by the manufacturer to calculate the concentration of SPIONs.
Note: As the temperature increases, the coating surrounding the SPIONs evaporates, and the mass of iron in the pan is recorded.
Alternatives: (i) Several other TGA instruments without auto-sampler are available and can be used to calculate the concentration of SPIONs. (ii) If a TGA instrument is not available or accessible, take 1 mL of SPIONs suspended in hexane on a weighing boat, evaporate the hexane completely, and measure the dry weight of the SPIONs.
CAUTION: SPIONs in particulate forms are highly toxic, and therefore wearing appropriate PPE is a must.
Measuring SPION Size and Shape
Note: The size and shape of newly synthesized SPIONs are crucial for proper functionalization and for subsequent effective internalization by the cells. Spherical particles have a large surface area to volume ratio, and therefore their surfaces can be utilized maximally. On the other hand, maintaining a small core size effectively retains the superparamagnetic property of the particles and aids in the internalization of the SPIONs by the cells. It is, therefore important to perform a quality check and determine the size and shape of the SPIONs following its synthesis and after functionalization.
Transmission Electron Microscope (TEM) is used for measuring the size and shape of SPIONs.
CAUTION: SPIONs in particulate forms are highly toxic, and therefore wearing appropriate PPE is a must.
-
34.
Place a small drop of SPIONs suspended in hexane on an amorphous carbon-coated copper grid. Do this on 4–5 grids.
-
35.
Wait for 5–10 min at room temperature (∼20°C) until the solvent evaporates.
-
36.
Place the carbon-coated copper grids on the sample holder and insert it into the TEM.
-
37.
Acquire TEM images from each of the several independent grids of the same sample.
-
38.
Measure the diameter of SPIONs from each of those grids and take an average of at least 100 particles to get an estimate of the shape and size distribution.
Alternatives: Dynamic Light Scattering (DLS, also known as Photon Correlation Spectroscopy or Quasi-Elastic Light Scattering) can be used in addition or if TEM is not readily available/accessible to determine the shape, size (hydrodynamic radius) distribution of SPIONs.
CAUTION: If a DLS instrument is used for determining the size distribution of SPIONs suspended in hexane, use only a glass cuvette for performing such measurements. It is important to clean the glass cuvette used for the measurement with a small volume (10 mL) of the piranha solution (as described in the section “Cleaning and preparation of glassware”) before and after the measurement. A disposable/plastic cuvette can be used for measuring the hydrodynamic radius of functionalized DMSA-coated SPIONs suspended in water.
Functionalization of SPIONs (Day 1)
The SPIONs synthesized using the thermal decomposition protocol have an iron core surrounded by oleic acid molecules. The oleic acid prevents the SPIONs to aggregate and renders them hydrophobic. For their use on living cells, SPIONs need to become hydrophilic. This step of SPION functionalization makes them hydrophilic and, at the same time, imparts a charge to SPIONs.
CAUTION: All steps are to be carried out under a fume hood. Wearing PPE is a must.
-
39.
Take 10 mg of SPIONs resuspended in hexane and transfer to a 50 mL glass centrifuge tube.
-
40.
Evaporate the hexane completely.
CAUTION: SPIONs in particulate forms are highly toxic, and therefore wearing a PPE is a must.
-
41.
Re-suspend SPIONs in 1 mL of extra-dry toluene.
-
42.
Weigh 200 mg of meso-2,3-dimercaptosuccinic acid (DMSA) in a 15 mL Falcon tube and re-suspend it by adding 1 mL of anhydrous, dimethyl sulfoxide (DMSO).
-
43.
Add the DMSA-DMSO mixture to the glass centrifuge tube containing SPIONs resuspended in toluene.
-
44.
Place the tube containing the reaction mixture on an orbital shaker (450 × g) placed at room temperature (∼20°C).
-
45.
Let the reagents react for 72 h.
Incubation period: 72 h
Functionalization of SPIONs (Day 3)
Note: After 72 h of shaking, two phases are formed. The top phase contains unreacted DMSA and DMSO, whereas the bottom phase contains the DMSA-coated (functionalized) SPIONs.
-
46.
Centrifuge the tube containing the reaction mixture at 4,100 × g for 10 min.
-
47.
Discard the top phase containing unreacted DMSA and DMSO.
-
48.
Re-suspend the bottom phase containing DMSA-coated SPIONs in 2 mL of milliQ water.
Note: As the SPIONs were in a hydrophobic environment until now, they may not re-suspend immediately. Vortex to uniformly suspend them.
-
49.
Place an LS Column on a SuperMACS™ II Separator (magnetic field).
Alternatives: The MidiMACS™ or QuadroMACS™ separators can be used as an alternative.
-
50.
Equilibrate the LS column by adding 1 mL of milliQ water.
-
51.
Collect and discard the flow through.
-
52.
Add 2 mL of resuspended DMSA-coated SPIONs on top of the LS column.
-
53.
Let the SPIONs enter into the column.
Note: The presence of trace amounts of DMSO in DMSA-coated SPIONs can clog the flow through of the column. If this occurs, pipet the SPIONs up/down in the column to ease the flow.
-
54.
Collect and discard the flow through.
-
55.
Wash the column once with 1 mL of milliQ water.
-
56.
Remove the LS column from the SuperMACS™ II Separator and place it on a stand (away from the magnet).
-
57.
Add 1 mL of milliQ water and insert the plunger that comes along with the LS column to force-collect the magnetically bound SPIONs.
-
58.
Add 1 mL of milliQ water and insert the plunger to force-collect the remaining magnetically bound SPIONs.
-
59.
Centrifuge the tube containing DMSA-coated SPIONs at 4,100 × g for 10 min.
-
60.
Measure the pH and adjust it to pH 7.0.
Note: Following functionalization, the pH of the DMSA-coated SPIONs is acidic (usually around pH 3). Use 1 M NaOH to adjust carefully to pH 7.
-
61.
Measure the concentration of the DMSA-coated SPIONs using TGA (as described above).
Lysosome Isolation: Part 1: Pulse-Chase Methodology
Note: A Pulse and a Chase method is used for loading lysosomes with DMSA-SPIONs. The pulse involves the period of incubation of DMSA-coated SPIONs with the cells, while the chase period is the time taken by DMSA-coated SPIONs to reach lysosomes within the cells.
-
62.
Before use, check the confluency and health of the cells under a bright-field microscope.
-
63.
Collect and discard cell culture media.
-
64.
Add 5 mL of pre-warmed PBS-/- to culture dishes and swirl the plates gently. Collect and discard the PBS.
-
65.
Re-suspend the DMSA-coated SPIONs to a concentration of 200 μg per mL in a serum-free culture medium.
-
66.
Slowly add 5 mL of DMSA-SPION containing medium to the culture dishes.
Note: The final amount of DMSA-SPIONs on each culture dish is therefore 1 mg.
-
67.
Place the culture dishes back in the incubator (37°C) for 15 min (Pulse).
-
68.
Take out, collect, and discard the DMSA-SPION containing medium.
-
69.
Repeat step 64.
-
70.
Add 5 mL of fresh medium (supplemented with 10% FBS) to the culture dishes.
-
71.
Place the culture dishes back in the incubator at 37°C for 4 h (Chase).
-
72.
Collect and discard the media.
-
73.
Wash the cells three times with 5 mL of acidic buffer (0.2 M glycine; “Composition of acidic buffer”).
Note: This washing step removes SPIONs that are bound tightly to the cell surface. However, when using neuronal cultures for lysosome isolation, this washing step might result in cell detachment and therefore, can be left out (optional).
Note: At this point, the cells can be fixed and processed for TEM.
-
74.
Place the culture dishes on ice and add 5 mL of ice-cold PBS-/-.
-
75.
Scrape the cells and transfer them to a 15 mL Falcon tube. Use one tube for each culture dish. Centrifuge at 180 × g for 10 min (4°C).
-
76.
Discard the supernatant and place the tube containing the cell pellets on ice.
Part: 2 Homogenization and Magnetic Purification of Lysosomes
Note: As the SPIONs are extremely small (∼10 nm), they exhibit superparamagnetic properties, i.e., they can be magnetized instantly by introducing a magnetic field, and this property is instantly lost upon removing the magnetic field. In this protocol, we use this superparamagnetic property to isolate and enrich for lysosomes.
-
77.
Add 1 mL of ice-cold homogenizing buffer (HB; “Composition of homogenizing buffer (HB)”) to cell pellets collected from each 100 mm diameter culture dish.
Note: The cell homogenizer or cell cracker has a void volume of ∼500–600 μL. Therefore for having a uniform homogenization, the cells need to be resuspended in at least 1 mL.
-
78.
Re-suspend the cell pellets and make sure that there are no clumps.
-
79.
Use 1 mL syringe to collect the cell suspension and mount the syringe to one end of the cell homogenizer placed on ice.
-
80.
Place an empty 1 mL syringe on the other end of the cell homogenizer.
-
81.
Slowly exert pressure on the syringe containing the cell suspension to force it through the cell homogenizer to the other syringe and back. This constitutes one passage.
-
82.
Repeat step 81 to a total of ten passages to ensure uniform homogenization of the cells.
-
83.
Carefully remove one syringe and transfer the total cell lysate (TCL) to a fresh 15 mL Falcon tube.
-
84.
Use the other mounted syringe to collect the remaining TCL that is present in the bore of the cell homogenizer and transfer to the same Falcon tube.
-
85.
Repeat step 79–84 for the other three cell pellets.
-
86.
Centrifuge the TCL at 800 × g for 10 min (4°C).
-
87.
Carefully collect the post-nuclear supernatant (PNS) without disturbing the pellet and pool all PNS in a fresh Falcon tube placed on ice.
Note: Steps 88–94 are to be performed at 4°C (for instance, in a cold room).
-
88.
Place an LS Column in a SuperMACS™ II Separator (magnetic field).
Alternatives: The MidiMACS™ or QuadroMACS™ separators can also be used.
-
89.
Equilibrate the LS column by adding 1 mL of HB. Collect and discard the flow through.
-
90.
Slowly add the PNS on top of the LS column (maximum 7 mL) and let the PNS pass through the column.
Note: As the SPIONs taken up by the cells are superparamagnetic, they will instantly bind together with the organelle in which they are present to the column matrix in the presence of the magnetic field. The other non-magnetic portion of the PNS will flow through the column.
-
91.
Collect and discard the flow through. Wash the column once with 5 mL of HB.
-
92.
Remove the LS column from the SuperMACS™ II Separator (magnetic field) and place it on a stand (away from the magnet).
-
93.
Add 3 mL of HB and insert the plunger that comes along with the LS column to force-collect the bound/magnetic fraction (containing the lysosomes) in a fresh Falcon tube.
-
94.
Repeat step 93 but with 1 mL HB, and pool with the previous 3 mL.
-
95.
Transfer the bound/magnetic fraction in a clean ultracentrifuge tube and centrifuge for 30 min at 20,000 × gavg (4°C).
-
96.
After centrifugation, discard the supernatant. The resulting pellet contains the isolated lysosomes.
Note: The purity of the isolated lysosomal fraction can be easily assessed by semi-quantitative Western blot, comparing equal amounts of protein for PNS/TCL and isolated lysosomes. A very low percentage recovery (mostly <2%) of marker proteins for mitochondria, ER, cytosol, peroxisomes, and cytoskeletal elements underscores very low levels of contamination and, therefore, a strongly enriched lysosomal fraction. It is important that marker proteins of contaminating organelles are absent or significantly de-riched (<2%) in the lysosomal fraction prior to subsequent omics analysis.
-
97.
Re-suspend the pellet in a small volume (100–200 μL) of HB or PBS or any other mass spectrometry compatible buffers.
Note: If the isolated lysosomes are to be used for biochemical or functional assays, re-suspend the pellets in HB. This will better preserve the intactness of the isolated lysosomes. If the isolated lysosomes are to be processed for proteomics or lipidomics analysis, re-suspend the pellets in 50 mM Triethylammonium bicarbonate (TEAB).
Note: Lysosomal integrity can be assessed in different ways. For instance, (Tharkeshwar et al., 2017) measured β-hexosaminidase activity to estimate the latency (intactness) and reported that close to 70% of the magnetically isolated lysosomes remained intact. The isolated lysosomes also remained acidic, as measured with Lysotracker and underscoring a functional proton pump. For more technical details, we refer to (Tharkeshwar et al., 2017).
Expected Outcomes
Starting with SPION synthesis, functionalization, and characterization, we have described in detail the methodological steps of the lysosome isolation procedure. Starting with about 1 × 106 cells on four 100 mm diameter dishes, this protocol can yield for HeLa cells 70–100 μg of highly enriched lysosomes (measured by protein content) that can be directly used for functional studies, biochemical and omics analysis. Of note, when starting from smaller cell types (e.g., HEK293, SH-SY5Y), the yield might be lower (40–60 μg). The quality and the quantity of SPIONs synthesized and the lysosomes purified can be assessed at several stages in the protocol, as in Figure 4.
-
i)
Figures 4A and 4B: TEM of oleic acid-coated SPIONs.
-
ii)
Figure 4C: TEM of HeLa cells incubated with DMSA-coated SPIONs ((Pulse 15 min; Chase 1 h).
-
iii)
Figure 4D: TEM of HeLa cells incubated with DMSA-coated SPIONs (Pulse 15 min; Chase 4 h).
-
iv)
Figure 4E: Magnetically purified lysosomal pellet.
-
v)
Figure 4F: TEM image of magnetically purified lysosomes.
Figure 4.

Quality Control Steps in Evaluating the Purity of Synthesized SPIONs and Isolated Lysosomes
(A and B) TEM images of synthesized SPIONs (scale bar in [A], 100 nm; scale bar in [B], 10 nm).
(C and D) TEM images and zoom (corresponding white square) of HeLa cells incubated with DMSA-SPIONs: (C) Pulse, 15 min; Chase, 1 h and (D) Pulse, 15 min; Chase, 4 h.
(E and F) Magnetically purified lysosomal pellet (E) and TEM image of the magnetically purified lysosomal pellet (F).
The protocol described here can be applied to any adherent cell types, including primary, iPSC-derived neuronal cultures, and can be up-scaled to suit the user’s requirements. In comparison with other existing subcellular organelle and lysosome isolation methodologies (reviewed in detail by (Tharkeshwar, Gevaert and Annaert, 2018)), our DMSA-SPION based lysosome isolation provides highly enriched lysosomes, thus offering increased spatial resolution when coupled with mass spectrometric analysis. The “Before you begin” section contains considerations that are important when one aims to make adaptations to the protocol. If steps described are followed properly, the research protocol provides highly purified lysosomes that can be subsequently analyzed by mass spectrometry. As such, changes in the biomolecular composition of lysosomes as occurs in lysosomal storage diseases can be identified, as we did in our study (Tharkeshwar et al., 2017). Likewise, the selective abundances of proteins on lysosomes can be qualitatively assessed as we did in our Sannerud et al., study (Sannerud et al., 2016). The DMSA-SPION isolated lysosomes are compatible for mass spectrometric analysis of the proteome and lipidome but is potentially extendable to N-glycoproteomics. Further, as the isolated lysosome is intact, functional analysis is also possible on magnetically isolated fractions, including lysosomal hydrolase activities, proton pump activity, and pH (as shown in Tharkeshwar et al, 2017). We performed proteomic analysis on the isolated lysosomal fraction and total cell extract and used Gene Ontology (GO) analysis (with a minimum 2-fold enrichment, a p-value ≤ 0.05) to analyze/compare protein enrichment levels. In doing so, we found that in the lysosome fraction, 36% of identified proteins (1136/3196) are highly significantly annotated with the GO term “membrane-enclosed lumen” (p-value: 2.6E-130) followed by GO terms including “envelope,” “organelle membrane,” and “endomembrane system” (p-values: 1.30E-60, 2.50E-45, and 1.40E-19, respectively).
Limitations
One current limitation of the protocol is that it has been optimized only for adherent cell types. Given the fact that the SPIONs are taken up by the cells via their endocytic machinery, a sufficient period of contact between the cells and SPIONs is required. As mentioned on several occasions in the protocol, the efficiency and the quantity of lysosomes purified is dependent on the cell line and its health status. For instance, depending on the morphology of the cell type under investigation (for instance, large versus small cell types), optimization is needed with respect to the homogenization step using the ball-bearing cell cracker. Disease-related conditions (such as (but not limited to) neurodegenerative diseases and lysosomal storage diseases) may strongly alter endolysosomal transport kinetics, including fusion events between, for instance, late endosomes and lysosomes or autophagosomes and lysosomes. This can be remediated by increasing chase periods. However, once such parameters are optimized and defined, the purity and yield remain consistent in repetitive isolations.
Troubleshooting
Problem
Low concentration of SPIONs after synthesis and functionalization
Potential Solution
Typically, SPIONs should look very homogeneous, and there should be no aggregates. The presence of large aggregates influences the overall yield at the end. It mainly happens when the chemicals used during the synthesis and functionalization protocol carry water molecules. Check the storage conditions of the chemicals used and make sure they are stored in a desiccator before and after use. As an alternative, the lysosome isolation protocol described here can be performed using commercially available magnetic particles such as DexoMAG® (Liquids Research Limited, UK). However, and compared to our protocol, a much higher particle concentration is needed, and the long pulse-chase period significantly lengthens the experimental procedure to reach a similar quality of isolated lysosomes (Walker and Lloyd-Evans, 2015). Moreover, it cannot be excluded that cells may exert more stress when challenged with much higher concentrations of iron oxide nanoparticles, and this for a much more extended time period.
Problem
Low yield of purified lysosomes
Potential Solution
Lower yield of purified lysosomes might be caused by several reasons, and it requires a thorough and step-wise quality check of the isolation procedure. (i) Make sure the functionalized DMSA-coated SPIONs that are used do not aggregate. The presence of aggregates would hinder the SPIONs from being taken up by the cells. (ii) Monitor the endocytic uptake of SPIONs by the cells during the pulse-chase period, for instance, using TEM of cells fixed at different time points. If only a few endosomal compartments contain DMSA-SPIONs, increase the length of the pulse period or increase the concentration of DMSA-SPIONs (or both). (iii) The isolates contain other contaminating organelles along with the purified lysosomes. When assessed by Western blot, an isolated lysosomal fraction must show strong enrichment for any lysosomal marker protein used, whereas markers proteins from other subcellular organelles such as mitochondria, endoplasmic reticulum, peroxisomes, cytoskeletal elements, nucleus etc. must be clearly de-enriched (<1 fold relative to the PNS or total cell extract) or even absent. Enrichments of >1 might indicate too high contamination, and we recommend in that case not to use such samples for further analysis. (iv) The fractionation/homogenization is suboptimal. This can be solved by comparing the homogenization using tungsten carbide balls of different diameters, and/or by altering the number of passages and evaluate the presence of viable cells (thus, not homogenized) using trypan blue staining.
Problem
Modifications and optimizations of the protocol when using other cell types and culture dishes.
Potential Solution
The protocol described here is applicable for any other adherent cell types, including neurons, and can be carried out using other types of culture dishes (e.g., six-well plate). However, this would require specific modifications and optimizations in the protocol. When doing so, it is important to pay attention to parameters such as cell size, growth rate, doubling time in case of dividing cells, affinity toward surface attachment etc. Based on our experience, for diving cells, we recommend plating cells at a density of around 30,000 cells/cm2. iPSC-derived neurons can be plated in their progenitor stage and can be cultured until they are mature before starting with the magnetic isolation procedure. In either case, it is important to ensure that the cells have adhered to the culture dish. Certain cell types, including primary and iPSC-derived neurons, may not favor attachment to plastic surfaces. In such scenarios, culture dishes can be coated with poly-L-lysine (PLL) prior to cell plating to enhance their attachment. In the case of neuronal cells, it is also important not to use a cell scraper to collect cells (as this will particularly rupture neurites as well, resulting in leakage of organelles) but to use an enzymatic cell detachment solution (such as Accutase). If smaller culture dishes are used, the volume of DMSA-SPION containing medium has to be scaled down accordingly (for instance, 2 mL per well in a six-well plate). The yield will be accordingly lower as well. As stated above, for each cell type, it is important first to monitor the endocytic update of SPIONs to assess or adapt the amount of SPIONs, pulse, and chase time intervals.
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Wim Annaert (wim.annaert@kuleuven.be).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
This study did not generate any unique datasets or code.
Acknowledgments
W.A. is financially supported by VIB, KU Leuven (C16/15/073), the FWO (S006617 N, G078117 N; G056017 N; G0C4220N; G0C3620N), the FWO-Hercules Foundation (AKUL/11/30, 13/39), and SAO (S# 20180020). The authors thank Imec for providing instrumentation and infrastructural support for carrying out SPION synthesis and characterization, the VIB BioImaging core (Electron and Light Microscopy Platforms), KU Leuven Department of Neurosciences, VIB – Center for Brain & Disease Research. The authors thank D.T.G. Raj for initial contributions.
Author Contributions
Methodology, A.K.T.; Investigation, A.K.T.; Validation, A.K.T. and D.D.; Writing and Visualization, A.K.T. and W.A.; Supervision, W.A.; Funding Acquisition, W.A.
Declaration of Interests
The authors declare no competing interests.
Contributor Information
Arun Kumar Tharkeshwar, Email: arun.tharkeshwar@kuleuven.be.
Wim Annaert, Email: wim.annaert@kuleuven.be.
References
- Sannerud R., Esselens C., Ejsmont P., Mattera R., Rochin L., Tharkeshwar A.K., De Baets G., De Wever V., Habets R., Baert V. Restricted location of PSEN2/γ-secretase determines substrate specificity and generates an intracellular Aβ Pool. Cell. 2016;166:193–208. doi: 10.1016/j.cell.2016.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider C.A., Rasband W.S., Eliceiri K.W. NIH Image to ImageJ: 25 years of image analysis. Nature methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S., Zeng H. Size-controlled synthesis of magnetite nanoparticles size-controlled synthesis of magnetite nanoparticles. J. Am. Chem. Soc. 2002;124:8204–8205. doi: 10.1021/ja026501x. [DOI] [PubMed] [Google Scholar]
- Tharkeshwar A.K., Trekker J., Vermeire W., Pauwels J., Sannerud R., Priestman D.A., Te Vruchte D., Vints K., Baatsen P., Decuypere J.P. A novel approach to analyze lysosomal dysfunctions through subcellular proteomics and lipidomics: The case of NPC1 deficiency. Sci. Rep. 2017;7:41408. doi: 10.1038/srep41408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tharkeshwar A.K., Gevaert K., Annaert W. Organellar omics—a reviving strategy to untangle the biomolecular complexity of the cell. Proteomics. 2018:1700113. doi: 10.1002/pmic.201700113. [DOI] [PubMed] [Google Scholar]
- Walker M.W., Lloyd-Evans E. A rapid method for the preparation of ultrapure, functional lysosomes using functionalized superparamagnetic iron oxide nanoparticles. Methods Cell Biol. 2015;126:21–43. doi: 10.1016/bs.mcb.2014.10.019. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This study did not generate any unique datasets or code.