

Summary
Dual RNA-sequencing is a powerful technique to assess both bacterial and host transcriptomes in an unbiased way. We developed a protocol to perform Dual RNA-seq on in vivo-derived macrophage populations infected with Mycobacterium tuberculosis. Here, we provide a practical step-by-step guide to execute the protocol on Mtb-infected cells from a murine infection model. Our protocol can also be easily applied to perform Dual RNA-seq on in vitro-derived cells as well as different Mtb-infected host cell types.
For complete details on the use and execution of this protocol, please refer to Pisu et al. (2020)
Graphical Abstract
Highlights
-
•
Dual RNA-seq of M. tuberculosis-infected cells
-
•
Simultaneous profiling of host and pathogen transcripts from an in vivo infection model
-
•
Gain in-depth understanding of host-pathogen interactions at the molecular level
Dual RNA-sequencing is a powerful technique to assess both bacterial and host transcriptomes in an unbiased way. We developed a protocol to perform Dual RNA-seq on in vivo-derived macrophage populations infected with Mycobacterium tuberculosis. Here, we provide a practical step-by-step guide to execute the protocol on Mtb-infected cells from a murine infection model. Our protocol can also be easily applied to perform Dual RNA-seq on in vitro-derived cells as well as different Mtb-infected host cell types.
Before you Begin
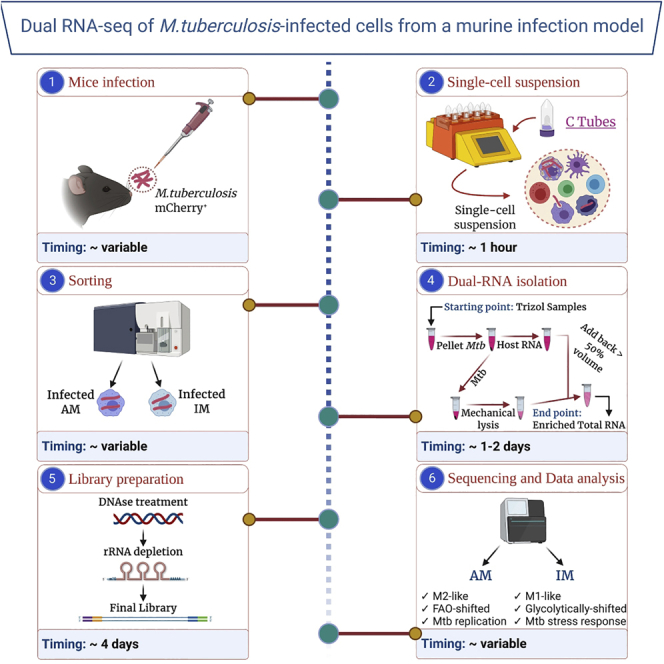
The main steps of this protocol include mice infection, generation of a single-cell suspension, isolation of infected cells by fluorescence activated cell sorting, RNA extraction, library preparation, sequence, and analysis. It is intended to provide an overview of these steps and the time, reagents, and equipment required as well as to highlight potential problems and alternative solutions.
Before starting the protocol make sure the reagents required to perform the different steps are ready to use.
CRITICAL: For mice infection it is important to consider background, age, and sex of the mice such as that they are matched as closely as possible between replicates. We typically use 6–8 weeks old female mice on a BL/6 background. All mice used in this protocol are maintained in a specific pathogen free animal biosafety level 3 facility. We recommend pooling n=3 mice/sample to minimize biological variability. Examination of the Dual RNA-seq data generated with this protocol provides an in-depth understanding of the host-pathogen interactions at the transcriptional level.
Prepare the bacterial stocks used for mice infection.
-
1.
Frozen bacterial stocks. We typically freeze 1 mL aliquots of mid-log phase cultures of our bacterial strains at -80˚C in 7H9/OADC + 10% glycerol and titer them prior to infection by CFU counts.
-
2.
For the original Dual RNA-seq paper we used the fluorescent reporter smyc′::mCherry on an Erdman background. Further details on basic bacterial cell culture methods should be sought elsewhere.
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CD64 - PerCP/Cyanine5.5 | Biolegend | Cat# 139307, RRID:AB_2561962 |
| Mertk - PE | Thermo Fisher | Cat# 12-5751-82, RRID:AB_257262 |
| SiglecF – BB515 | BD Bioscience | Cat# 564514, RRID:AB_2738833 |
| Fc Block (CD16/32) mouse | eBioscience | Cat# 14-0161-82 RRID: AB_467133 |
| Bacterial and Virus Strains | ||
| M. tuberculosis Erdman mCherry | Originally from Tanya Parish lab https://doi.org/10.1371/journal.pone.0009823 | N/A |
| Critical Commercial Assays | ||
| Ribo-Zero Gold rRNA Removal Kit (Epidemiology) | Illumina | Cat# MRZE724 |
| Glycoblue | Thermo Fisher | Cat# AM9515 |
| Turbo-DNA free kit | Ambion | Cat# AM1907 |
| NEBNext® Ultra™ II Directional RNA Library Prep Kit for Illumina | New England Biolabs | Cat# E7760 |
| NEBNext® Multiplex Oligos for Illumina | New England Biolabs | One or more of Cat#s E7335, E7500, E7710, E7730 |
| SPRIselect beads | Beckman Coulter | Cat# B23317 |
| Experimental Models: Organisms/Strains | ||
| C57BL/6J mice | The Jackson Laboratory | Cat# JAX:000664, RRID:IMSR_JAX:000664 |
| Software and Algorithms | ||
| DESeq2 | (Love et al., 2014) | N/A |
| ApeGLM | (Zhu et al., 2019) | N/A |
| GSEA | Broad Institute http://software.broadinstitute.org/gsea/index.jsp | N/A |
| EnrichmentMap | (Reimand et al., 2019) | N/A |
| FastQC (v. 0.11.5) | https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ | N/A |
| Flexbar (v. 3.4) | (Roehr et al., 2017) | N/A |
| Bowtie2 | (Langmead and Salzberg, 2012) | N/A |
| Hisat2 (v. 2.1.0) | (Kim et al., 2015) | N/A |
| HTSeq (v. 0.11.0) | (Anders et al., 2015) | N/A |
| Other | ||
| BD Tuberculin Syringe 25G | BD | Cat# E96242800JD |
| Isoflurane | Patterson | Cat# 14043070406 |
| GentleMacs C tubes | Miltenyi Biotech | Cat# 130-093-237 |
| Mini-Beadbeater with 8 speed controller | Biospec | Cat# B020-1G2 |
| Collagenase IV | Worthington | Cat# LS004186 |
| Cell Strainers | VWR | Cat# 10199-656 Cat# 10199-655 |
| ACK lysis buffer | Lonza | Cat# 10-548E |
| EDTA | Invitrogen | Cat# AM9260G |
| Hepes | Gibco | Cat# 15630080 |
| Trizol | Invitrogen | Cat# 15596026 |
| Zirconia Beads | Biospec | Cat# 11079101z |
| Chloroform | Sigma | Cat# MCX10546 |
| 1.5 mL RNase-free tubes | Ambion | Cat# AM12400 |
| 2 mL Eppendorf Tubes | Thermo Scientific | Cat# 21-403-200 |
| Isopropanol | Sigma | Cat # I9516-500ML |
| Nuclease Free Water | Ambion | Cat# AM9938 |
Materials and Equipment
Alternatives: This protocol requires the use of a Qubit to measure the concentration of total RNA recovered from infected macrophages. If a Qubit is not available, it is possible to skip this step and directly proceed to analysis with the Fragment Analyzer to assess both quality and quantity of the extracted RNA before the rRNA depletion step. We strongly suggest analyzing the RNA quality with a Fragment Analyzer before proceeding further.
Alternatives: This protocol describes the generation of a single-cell lung suspension using the GentleMacs instrument from Miltenyi Biotech. If a GentleMacs is not available, it is possible to generate a single-cell suspension manually mincing the lung with a scissor and incubating for 1 h in the presence of collagenase IV as described in the step-by-step part of the protocol.
Alternatives: this protocol was originally performed using the Ribo-Zero Gold Epidemiology kit from Illumina for the rRNA-depletion step. Unfortunately, Illumina have discontinued this kit. However, we were recently able to successfully remove rRNA (which represents ∼95% of the extracted total RNA) using both Illumina and NEB kits (Ribo-Zero Gram+ and Ribo-Zero H/M/R kit from Illumina and NEBNext rRNA Depletion Kit H/M/R [cat# E6350 or improved version E7405] and NEBNext rRNA Depletion Kit (Bacteria) [cat# E7860] from New England Biolabs) by combining the beads from the two kits (mouse and bacteria) in a 1:1 ratio. We typically get ∼15% of reads matching to rRNA when performing depletion with the combined kits versus ∼5% when using the previously available Ribo-Zero Gold Epidemiology kit.
Infection Buffer
| Reagent | Final Concentration (mM or μM) | Amount |
|---|---|---|
| PBS 1× | n/a | 49.975 mL |
| Tween 80 | 0.05% (v/v) | 25 μL |
| Total | n/a | 50 mL |
We filter-sterilize the infection buffer and store it at 20°C–25˚C until use.
Staining Solution
| Reagent | Final Concentration (mM or μM) | Amount |
|---|---|---|
| PBS 1× | n/a | 95 mL |
| FBS | 5% (v/v) | 5 mL |
| Total | n/a | 100 mL |
We filter-sterilize the staining solution and store it at 4˚C.
Dissociation solution
| Reagent | Final Concentration (mM or μM) | Amount |
|---|---|---|
| Staining Solution | n/a | 3.98 mL |
| Collagenase IV | 250 U/mL | 20 μL |
| Total | n/a | 4 mL |
We make stocks of Collagenase IV at 50,000 U/mL and store at −20˚C until use. Right before collecting mouse lungs we prepare the dissociation solution adding staining solution + collagenase IV directly into the GentleMacs C tubes under sterile conditions.
Sorting Buffer
| Reagent | Final Concentration (mM or μM) | Amount |
|---|---|---|
| PBS 1× | n/a | 47.75 mL |
| FBS | 1% (v/v) | 0.5 mL |
| EDTA | 5 mM | 0.5 mL |
| Hepes | 25 mM | 1.25 mL |
| Total | n/a | 50 mL |
We prepare the sorting buffer the day before sorting. We filter-sterilize the sorting buffer and store at 4˚C until use.
75% Ice-cold Ethanol
| Reagent | Final Concentration (mM or μM) | Amount |
|---|---|---|
| Ethanol | 75% | 75 mL |
| Nuclease Free Water | 25% | 25 mL |
| Total | n/a | 100 mL |
We usually divide the 75% ethanol in different aliquots in Falcon Tubes and store it at −20˚C until ready to use.
Step-By-Step Method Details
Mouse Infection
Timing: ∼2 h
In this step we infect the mice intranasally with 103 CFU of our bacterial strains.
-
1.
Thaw the bacterial aliquot for mice infection at 20°C–25˚C (room temperature), passage it through a BD Tuberculin Syringe with a 25G-needle for ∼15× (to breakup bacterial clumps) and dilute in infection buffer to a final concentration of 4 × 104 bacteria/mL (103 bacteria in 25 μL of infection buffer).
Note: For cell sorting in the later part of the protocol, we add an additional mouse infected with WT Mtb, which will be stained and used as an FMO (fluorescence minus one) control for the mCherry Mtb signal.
-
2.
Mice are anesthetized using an isoflurane and oxygen mixture (5% isoflurane in oxygen at 4.5 liters/min; VIP 3000 isoflurane vaporizer) for ∼2 min, until the animals are sedated, and the breath slows.
-
3.
25 μL (103 bacteria) are then administered intranasally to each mouse.
Note: Mice infected with different strains are kept in different cages for the duration of the infection. In the original Dual RNA-seq paper we infected the mice and kept them in cages for 14 days as we were interested in examining a dynamic timepoint in between innate immunity and onset of the adaptive immune response. Typical timepoints examined during an Mtb murine infection are 2 weeks, 3 weeks, 4 weeks (covering the transition from the innate to the adaptive immune response) and 6–8 weeks (for steady-state or chronic infection).
Generation of a Single-Cell Suspension and Antibody Staining
-
4.
Prepare (n= number of mice in the experiment) GentleMacs C tubes filled with 4 mL of the dissociation solution.
-
5.
Euthanize the mice using an approved procedure.
-
6.
Spray the mice with 70% ethanol. Figure 1A
Figure 1.

Generation of a Single-Cell Lung Suspension
(A) Mouse is sprayed with 70% Ethanol.
(B) Skin is removed and chest walls exposed.
(C) Removal of the chest walls to access the lobes of the lung.
(D) Lobes are removed.
(E) GentleMACS C Tube with the lung in the dissociation solution.
(F) Manual dissociation of the mouse lung in a 6 well-plate.
(G) Lungs are minced with scissors.
(H) Minced lungs.
(L) The lung is transferred to a 70uM cell strainer, using a wide-bore pipette tip.
(M) Lungs are grinded against the mesh of the cell strainer.
-
7.
Using a sharp pair of scissors and forceps, remove the skin, opening from the diaphragm to expose the chest walls. Figure 1B
-
8.
Carefully open the thorax performing a midline incision, cutting away the diaphragm and removing the lateral chest walls to expose the lungs. Figure 1C
-
9.
Using forceps and scissors, remove the different lobes, paying attention to not perforate the heart. Figure 1D
-
10.
Place the lobes from each mouse in a single GentleMacs C tube. Figure 1E
-
11.
Run the appropriate GentleMacs protocol for your application in order to obtain a single-cell suspension. For the previous Dual RNA-seq paper we used the “37C_m_LDK_1” program.
Note: if a GentleMacs dissociator is not available, proceed with the manual generation of the single-cell suspension from the mouse lungs:
-
a.Place lung lobes in a well of a 6-well plate. Figure 1F
-
b.Using scissors, mince the lobes in very small pieces until they become mostly homogenous and smooth. Figure 1G
-
c.Prepare (n=number of mice) 15 mL Falcon tubes filled with 4 mL of dissociation solution.
-
d.Recover the entire mass of the minced lung using 1–2 mL of dissociation solution and transfer back to the 15 mL Falcon tube, using a wide-bore pipette tip.
-
a.
Note: cut the end of the 1 mL tips to avoid blockage. Figure 1H
-
e.Incubate at 37˚C, with the falcon tubes lying flat in a shaking incubator (60 rpm) for 60 min.
-
e.
Note: Because of the success of the current protocol we have not explored whether variations in temperature or incubation time improves cell fitness.
-
f.At the end of the incubation, vortex the lung suspension.
-
f.
-
12.
Pass the lung suspension through a 70 uM cell strainer and filter. Figure 1L
Note: if you performed the manual dissociation, use the internal part of a 10 mL syringe or an appropriate instrument to push the remaining pieces of the lobes through the nylon mesh of the cell strainer. Add staining solution as appropriate to let the suspension flow through the strainer and into the 50 mL Falcon tube. Figure 1M
-
13.
Spin down 5 min at 400 × g.
-
14.
Slowly remove the supernatant, paying attention to not touch the cell pellet at the bottom of the Falcon tube.
-
15.
Resuspend in 1 mL of ACK lysis buffer to lyse red blood cells.
-
16.
Incubate for 5 min at 20°C–25°C
-
17.
Immediately add 9 mL of staining solution to the lung suspension.
-
18.
Spin down 5 min at 400 × g.
-
19.
Slowly remove the supernatant. Resuspend in 4 mL of staining solution.
-
20.
Pass the lung suspension through a 40 uM cell strainer and filter.
-
21.
Spin down 5 min at 400 × g.
Optional: Remove the supernatant and resuspend in 1 mL of Fc blocking solution (Staining solution + Fc block (anti CD16/32 for mouse)). Transfer to a 2 mL Eppendorf tube and incubate for 15 min at 4˚C. Spin down at 500 × g for 3 min.
-
22.
In the meantime, prepare the antibody mixture for staining. (In our previous Dual RNA-seq paper we used a mixture of CD64, MertK and SiglecF antibodies to distinguish between alveolar and interstitial macrophages at a concentration of 1:200).
-
23.
Remove the supernatant and resuspend the lung pellet with the antibody mix in staining solution at the appropriate concentration.
-
24.
Incubate for 20 min in the dark at 20°C–25˚C.
-
25.
Spin down at 1200 × g for 3 min.
-
26.
Remove the supernatant and wash twice with 1 mL of PBS 1×.
-
27.
Resuspend in sorting buffer at an appropriate dilution based on the characteristics of your sorter, passing the lung suspension through a 40 uM cell strainer right before sorting, to avoid the formation of clumps that may clog the nozzle of the instrument.
Sorting
-
28.
Fill 2 mL Eppendorf tubes (one for each sample) with ∼700 μL of Trizol reagent to collect the sorted cells.
Note: This amount depends on the configuration of your machine. We usually get about ∼150 μL of sorting volume for 30k cells.
-
29.
Prepare the sorting gates, acquire and start sorting keeping the samples at 4˚C during the process, in order to minimize unwanted changes in the expression profile of both host and bacteria.
-
30.
Sort the infected cells directly into Trizol.
-
31.
At the end of the sorting process immediately mix the samples, manually rocking the tubes 5 times to allow dissociation of the nucleoprotein complexes of the host cells.
-
32.
Incubate for 5 min at 20°C–25˚C.
-
33.
Place at −80˚C or proceed to the following section of the protocol.
Pause Point (at this stage we usually leave the samples at −80˚C for not more than 2 days)
RNA Extraction
Following sorting into Trizol, the host cell RNA will be free in the supernatant, while the bacteria will be for the most part still intact. The rationale is to use a high speed centrifugation step to separate bacteria and cell debris as a pellet at the bottom of the tube. Because of the limited number of infected host cells recoverable from an in vivo experiment, we developed what we call a “one tube procedure” for RNA extraction, in order to minimize loss of material. Therefore, following centrifugation, the Trizol supernatant should be removed carefully, to minimize the potential loss of bacteria. Following the removal of the Trizol supernatant containing the host RNA, we perform mechanical disruption of the pelleted bacteria and add back part of the Trizol supernatant, to generate a more favorable bacteria/host RNA ratio.
We were able to recover sufficient RNA to generate a library from as low as 20k infected host cells.
Note: all of the reagents that we use in this step are certified DNase and RNase free.
-
34.
Thaw Trizol samples at 20°C–25˚C, if frozen.
-
35.
Centrifuge samples at high speed (≥12,000 × g) for 20 min.
-
36.
Slowly remove ∼90% of the volume of the Trizol supernatant (containing host RNA) and set aside in a new 2 mL Eppendorf tube, leaving ∼150 μL behind.
Note: Leave a small amount of Trizol behind to avoid disturbing the bacteria/cell debris pellet. For 2 mL Eppendorf tubes we typically leave behind ∼150 μL.
Figure 2.

Resuspending the Bacterial/Cell Debris Pellet in Trizol and Zirconia Beads
(A) ~150ul of Trizol supernatant is left behind in the tube containing the bacterial/cell debris pellet.
(B) 150ul of Zirconia Beads and 400ul of fresh Trizol are added to the same tube.
-
37.
Add 150 μL of Zirconia beads to the tube containing the bacterial pellet.
-
38.
Add 400 μL of fresh Trizol to the same tube (Figure 2B).
-
39.
Perform bead beating using the homogenizer setting (BioSpec – Mini-Bead Beater with 8 speed controller) or at max speed for 2 cycles of 1 min, resting on ice 2 min between the cycles.
-
40.
After bead beating, mix well by manually inverting the tubes 5 times.
-
41.
Let the samples rest for 2 min at 20°C–25˚C.
-
42.
Re-add part of the volume of the Trizol containing the host cell RNA that you set aside in step 36 to the tube with the zirconia beads.
-
a.For in vivo experiments and recovery of ∼30k infected cells we typically add back 70% of the Trizol supernatant. We usually achieve between 2%–3% of reads mapping to Mtb.
-
b.For in vitro experiments (BMM or HMDM infected at an MOI of 4:1), we sort 45k infected cells and put back ∼40%–50% of the Trizol supernatant. We usually achieve between ∼8% to ∼10% of reads mapping to Mtb.
-
a.
Note: The goal here is to find a balance between adding back sufficient host RNA to have the critical mass of rRNA-depleted mRNA needed for library preparation and avoiding having too much host RNA that would lead to a low percentage of reads mapping to Mtb.
-
43.
Mix well by inverting the tubes 5–6 times.
-
44.
Add the appropriate amount of Chloroform to each sample (200 μL for 1 mL of Trizol).
-
45.
Mix, shaking vigorously for 15 s.
-
46.
Incubate 2 min at 20°C–25˚C.
-
47.
Centrifuge 15 min at 12,000 × g.
-
48.
Carefully transfer the aqueous phase to a new 1.5 mL RNase-free eppendorf tube.
-
49.
Add 2 μL of Glycoblue to each sample (follow manufacturer instructions: flick well but do not pipette to mix, quick spin, and put the tubes on ice.)
-
50.
Add the appropriate amount of Isopropanol to each sample (flick well to mix, do not pipette). For 1 mL of Trizol, add 500 μL of Isopropanol.
-
51.
Wipe out the tubes and take out of BSL3.
-
52.
Incubate at least 1 h at −80˚C (at this stage we typically leave the samples at −80˚C).
-
53.
Thaw the frozen samples at 20°C–25˚C.
-
54.
Centrifuge 20 min at 12,000 × g, at 4˚C.
-
55.
Remove the supernatant using a wide-bore pipette tip. You should now be able to see the nucleic acid pellet colored in blue at the bottom of the tube.
-
56.
Wash with 75% ice-cold ethanol. Flick to loosen the pellet. For 1 mL of Trizol, use 1 mL of 75% ice-cold ethanol.
-
57.
Centrifuge 15 min at 7,400 × g, at 4˚C.
-
58.
Pipette off as much ethanol as possible without disturbing the pellet.
-
59.
Air dry the pellet in a clean place.
-
60.
Resuspend each RNA pellet vigorously in 12 μL of Nuclease free water.
-
61.
Incubate at 20°C–25˚C for 2 min, flick to mix and store on ice (if proceeding to the next step) or at −80˚C for long term storage.
DNase Treatment, rRNA Removal, and Library Construction
In many cases the following steps will usually be performed by the RNA-seq core of your institution. For the most part we follow the standard manufacturer protocols with very slight modifications as detailed here:
-
1.
Dnase treatment: we use the commercially available Turbo-DNA free kit (Invitrogen), following manufacturer instructions with no modifications.
-
2.rRNA removal:
-
a.After DNase treatment, quantify the amount of total RNA recovered. Ex: from 30k sorted infected macrophages we usually recover ∼50/60 ng of total RNA.
-
a.
Note: We strongly suggest the use of the Qubit instrument for this step (RNA high sensitivity kit), as quantification with the Nanodrop of very low amounts of RNA is imprecise.
-
b.Analyze the quality of the RNA by Fragment Analyzer.
-
b.
Figure 3.

Examples of a Fragment Analyzer Track
(A) In the top right corner is the expected result of the Fragment Analyzer track when following this protocol. RQN values <6 may lead to inefficient rRNA removal when using the Ribo-Zero or similar kits.
(B) Example of a Bioanalyzer track of the final library from one of our low input dual RNA-seq samples.
Note: Highly degraded RNA (RIN <6) will strongly decrease the efficiency of the rRNA-depletion step.
-
c.rRNA removal is performed using 50–100 ng of total RNA input and a minor modification to the manufacturer protocol for the Ribo-Zero Epidemiology Gold rRNA removal kit (Illumina) https://cornell.box.com/s/ajqggp6xpkg768456mzflgph2qlrsbj4. Briefly, 90 μL bead stock is used per sample in step A, while 2 μL each of reaction buffer and removal solution in a 20 μL reaction volume are used in step B, as detailed in the manufacturer’s protocol. The final rRNA-depleted samples are purified by ethanol precipitation following the Ribo-Zero protocol.
-
d.Alternatively, a 1:1 mix of depletion solutions from the H/M/R and Bacteria NEBNext rRNA Depletion Kits can be used to remove rRNA. The final rRNA-depleted samples are purified using RNA Sample Purification Beads.
-
c.
-
3.
Library preparation: Sequencing libraries are generated using the NEBNext Ultra II Directional RNA Library Prep Kit for Illumina (New England Biolabs) using half of the rRNA-depleted sample (no quantification), with 15 min fragmentation time in first-strand reaction buffer (Protocol Chapter 2 [NEBNext rRNA Depletion Kits; 1:1 mix of depletion reagents] or Chapter 4 [Illumina Ribo-Zero Epidemiology Depletion]). We follow the manufacturer instructions with 5-fold adapter dilution, except that only half of the cDNA sample is used as input for PCR (12 cycles) to allow for a second amplification reaction if needed to adjust the barcode index or the number of PCR cycles. The total yield per library is typically ∼20–100 ng (1–5 ng/μL in 20 μL, determined with HS DNA Qubit assay). Higher or lower total yields may require adjustments in the number of PCR cycles. The library size distribution is determined on a Fragment Analyzer. Figure 3B
-
4.
Sequencing: Libraries are pooled based on estimated molarity and sequenced on a NextSeq500 (Illumina) in multiple rounds until the desired sequencing depth for bacterial reads is reached (target ∼1 M Mtb reads). Libraries are also compatible with sequencing on other Illumina instruments, however, adjustments may be recommended during library preparation for longer read lengths (less fragmentation time, adjustments to bead cleanup steps); see NEBNext Ultra II Directional RNA Library Prep Kit, Appendix A).
Expected Outcomes
The amount of total RNA recovered correlates with the number of infected cells that have been processed: for ∼30k cells we usually recover ∼50–60 ng of total RNA.
Quality of RNA should be assessed by Fragment Analyzer. Ideally, the Fragment Analyzer track should show very small amount of RNA degradation (clear rRNA picks) and should be free of genomic DNA contamination.
Libraries should have a total yield of 20–100 ng (1–5 ng/μL in 20 μL) with a size distribution centered on ∼300 bp (anticipating 75nt SE reads).
You should expect to obtain between 2%–3% of the total reads to align to the Mtb transcriptome for an in vivo experiment, and higher numbers for in vitro experiments when using MOI > 2:1.
Quantification and Statistical Analysis
Data Analysis
Here we provide an in-depth description of the pipeline we used for the analysis of the sequencing files in the original Dual RNA-seq paper. After sequencing, Mtb and host reads are mixed together in the same FASTQ file for each sample. Furthermore, even after rRNA-depletion a certain number of unwanted rRNA reads are present in the sample files. The strategy we use is to first separate and quantify the rRNA reads present in each sample (this is useful as a benchmark to evaluate the efficiency of the rRNA-depletion step). After the rRNA reads are removed, for each sample, we proceed to separate Mtb and host cell reads into two different FASTQ files . At this point, reads for each organism are aligned against the respective reference genome and we count the number of times a sequence from each gene was present in our data. We then use the framework provided by the DESeq2 package for the differential gene expression analysis.
-
1.
Perform quality control of the fastq files with FastQC.
-
2.Remove low quality reads and trim Illumina Adapter. We used Flexbar 3.4 for this step with the following parameters:
-
a.--adapter-min-overlap 7 --max-uncalled 2 --min-read-length 50 -aa TruSeq -q TAIL -qf i1.8 -qt 20 --qtrim-post-removal
-
a.
-
3.Remove rRNA reads using Bowtie2 and a custom fasta file containing rRNA regions from both organisms. We used the following settings in Bowtie2 to separate rRNA and mRNA reads:
-
a.--sensitive --al rRNA.fastq --un mRNA.fastq
-
a.
-
4.Separate Mtb and host cells reads with Bowtie2:
-
a.--very-sensitive --al TB.fastq --un Mouse.fastq
-
a.
-
5.Align Mtb and mouse reads using Hisat2. If you use a directional library preparation kit, specify the strandness of the reads. For our Dual-seq paper where we used the “NEBNext Ultra II Directional RNA Library prep kit” that would be “reverse.” For example:
-
a.hisat2 --rg-id=TB_AM1 --rg SM:TB_AM1 --rg LB:TB_AM1 --dta -x tb_index --rna-strandness R -U TB_AM1.fastq -S TB_AM1.sam
-
a.
-
6.
Sort, convert, and index the SAM and BAM files.
-
7.
Use HTSeq to generate a raw read counts matrix for each organism.
-
8.
Import the raw reads count matrix in R and perform differential expression using DESeq2 and APEGLM for log-fold change estimation.
Note: The entire pipeline, including linux commands and the R scripts that we used in the original Dual RNA-seq paper are available here: https://cornell.box.com/s/vkbz5n5egy6e7v4fzbowsd5hx9qtmxr9.
Statistical Testing
Statistical testing for the differential gene expression analysis (DGE) is performed as described (Love et al., 2014). Shrinkage of effect sizes (LFC estimates) is performed using the APEGLM method (Zhu et al., 2019). Genes with less than 10 raw counts across all samples are excluded from downstream analysis. Genes having a false discovery rate (FDR) <0.05 and a fold change >1.5 are considered significant. Visualization and clustering (PCA, heatmaps of sample-to-sample distances) are performed on variance stabilized counts (vst) (Anders and Huber, 2010) with the option “blind = TRUE” in the DESeq2 package in order to compare samples in an unbiased manner. Heatmaps for specific groups of genes are generated using the normalized counts obtained from the DESeq2 analysis, log-transformed and Z-scaled using the package heatmap2 in R.
The raw datasets needed to repeat the analysis published in Cell Reports are available on GEO: http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE132354.
Limitations
The protocol has been developed for the use with Mtb, a bacteria with a strong cell wall that is resistant to immediate dissociation in Trizol/GTC (guanidine thiocyanate). Although we have not yet tested this, it is possible that the protocol may work with other Gram+ bacteria, particularly if isolation of nucleic acids requires mechanical disruption of the cell wall, comparable to Mtb.
Our approach is based on adjusting the ratio of bacteria/host RNA, therefore the protocol will not work in all those situations where sorting and isolation of infected cells is not possible (ex: most human disease samples). Because the average amount of total RNA for each bacterial cell is ∼0.1 pg vs 20 pg of RNA for a host cell (Westermann et al., 2012), even small proportions of non-infected cells in your sample (ex:20%) will skew the bacteria/host RNA ratio in an unfavorable way and therefore recovering enough Mtb reads will require deep sequencing, with all the economic limitations associated with it. Finally, the protocol will not work with archived, PFA fixed materials.
Troubleshooting
Problem
Poor total RNA quality in the Fragment Analyzer track.
Potential Solution
A degraded Fragment Analyzer track may arise from a multitude of different issues. The followings are some aspects to take into consideration:
-
•
Make sure to remove the host RNA in step 36 of the RNA extraction protocol and set it aside on a RNase-free tube. Performing bead beating on a large amount of host RNA will lead to poor-quality isolation.
-
•
Make sure to use RNase-free certified reagents during the RNA extraction process and beyond.
-
•
Do not air dry the RNA pellet at 37˚C, but only at 20°C–25˚C and only for short periods of time (we usually air dry the pellet for 20 min at 20°C–25˚C ). Do not leave the RNA pellet at 20°C–25˚C for >1 h.
-
•
Keep the RNA on ice whenever possible and store it at −80˚C for long term storage.
-
•
Make sure to cleanse all pipettes and tips with a product capable of removing RNase contamination.
Problem
Low yields of total RNA extracted. Not enough rRNA-depleted mRNA to prepare a library for sequencing.
Potential Solution
In our experience low RNA yields arise in the following situations:
-
•
Removal of too much host RNA in step 42 of the RNA extraction protocol. You should consider the host RNA as a carrier for the bacterial RNA. With 30–40k infected cells, you only have ∼80–100k bacteria which equals to <2 ng of total RNA. Therefore, removing more >50% volume of the host RNA, will lead to poor total RNA yields for the mixed total RNA. We suggest to never remove more than 50% of host RNA.
-
•
Poor recovery of the sorted cells in the collection tubes. We suggest to accurately control and check the deflection points of your sorting instrument, to make sure all the sorted droplets end up in the collection tubes. This is very important when you are only collecting 30k infected cells.
-
•
Losing the RNA pellet during the precipitation step. We suggest to use the Glycoblue reagent in order to help visualize the nucleic acids pellet.
-
•
RNase contamination. Check that all reagents are certified DNase and RNase free.
Problem
Small insert size in Illumina libraries, or reads that extend through the insert or overlap (for PE reads).
Potential Solution
Use the recommended modifications in the NEBNext Ultra II Directional RNA Library Prep Kit protocol, Appendix A, to increase library insert size.
Problem
Significant peak at ∼135 bp indicating >10% adaptor dimer (molarity) in the Illumina library.
Potential Solution
Use more RNA input for rRNA depletion or for library preparation, or increase the fold dilution of the NEBNext adaptor.
0.8X SPRI beads cleanup might also help to exclude any fragment below 200 bp. However, additional cleanup step may result in loss of material. If working with low yield libraries and using the NEBNext library preparation kit presented in this protocol, we suggest increasing the fold dilution of the NEBNext adaptor.
Problem
Illumina library total yields <1 ng (<0.5 ng/μL in 20 μL) or >400 ng (>20 ng/μL in 20 μL) for libraries of size ∼250–350 bp (adjust accordingly for larger insert sizes).
Potential Solution
Adjust PCR cycle number. Because adding more PCR cycles always introduces some PCR bias and we do not have UMIs to identify and remove reads that originate from PCR duplicates, we process the samples starting from the one with the lower amount and we keep the PCR cycle number a constant (as well as input amount) for the entire project so that any bias is as similar as possible across all samples in the experiment.
Problem
Low percentage of reads mapping to the Mtb transcriptome.
Potential Solution
-
•
Make sure to not lose the bacterial/cell debris pellet in step 36 of the RNA extraction protocol. Remove the tubes from the centrifuge very carefully, mark the spot where the bacterial pellet is supposed to be and slowly remove the supernatant leaving behind a small amount of Trizol.
-
•
Make sure to use an FMO control for cell sorting. This is essential for setting up the sorting gate for the mCherry signal: contamination of cells that are not infected with the bacteria during cell sorting (whose signal arise from autofluorescence or from spillover from other channels) will lead to low percentage of reads mapping to Mtb.
-
•
Make sure to select the correct strandness of the reads in the data analysis steps.
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, [David G. Russell] (dgr8@cornell.edu).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
The raw datasets needed to repeat the analysis published in Cell Reports are available on GEO: http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE132354.
The entire pipeline, including linux commands and the R scripts that we used in the original Dual RNA-seq paper are available here: https://cornell.box.com/s/vkbz5n5egy6e7v4fzbowsd5hx9qtmxr9.
Acknowledgments
The work was supported by grants AI118582 and AI134183 to D.G.R. from the National Institutes of Health, USA. The authors wish to thank Linda Bennett for technical and organizational support.
Author Contributions
D.P., L.H., and D.G.R. designed the study. D.P., B.R.N.L., and L.H. conducted experiments. D.P., L.H., J.K.G., and D.G.R. analyzed and interpreted the results. D.P., D.G.R., and J.K.G. drafted and edited the manuscript.
Declaration of Interests
The authors declare no competing interests.
Contributor Information
Davide Pisu, Email: dp554@cornell.edu.
David G. Russell, Email: dgr8@cornell.edu.
References
- Anders S., Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11 doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S., Pyl P.T., Huber W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D., Langmead B., Salzberg S.L. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods. 2015;12:357–360. doi: 10.1038/nmeth.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisu D., Huang L., Grenier J.K., Russell D.G. Dual RNA-seq of Mtb-infected macrophages in vivo reveals ontologically distinct host-pathogen interactions. Cell Rep. 2020;30:335–350 e334. doi: 10.1016/j.celrep.2019.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimand J., Isserlin R., Voisin V., Kucera M., Tannus-Lopes C., Rostamianfar A., Wadi L., Meyer M., Wong J., Xu C. Pathway enrichment analysis and visualization of omics data using g:Profiler, GSEA, Cytoscape and EnrichmentMap. Nat. Protoc. 2019;14:482–517. doi: 10.1038/s41596-018-0103-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roehr J.T., Dieterich C., Reinert K. Flexbar 3.0 - SIMD and multicore parallelization. Bioinformatics. 2017;33:2941–2942. doi: 10.1093/bioinformatics/btx330. [DOI] [PubMed] [Google Scholar]
- Westermann A.J., Gorski S.A., Vogel J. Dual RNA-seq of pathogen and host. Nat. Rev. Microbiol. 2012;10:618–630. doi: 10.1038/nrmicro2852. [DOI] [PubMed] [Google Scholar]
- Zhu A., Ibrahim J.G., Love M.I. Heavy-tailed prior distributions for sequence count data: removing the noise and preserving large differences. Bioinformatics. 2019;35:2084–2092. doi: 10.1093/bioinformatics/bty895. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The raw datasets needed to repeat the analysis published in Cell Reports are available on GEO: http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE132354.
The entire pipeline, including linux commands and the R scripts that we used in the original Dual RNA-seq paper are available here: https://cornell.box.com/s/vkbz5n5egy6e7v4fzbowsd5hx9qtmxr9.