

Key Points
CLL increases the pool of ST-HSCs in the peripheral blood.
Differentiation of CLL-derived peripheral blood HSPCs is skewed toward the myeloid lineage.
Visual Abstract

Introduction
Chronic lymphocytic leukemia (CLL) is an indolent B-cell malignancy invariably infiltrating the bone marrow. Although treatment options for patients with advanced disease have significantly improved in the past years, the disease remains incurable, and after emergence of therapy, resistant disease patients succumb to infections because of secondary bone marrow failure. The underlying mechanisms impairing normal hematopoiesis in patients with CLL are poorly defined.
Mature, differentiated B cells, either pre- or postgerminal center, are considered the cell of origin of unmutated and mutated CLL, respectively.1 However, this paradigm has been challenged based on the observation of CLL-recurrent mutations present in a fraction of hematopoietic stem cells (HSCs),2 which demonstrated a differentiation bias toward the lymphoid lineage in a xenotransplantation model.3 To what extent this skewed differentiation of HSCs contributes to bone marrow failure of patients with advanced disease remains unknown. Other mechanisms contributing to suppressed hematopoiesis in CLL include the secretion of tumor necrosis factor-α by malignant B cells, which not only inhibits the growth of HSCs in vitro4,5 but also has complex effects on HSCs and progenitor cell function in vivo, including enhancing myelopoiesis. In support of this hypothesis, higher levels of plasma tumor necrosis factor-α correlate with anemia in patients with CLL. In addition, as shown by us6 and others,7 “disease-induced” activation of NF-κB in bone marrow stroma cells further contributes to an inflammatory microenvironment and potentially aggravates bone marrow failure.
We hypothesized that the inflammatory microenvironment induced by bone marrow–infiltrating CLL cells negatively affects normal hematopoiesis by skewing hematopoietic stem and progenitor cell (HSPC) lineage commitments and residence in the bone marrow.
Methods
We assessed the frequency of HSPCs in the peripheral blood (PB) of 69 individual patients with the diagnosis of CLL, and 21 age-matched healthy controls. All individuals consented to the use of PB samples for research purposes and in accordance with the Declaration of Helsinki. These studies were approved by the Cambridgeshire Research Ethics Committee (07/MRE05/44). PB samples were collected in the afternoon, when circulation of hematopoietic progenitors is known to reach its peak because of circadian oscillation.8 To assess the absolute numbers of CD34+ cells, nonpurified peripheral blood mononuclear cells from 5 mL PB were stained and gated for CD34 positivity and CD19 negativity to quantify the total amount of circulating hematopoietic progenitor cells, using precision counting beads (Biolegend) and flow cytometry. Relative frequencies of HSPC subsets were assessed on CD34+-enriched cells using beads (Miltenyi Biotec) and following the gating strategy depicted in supplemental Figures 1 and 2.
To assess differentiation capacity of HSPCs, 1 × 103 or 6 × 103 CD34+ cells were plated in duplicate in complete semisolid methylcellulose (Methocult H4435; STEMCELL Technologies). Differentiated colonies were counted and scored based on morphology 14 days after culture in a humid environment at 37°C using STEMVision camera and software (STEMCELL Technologies).
Results and discussion
The average age of all patients was 69 years (range 40 to 92 years) with a male:female ratio of 1.8:1 (patient characteristics are listed in supplemental Table 1). Patients were either treatment naive or relapsed (average number of previous treatments = 2). The average age of healthy controls was 66 (range 32 to 85 years).
We first quantified the absolute numbers of circulating HSPCs and found similar numbers of CD34+ cells to age-matched healthy individuals (Figure 1A). Similarly, no differences were observed between different Binet groups (Figure 1B) or treatment-naive vs previously treated patients (Figure 1C). Subsequently, differences in the relative frequency of HSC subpopulations were assessed by flow cytometry using lineage markers and CD34, CD38, CD45, CD10, CD7, CD49f, and CD90 (supplemental Table 2). Our analyses revealed that common myeloid and myeloerythroid progenitors (CMPs-MEPs) (CD19−CD34+CD38+CD45RA−CD10−CD7−) were more abundant in the PB of patients with CLL (Figure 1D-E). Conversely, myelolymphoid progenitors (CD19−CD34+CD38−CD45RA+) were significantly decreased in patients’ blood compared with healthy controls. The HSC pool (CD19−CD34+CD38−CD45RA−), defined as a pool of HSC and multipotent progenitors, is known to contain a heterogeneous pool of cells with different self-renewal and differentiation capacities,9-11 was also more abundant in patients’ blood. In particular, short-term HSCs (ST-HSCs) (CD19−CD34+CD38−CD45RA−CD90−CD49f−), multipotent cells with limited self-renewal and repopulation capacity,10 and differentiation bias toward the erythroid lineage,11,12 were found in higher frequencies in the PB of patients with CLL (Figure 1D-E), whereas frequencies of long-term HSCs (CD19−CD34+CD38−CD45RA−CD90+CD49f+), cells with multipotent long-term repopulation capacity,10,13 were comparable to healthy controls. In addition, we did not observe significant differences within the group of treatment-naive patients stratified by Binet stage with the exception of slightly fewer total HSCs in the Binet C group (supplemental Figure 3). Upon dividing the entire cohort of patients based on the treatment status, we observed similar changes between naive and previously treated patients with CLL (Figure 1F). Notably, PB white blood cell counts did not correlate with higher frequencies of ST-HSCs (Figure 1G). In addition, we did not observe differences between patient samples with Del13q, normal karyotype, or presence of TP53 (supplemental Figure 4). Conclusively, these results suggest that CLL cells impair bone marrow residency of ST-HSC and MEP progenitor cells, irrespective of previous treatments. The absence of a correlation between full blood counts and HSPCs indicates that these changes are already present at an early disease stage.
Figure 1.
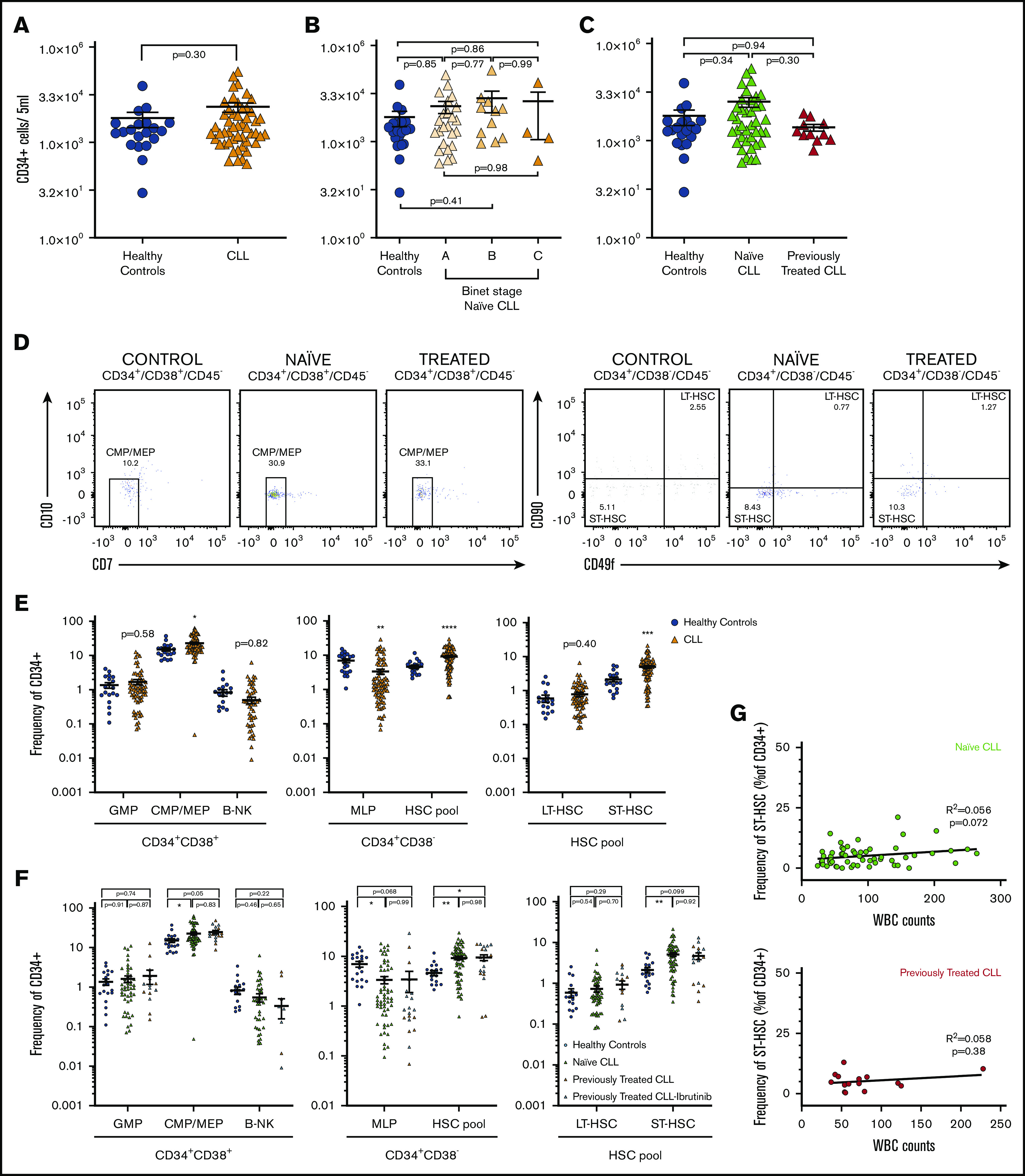
Altered frequencies of PB HSPCs in patients with CLL. (A-C) Absolute numbers of global (CD34+) hematopoietic progenitors from nonpurified peripheral blood mononuclear cells isolated from PB. (A) Comparison between aged-matched controls (n = 20) and patients with CLL (n = 54) is shown. Unpaired Student t test. (B) Quantification of CD34+ cells in treatment-naive patients, separated by Binet stage (ordinary 1-way analysis of variance [ANOVA] followed by unpaired Student t tests). Binet stage, A = 25; B = 11; C = 4. (C) Abundance of CD34+ progenitors in the CLL cohort by treatment type (ordinary 1-way ANOVA followed by unpaired Student t tests). Controls, N = 20; naive patients with CLL, n = 40; ibrutinib-treated, n = 3; previously treated, n = 11. (D) Representative fluorescence-activated cell sorting plots of CMPs-MEPs and ST-HSC abundance of freshly isolated CD34+ cells. Values indicate abundance of the specific population, as relative frequency of total CD34+ pool. (E) Dot-plot showing relative abundance of specific hematopoietic progenitors in healthy controls (n = 20) and the entire patient cohort (n = 89) (unpaired Student t test controls). (F) Dot-plot showing relative abundance of specific hematopoietic progenitors in healthy controls (n = 20) and in patients with CLL by treatment group (ordinary 1-way ANOVA); naive patients, n = 66; previously treated patients, n = 23. (G) Linear regression plots showing correlation of ST-HSC frequencies (as %CD34+) to white blood cell counts in naive patients (n = 58) (above), and previously treated patients (n = 15) (below). P values from unpaired Student t tests or 1-way ANOVA are denoted with asterisks according to the following: ****P < .0001, ***P < .001, **P < .01, *P < .05. GMP, granulocyte monocyte progenitors; B-NK, B-NK progenitors; WBC, white blood cell.
We next investigated the differentiation properties of PB-derived CD34+ cells. CLL-derived and healthy CD34+ progenitors showed similar proliferation capacity and generated similar numbers of differentiated colonies (Figure 2A). We also did not observe differences in colony formation between CD34+ cells isolated from patients with different Binet groups (Figure 2B) or between treatment-naive and previously treated patients (Figure 2C). However, further characterization of colonies identified that CD34+ progenitors from patients with CLL generated a significantly higher proportion of myeloid colonies at the expense of erythroid colonies compared with controls (Figure 2D-E). This skewed differentiation of CD34+ cells was predominantly driven by treatment-naive patients compared with previously treated patients (Figure 2F). These data demonstrate that higher frequencies of ST-HSCs and CMP-MEPs in the PB of patients with CLL are associated with a skewed differentiation into myeloid cells in vitro, and progenitor cells are less likely to produce erythrocytes. Interestingly, we found that this phenotype was predominantly driven from CD34+ cells from normal-karyotype patients, whereas cells from patients with Del13q showed a more balanced differentiation output, similar to progenitors derived from healthy controls (Figure 2G).
Figure 2.
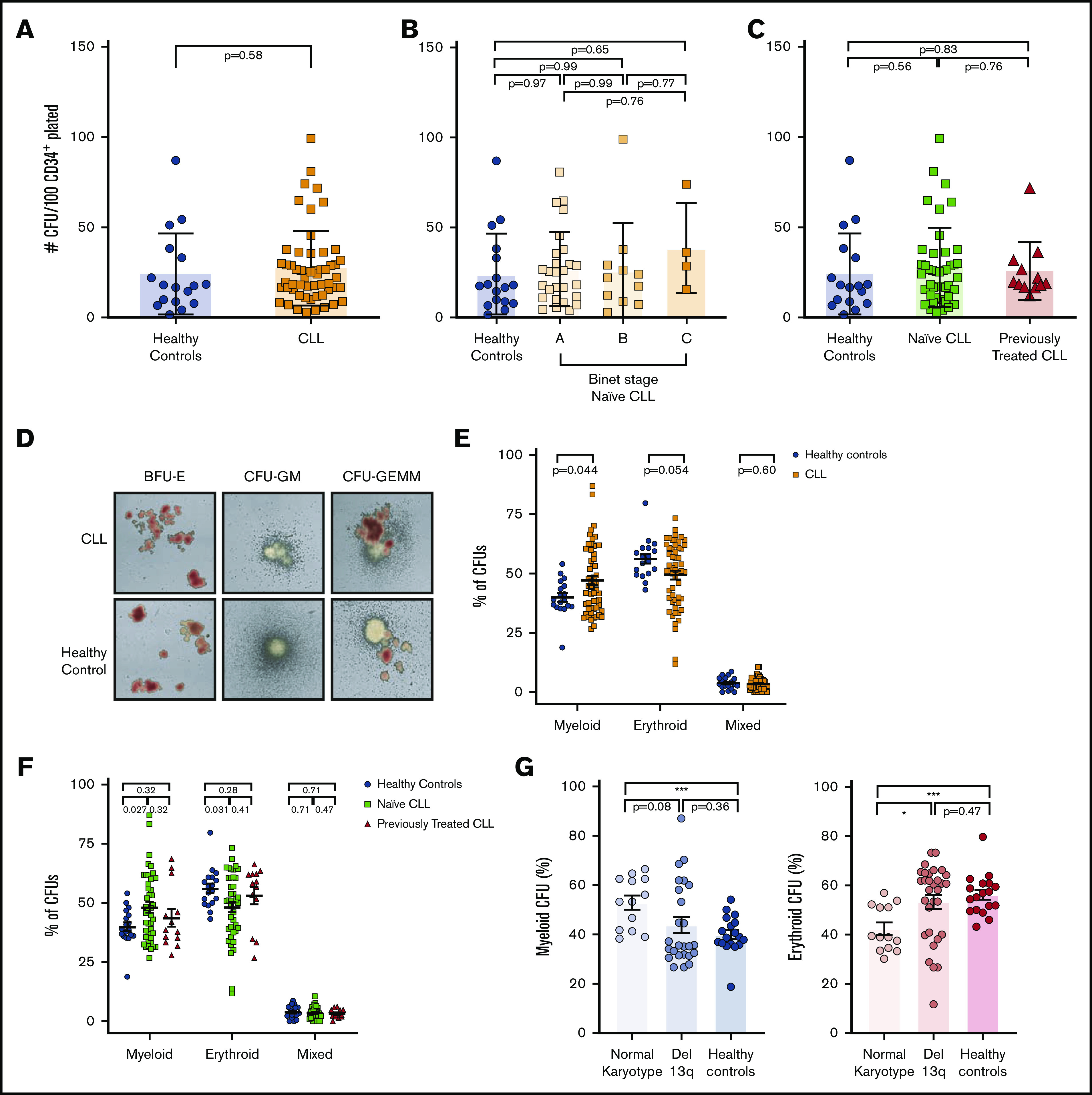
Functional characterization of PB CD34+progenitor cells from patients with CLL. (A-C) Bar plots showing the absolute number of colony-forming units (CFU) generated in methylcellulose assays by PB CD34+ of (A) healthy controls (n = 17) and of patients with CLL (n = 55) (unpaired Student t test; P < .05). Comparison between healthy controls and Binet groups (B) or treatment-naive patients with CLL vs treated patients (C) (ordinary 1-way ANOVA followed by unpaired Student t tests). Differentiated colonies were counted and scored based on morphology 14 days postculture in humid environment at 37°C. Colonies were analyzed using STEMVision (STEMCELL Technologies). (D) Representative pictures of CFUs generated by healthy or CLL-derived hematopoietic progenitors. BFU-E, burst forming unit–erythroid; CFU-GEMM, colony-forming unit–granulocyte, erythrocyte, macrophage, megakaryocyte; CFU-GM, colony-forming unit–granulocyte, macrophage. (E-F) Dot-plots showing the composition of CFUs generated by healthy controls and patients with CLL (E) or by naive patients with CLL or treated patients with CLL (F) (1-way ANOVA followed by unpaired Student t test). (G) Histograms showing myeloid (left) or erythroid (right) CFU frequencies in CLL samples clustered based on chromosomal abnormalities. P values from unpaired Student t tests or 1-way ANOVA are denoted with asterisks according to the following: ***P < .001, *P < .05.
To our knowledge, this is the first study using high-resolution flow cytometry identifying significantly higher frequencies of PB ST-HSCs and CMP-MEPs in patients with CLL, associated with a skewed differentiation into myeloid colonies at the expense of erythroid cells. These findings likely reflect first signs of bone marrow failure and exhaustion of normal stem and progenitor cells and complement recently published data, indicating that the bone marrow of patients with CLL is depleted in phenotypically defined HSC, multipotent progenitor, CMP, and common lymphoid progenitors.14 It is therefore reasonable to assume that disease-induced alterations in bone marrow niches contribute to the redistribution of HSPCs, which then no longer participate in normal blood production.
Although these conclusions are drawn from in vitro experiments and whether skewing is also present in physiological niches in vivo remains unknown, they are supported by clinical findings in this patient population. Early- to intermediate-stage patients almost never present with neutropenia, whereas mild anemia is a common finding in this patient population. In contrast, myelosuppression is more common in previously treated patients, and the absence of a differentiation bias of PB CD34+ cells in this group may be a result of direct cytotoxic effects from therapies on normal HSCs. In addition, the observation that progenitor cells from Del13q patients behave more similarly to normal healthy control cells with regard to their differentiation potential may reflect the better overall prognosis of this group compared with other cytogenetically defined subgroups.15 Whether the skewed differentiation of PB HSPCs in patients with CLL reflects compensation mechanisms of normal progenitor cells in response to an abnormal cytokine and niche environment or relates to recurrent genetic abnormalities found in a subset of HSCs remains to be investigated. Our data provide further insights on the mechanisms causing cytopenia in patients with CLL and suggest that a pathologic mobilization of ST-HSCs is a contributing factor.
Supplementary Material
The full-text version of this article contains a data supplement.
Acknowledgments
The authors express their deepest gratitude to the patients who donated blood for this research. Samples were obtained with assistance from the Cambridge Blood and Stem Cell Biobank, funded by the Cambridge Cancer Centre and Cambridge Stem Cell Institute.
This work was funded by the Cancer Research UK (CRUK; C49940/A17480). I.R. is a senior CRUK fellow. This work was supported by a Sir Henry Dale fellowship from Wellcome/Royal Society (107630/Z/15/Z) (E.L.). Research in E.L.’s laboratory is supported by Wellcome, BBSRC, EHA, and Royal Society. Research in I.R.’s and E.L.’s laboratories is supported by core support grants by Wellcome and MRC to the Wellcome-MRC Cambridge Stem Cell Institute.
Footnotes
The manuscript does not contain high-throughput data; all reagents and methods have been disclosed.
Authorship
Contribution: A.S. performed all experiments and analyzed data; C.A. and C.B. performed fluorescence in situ hybridization analyses with support from A.G.; E.C. contributed to functional assays and phenotyping panel design; A.W. and J.E.B. recruited patients to this study; and E.L. and I.R. planned this study and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Ingo Ringshausen, University of Cambridge, Wellcome Trust/MRC Cambridge Stem Cell Institute & Department of Haematology Jeffrey Cheah Biomedical Centre, Cambridge Biomedical Campus Puddicombe Way, Cambridge CB2 0AW, United Kingdom; e-mail: ir279@cam.ac.uk.
References
- 1.Seifert M, Sellmann L, Bloehdorn J, et al. Cellular origin and pathophysiology of chronic lymphocytic leukemia. J Exp Med. 2012;209(12):2183-2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Damm F, Mylonas E, Cosson A, et al. Acquired initiating mutations in early hematopoietic cells of CLL patients. Cancer Discov. 2014;4(9):1088-1101. [DOI] [PubMed] [Google Scholar]
- 3.Kikushige Y, Ishikawa F, Miyamoto T, et al. Self-renewing hematopoietic stem cell is the primary target in pathogenesis of human chronic lymphocytic leukemia. Cancer Cell. 2011;20(2):246-259. [DOI] [PubMed] [Google Scholar]
- 4.Michalevicz R, Porat R, Vechoropoulos M, et al. Restoration of in vitro hematopoiesis in B-chronic lymphocytic leukemia by antibodies to tumor necrosis factor. Leuk Res. 1991;15(2-3):111-120. [DOI] [PubMed] [Google Scholar]
- 5.Tsopra OA, Ziros PG, Lagadinou ED, et al. Disease-related anemia in chronic lymphocytic leukemia is not due to intrinsic defects of erythroid precursors: a possible pathogenetic role for tumor necrosis factor-alpha. Acta Haematol. 2009;121(4):187-195. [DOI] [PubMed] [Google Scholar]
- 6.Lutzny G, Kocher T, Schmidt-Supprian M, et al. Protein kinase c-β-dependent activation of NF-κB in stromal cells is indispensable for the survival of chronic lymphocytic leukemia B cells in vivo. Cancer Cell. 2013;23(1):77-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Paggetti J, Haderk F, Seiffert M, et al. Exosomes released by chronic lymphocytic leukemia cells induce the transition of stromal cells into cancer-associated fibroblasts. Blood. 2015;126(9):1106-1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Méndez-Ferrer S, Lucas D, Battista M, Frenette PS. Haematopoietic stem cell release is regulated by circadian oscillations. Nature. 2008;452(7186):442-447. [DOI] [PubMed] [Google Scholar]
- 9.Majeti R, Park CY, Weissman IL. Identification of a hierarchy of multipotent hematopoietic progenitors in human cord blood. Cell Stem Cell. 2007;1(6):635-645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Notta F, Doulatov S, Laurenti E, Poeppl A, Jurisica I, Dick JE. Isolation of single human hematopoietic stem cells capable of long-term multilineage engraftment. Science. 2011;333(6039):218-221. [DOI] [PubMed] [Google Scholar]
- 11.Belluschi S, Calderbank EF, Ciaurro V, et al. Myelo-lymphoid lineage restriction occurs in the human haematopoietic stem cell compartment before lymphoid-primed multipotent progenitors. Nat Commun. 2018;9(1):4100-4115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Notta F, Zandi S, Takayama N, et al. Distinct routes of lineage development reshape the human blood hierarchy across ontogeny. Science. 2016;351(6269):aab2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huntsman HD, Bat T, Cheng H, et al. Human hematopoietic stem cells from mobilized peripheral blood can be purified based on CD49f integrin expression. Blood. 2015;126(13):1631-1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Manso BA, Zhang H, Mikkelson MG, et al. Bone marrow hematopoietic dysfunction in untreated chronic lymphocytic leukemia patients. Leukemia. 2019;33(3):638-652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Döhner H, Stilgenbauer S, Benner A, et al. Genomic aberrations and survival in chronic lymphocytic leukemia. N Engl J Med. 2000;343(26):1910-1916. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.