

Abstract
Crohn's disease (CD) is a chronic inflammatory disease of the gastrointestinal tract that leads to substantial suffering for millions of patients. In some patients, the chronic inflammation leads to remodelling of the extracellular matrix and fibrosis. Fibrosis, in combination with expansion of smooth muscle layers, leaves the bowel segment narrowed and stiff resulting in strictures, which often require urgent medical intervention. Although stricture development is associated with inflammation in the affected segment, anti‐inflammatory therapies fall far short of treating strictures. At best, current therapies might allow some patients to avoid surgery in a shorter perspective and no anti‐fibrotic therapy is yet available. This likely relates to our poor understanding of the mechanism underlying stricture development. Chronic inflammation is a prerequisite, but progression to strictures involves changes in fibroblasts, myofibroblasts and smooth muscle cells in a poorly understood interplay with immune cells and environmental cues. Much of the experimental evidence available is from animal models, cell lines or non‐strictured patient tissue. Accordingly, these limitations create the basis for many previously published reviews covering the topic. Although this information has contributed to the understanding of fibrotic mechanisms in general, in the end, data must be validated in strictured tissue from patients. As stricture formation is a serious complication of CD, we endeavoured to summarize findings exclusively performed using strictured tissue from patients. Here, we give an update of the mechanism driving this serious complication in patients, and how the strictured tissue differs from adjacent unaffected tissue and controls.
Keywords: Crohn's disease, fibrosis, strictures
1. SEARCH STRATEGY
The information summarized in this review is from a PubMed search for relevant articles by conjoining three main search blocks with the Boolean operator ‘AND’. The first block is the disease group itself and contained (‘Inflammatory Bowel Diseases’ [Mesh] OR ‘Crohns’ OR ‘Crohn's’ OR ‘IBD’ OR ‘Inflammatory bowel disease*’). The second block is the disease condition (‘Fibrosis'[Mesh] OR ‘Constriction, Pathologic’[Mesh] OR ‘Stenos*’ OR ‘Strictur*’ OR ‘fibrotic’ OR ‘fibrosis’). The third block concerned the cellular and molecular mechanisms (‘Fibroblasts’[Mesh] OR ‘Fibroblast*’ OR ‘Extracellular matrix’[Mesh] OR ‘Intestinal Mucosa’[MeSH] OR ‘Myofibroblast*’ OR ‘Macrophages’[Mesh] OR ‘Macrophage*’ OR ‘Monocyte*’ OR ‘Leucocytes, Mononuclear’[Mesh] OR ‘Dendritic Cells’[Mesh] OR ‘Intercellular Signalling Peptides and Proteins’[Mesh] OR ‘Muscle Cells’[Mesh]). Filtered on 'human' and 'English', the search generated over 700 articles. After an initial screening, based on the content of the abstracts, around 250 articles remained that were imported into a reference library for a more thorough read‐through. About 50 articles remained that met the strict criteria of having at least one figure or paragraph in the article where strictured tissue was taken from a Crohn's patient and compared to non‐strictured tissue. Consequently, to meet the scope of this review, all data from animal or cell lines, as well as exclusively clinical, epidemiological, and radiological data, were omitted. Articles focused on blood markers or CD papers without comparisons to strictured tissue/cells were also excluded. To ensure that we did not miss relevant articles that were not captured by this search, we also screened previously published reviews and key papers for references.
2. BACKGROUND
2.1. CD progression into stricturing disease
Crohn's disease (CD) is a chronic relapsing inflammatory bowel disease affecting the gastrointestinal tract of millions of patients worldwide 1 that leads to substantial life‐long suffering. On the one hand, there are symptoms typically associated with the intestinal inflammation itself, such as abdominal pain, diarrhoea, fatigue and weight loss. On the other hand, are indirect effects of having the disease. These include side effects of drugs, permanent effects of surgery, several possible complications and increased risk of malignancies. 2 These indirect effects can be at least as debilitating as the underlying inflammation and are clearly part of the disease picture for patients. Indeed, for unknown reasons during the disease course, CD patients typically display one of three phenotypes: stricturing, penetrating, or non‐stricturing/non‐penetrating. 3 Each disease behaviour comes with a different set of complications that add up to the disease burden. The stricturing phenotype, characterized by progressive bowel wall thickening and lumen narrowing (called B2 phenotype 4 ), affects a substantial number of patients. It is associated with obstructive symptoms that often require acute surgical intervention. Indeed, strictures are one of the main indications for CD‐associated surgery. 5
2.2. B2 stricturing disease—current therapy and why better understanding is needed
Crohn's patients are heterogeneous in their clinical picture. Thus, while an anti‐inflammatory treatment(s) may alleviate fibrotic complications for some patients, it may have no effect on others. Recent data suggest that anti‐inflammatory therapies might be beneficial to avoid surgery for a subgroup of patients in a shorter perspective. 6 However, whether anti‐inflammatory therapy treats only the inflammatory component 7 , 8 of the stricture, or if it affects stricture development per se, is not known. Indeed, despite the introduction of biological treatments 20 years ago, a significant number of patients still need stricture‐associated surgery. Moreover, an anti‐fibrotic therapy per se is not currently available. Understanding the mechanisms leading to strictures will thus improve the quality of life for many patients by reducing the need for surgery. It may also lead to as yet unavailable treatments and thus positively impact patient care and disease outcome.
3. OVERVIEW OF STRICTURE FORMATION
Chronic intestinal inflammation is necessary for stricture formation. However, the factor(s) driving inflammation into fibrosis and strictures are poorly understood, as is the question of why not all patients with intestinal inflammation develop strictures.
3.1. Histological characteristics of strictures
Histologically, the strictured wall displays a twofold increase in mural thickness through disproportional changes within each layer (Figure 1: Normal margin to Stricture). 9 This leads to about six times less compliant wall. 10 Stricture formation is a slow process and only becomes apparent in late stages, which is why antecedent events are hard to study. Presumably, it is a dynamic process where mediators, cell populations, and physical conditions change over time; it is also intuitive to think that stricture location (ileum vs colon) likely matters.
Figure 1.
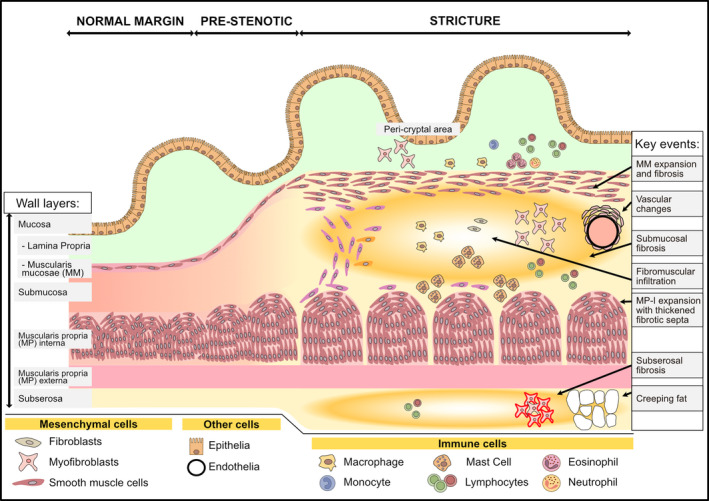
An overview of strictured tissue showing changes in intestinal wall layers and cell populations. A schematic of the most apparent histological changes in the intestinal wall during the progression from normal to strictured tissue is shown. This includes increased intestinal wall thickness due to expanded smooth muscle cells, and hence muscle layers, concomitant with fibrotic changes in the submucosa and subserosa. Histological features described in the text are highlighted in the right column as ‘Key events’ with the location of the event indicated by an arrow. See the text and subsequent figures for details
Although the mechanism behind stricture initiation and propagation is poorly understood, it involves two main parallel events, namely, expansion of the smooth muscle layers and a fibrosing process (Figure 1: stricture). Affected segments display increased collagen deposition in all layers. Typically, the submucosa is most affected (Figure 1: key events ‐ submucosal fibrosis), leading to a 2‐3‐fold thickening. 9 , 11 , 12 , 13 , 14 Significant fibrosis also affects the subserosa 11 , 13 , 15 , 16 (Figure 1: key events ‐ subserosal fibrosis). In the muscularis mucosae (Figure 1: key events ‐ MM expansion and fibrosis), fibrosis intermingles with distorted smooth muscle bundles. In contrast, fibrosis in the muscularis propria interna (Figure 1: key events: MP‐I expansion with thickened fibrotic septa) affects the intramuscular septa but leaves the organization of the smooth muscle well‐preserved. 9 , 15
Despite these changes, the increase in smooth muscle outweighs the increase in collagen; quantitatively, expansion of the muscle layers contributes more to the overall increase in thickness. 9 , 15 For example, Zhang et al reported a 17‐fold increase in muscularis mucosae thickness in ileal strictures, which accounted for nearly half of the wall thickening. 9 Chen et al reported a more modest increase in muscularis mucosae and instead highlighted the 2‐fold increase in muscularis propria as the most striking finding. Interestingly, muscular hyperplasia/hypertrophy in each layer was higher in the ileum, whereas fibrosis in the submucosa and muscularis propria was significantly higher in colonic lesions. 15 In addition, fibromuscular bundles, containing smooth muscle cells (SMCs), project out from muscularis mucosae and muscularis propria into the fibrotic submucosa. Sometimes, they create a bridge of hyperplastic muscle between the two layers (Figure 1: key events ‐ fibromuscular infiltration). 15
Other microanatomical features reported in strictured tissue are the remodelling of mucosal‐facing vascular walls in the submucosa, which show smooth muscle thickening and collagen deposition 9 (Figure 1: key events ‐ vascular changes). A feature of CD is the so‐called creeping fat (also known as mesenteric fat‐wrapping) (Figure 1: key events ‐ creeping fat). This is an accumulation of mesenteric fat around affected segments of the intestine. Indeed, there is increasing interest in how factors secreted from this adipose tissue affect the wall layers and stricture development. Creeping fat is particularly interesting as histologically, it appears to dissect into the subserosa and directly face the outer muscular layers. 17
3.2. Location of different cell types gives clues to the players in stricture pathogenesis
3.2.1. Cellular characteristics of strictures
Associated with the histological changes observed in strictures is accumulation of, and alterations in, the behaviour of cells of mesenchymal origin as well as certain immune cell populations. Mesenchymal cells, which include fibroblasts, myofibroblasts and SMCs, expand and change behaviour in response to environmental cues such as cytokine signalling from immune cells. However, the signalling is not necessarily unidirectional as mesenchymal cells also secrete multiple factors that impact surrounding immune cells. Moreover, all three mesenchymal cells can also secrete extracellular matrix components that fill the fibrotic tissue.
One of the keys to understanding possible interactions between immune cells and mesenchymal cells is to define their specific location in the strictured tissue. This is summarized in Figures 1, 2, 3, which provide a summary compiled from available studies using human tissue. Figure 1 gives an overview, while Figures 2 and 3 highlight the strictured wall with a focus on location of mesenchymal cells. However, there are some limitations to this compilation due to inconsistencies in the literature in the use of cell markers, and that tissues from different intestinal compartments (ileum versus colon) are lumped together.
Figure 2.
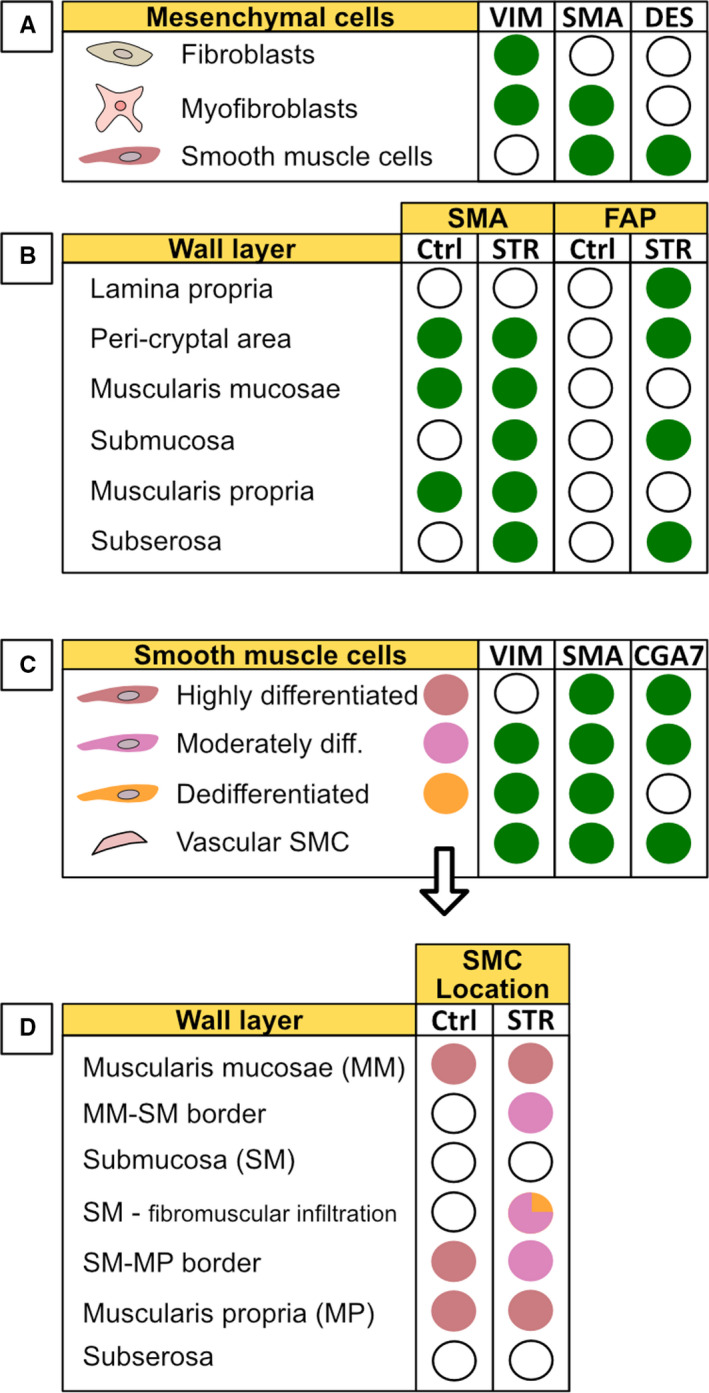
An overview of mesenchymal cell markers and their distribution in tissue. A, Fibroblasts, myofibroblasts and smooth muscle cells are traditionally distinguished by their staining pattern of the three markers vimentin, α‐SMA and desmin. B, Distribution of the additional markers SMA and FAP in the intestinal walls of strictured (STR) tissue compared to normal (non‐strictured) control (Ctrl) tissue. C, Staining pattern for smooth muscle cells with different phenotypes (based on the degree of differentiation) as described in the text. D, The distribution of smooth muscle cells with different degrees of differentiation in the intestinal wall layers in non‐strictured tissue (Ctrl) and strictured tissue (STR). (A‐C): Green filled circle indicates presence of a marker (positive staining). C,D, Filled circles indicate the presence of a particular SMC phenotype based on the colour coding defined in C. VIM, Vimentin; SMA, alpha‐smooth muscle actin; DES, Desmin; FAP, Fibroblast activating protein; Ctrl, normal non‐strictured tissue; STR, strictured tissue; diff, differentiated; CGA7, antibody that selectively recognizes contractile smooth muscle cells expressing both alpha‐ and gamma‐actin; SMC, Smooth muscle cell. The staining patterns reflect data from numerous publications and mostly reflect immunohistochemical observations
Figure 3.
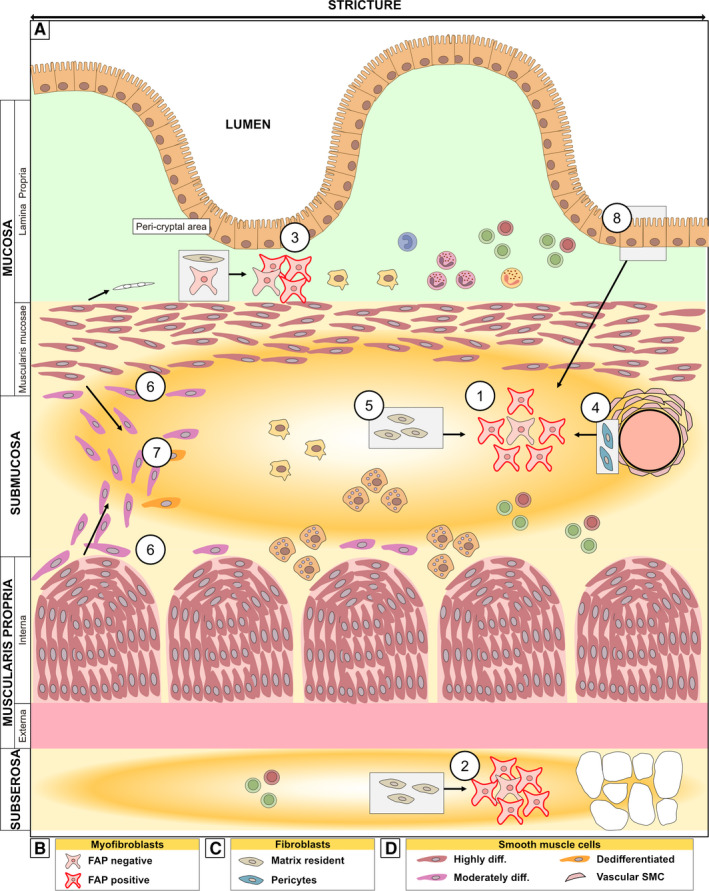
Schematic of the strictured wall with a focus on location of mesenchymal cells. A, The transition into strictured tissue involves an increase in extracellular matrix deposition, fibrosis (indicated by yellow/orange) particularly in the submucosa and subserosa, and expansion of the muscle layers along with infiltration of cells. There is accumulation of predominantly FAP+ myofibroblast in the layers most affected by fibrosis, namely, in ① the submucosa and ② subserosa. However, similar enrichment is also observed in the ③ peri‐cryptal area of the mucosa. Grey boxes show the condition in normal (non‐strictured) tissue with arrows showing the changes that occur inside strictured tissue. The accumulated myofibroblasts might have arisen from ④ pericytes, ⑤ matrix‐resident fibroblasts, or ⑧ epithelial cells (through epithelial‐to‐mesenchymal transition). Also depicted is ⑥ phenotypically changed SMCs along the border between the submucosa and the muscle layers and in the ⑦ fibromuscular infiltration in submucosa. (B‐D) shows the subtypes of the mesenchymal cells depicted in (A), as discussed. See the text for further details
3.2.2. Immune cells in strictures
The importance of immune cells in CD pathogenesis is well‐documented and uncontested. In particular, immune cells in the mucosal layer, particularly the lamina propria, are well‐studied in non‐strictured mucosa of CD patients. However, in strictured mucosa, the descriptions are patchier. Stricture mucosa shows an increased frequency of CD3+ cells (Figure 1: lymphocytes), but the relative abundance of T cells in strictures is significantly less than that in inflamed, non‐strictured mucosa. Eosinophils in the mucosa (Figure 1: eosinophils) of paediatric strictures are elevated, as they are in inflamed mucosa, but strictured tissue had a higher eosinophil burden. 18 An increased frequency of an IL36+ macrophage subset that co‐localizes with peri‐cryptal myofibroblasts has also been described 12 (Figure 1: peri‐cryptal area).
More detailed studies that address other specific immune cell subtypes in strictures (such as T‐cell subsets, B cells, NK cells, etc) are lacking. Moreover, in CD in general as well as in CD strictures, there is often a transmural inflammation with immune cells present to varying degrees in all intestinal wall layers (Figure 1: stricture). 15 , 19 In contrast to the mucosa, immune cells in deeper layers are poorly characterized, and even less is known about their function in strictures and cell‐cell interactions. Indeed, studies defining specific immune cell types below the mucosa of strictured tissue are relatively few or only broadly define the immune cell types present. For example, in immunohistochemistry analyses, such cells are often identified in broader terms like chronic inflammatory infiltrates or aggregates of lymphocytes or mononuclear cells. 15 , 16 , 19 , 20 However, there are some exceptions to this. For example, CD68+ macrophages are increased in frequency and occupy the fibrotic areas 21 (Figure 1: macrophages). In addition, there is an enrichment of mast cells in all layers, particularly in the submucosa and muscularis propria (Figure 1: mast cells). 22 , 23
In summary, information about the relative abundance of immune cells in stricture tissue is fragmentary or is broadly characterized. The functions and cell‐cell interactions of immune cells in the setting of strictures are poorly understood, especially below the mucosa.
3.3. Mesenchymal cells and their location in strictures
3.3.1. Mesenchymal markers and their distribution in the strictured tissue
The three major mesenchymal cell types central to strictures have been historically identified using a combination of three markers: the intermediate filament proteins vimentin (vim) and desmin (des) as well as the contractile protein alpha‐smooth muscle actin (α‐SMA). 24 Using these markers, fibroblasts are identified as vim+α‐SMA−Des− cells. In contrast, myofibroblasts are vim+α‐SMA+ Des− whereas SMCs are vim−α‐SMA+Des+ (Figure 2A). Thus, comparing the immunohistochemical staining pattern of α‐SMA in strictured vs non‐strictured tissue gives a rough indication of the major changes in mesenchymal subpopulations (Figure 2B: SMA column). This type of study has shown that, in strictures, α‐SMA stains the submucosa 13 and subserosa 16 in addition to the cell types stained in non‐stricture tissue (muscle layers, blood vessel walls and peri‐cryptal cells). Thus, there is an accumulation of myofibroblasts in layers most affected by fibrosis (Figure 3: ① and ②).
Although fibrosis in the mucosa is relatively scarce, strictured mucosa displays changes in the staining pattern of mesenchymal cells. There is increased vimentin staining, predominantly in the peri‐cryptal region, and lamina propria. 25 This occurs together with extra‐muscular staining of α‐SMA, 13 especially in the peri‐cryptal area. 12 , 15 Proliferating Des+α‐SMA+ cells (indicative of SMCs) also occasionally infiltrate the lamina propria. 15 Thus, there seems to be an enrichment of mesenchymal cells, also in the mucosa (Figure 3: ③). However, the VIM/α‐SMA/Des classification scheme discussed here is simplified and not specific, especially since vimentin and α‐SMA stain cell types other than those mentioned above. Thus, to help refine the picture of changes in mesenchymal cells in strictures, other markers have been used to further classify the cells as described below.
3.3.2. FAP
One such marker is Fibroblast Activating Protein (FAP) (Figure 3B). The function of FAP 26 , 27 is unclear in non‐tumorous conditions, and FAP is not a well‐defined marker for fibroblasts or myofibroblasts. However, FAP positivity is an interesting marker since it is essentially absent from both healthy intestine and purely inflamed (nonfibrotic) intestinal mucosa (Figure 2B: FAP column). 16 In contrast, FAP is present in the mucosa, submucosa and subserosa of strictured tissue, 16 , 21 , 28 , 29 and the majority of myofibroblasts (and/or fibroblasts) isolated from mucosa and submucosa of strictures show FAP positivity 28 , 29 (Figure 3: ①, ②, ③). These findings and for several other reasons, as discussed below, make FAP an interesting marker.
3.3.3. Pericytes
To distinguish them from resident fibroblasts in the extracellular matrix, the fibroblasts attached to blood vessel walls are called pericytes (Figure 3C). Pericytes have been proposed to be a source of myofibroblasts based on observations from strictures 16 and findings in other fibrotic diseases. 30 Under healthy conditions, pericytes are found around blood vessels (Figure 3: ④), in the submucosa and serosa. Interestingly, in strictures, there was an accumulation of myofibroblasts, but neither the pericytes nor the matrix‐resident fibroblasts were present. 16 This observation suggests that pericytes (Figure 3: ④), in addition to the matrix‐resident fibroblasts (Figure 3: ⑤), may contribute to the accumulated pool of myofibroblasts (Figure 3: ① and ③).
3.3.4. Smooth muscle cells (SMCs)
A combination of vimentin and two different antibodies that recognize α‐SMA (1A4 and CGA7) have been used to identify three different subtypes of SMCs inside strictures 23 : highly differentiated SMCs (vim−1A4+CGA7+), moderately differentiated (vim+1A4+CGA7+), and dedifferentiated SMCs (vim+1A4+CGA7−) (Figures 2C, and 3D). Using this classification, the authors showed that the muscularis mucosa and muscularis propria consisted of highly differentiated SMC, as expected. However, what is interesting was that less mature SMCs (moderately differentiated) were found along the borders between the submucosa and the two muscular layers (Figures 2D, and 3: ⑥). They were also found in the fibromuscular strokes infiltrating the submucosa (Figure 3: ⑦), where also the third phenotype (dedifferentiated SMCs) could occasionally be seen. 23 Within the field of vascular biology, it is postulated that smooth muscle cells can switch to a secretory phenotype with the ability to secrete extracellular matrix. 31 However, whether such a phenotype has any implications in stricture development is unknown.
3.3.5. Summary
Regarding mesenchymal cells in intestinal strictures, there is an increase in myofibroblasts, especially in the wall layers most affected by fibrosis, that are predominantly FAP+. Changes in the SMC phenotype can also be observed in the fibromuscular infiltration and in the linings of the muscle layers. These changes in mesenchymal cells occur parallel with alterations in the relative frequency of immune cells, particularly mast cells.
3.4. Expression of genes and proteins in fibrotic tissue
3.4.1. Introduction
Each of the cell populations residing in the intestinal wall differs in relative frequency, location and function. These populations communicate through direct cell‐cell interactions and/or by producing or responding to factors in the environment. Sensing mechanical and microbial signals adds further complexity to the environment. Thus, describing differences at the whole tissue level (analyses on endoscopic surgical or biopsies) with respect to transcribed genes or secreted factors is too unspecific to address the contribution of a particular cell type in this complex environment. However, whole tissue analyses give valuable insight into the result of cells working in concert. They also provide information to further decipher the cellular sources and temporal production of factors that impact the tissue. Thus, studying stricture tissue as a whole, and the cell populations that comprise the tissue, gives clues into stricture pathogenesis.
The goals of this section are twofold. First, differences between strictured and non‐strictured whole tissue are summarized (Figure 4A). Second, differences in specific intestinal wall layers in strictured tissue compared to the same layers in normal margin tissue are presented (Figure 4B). The comparisons of strictured tissue presented here are relative to non‐strictured margin from the same individual and/or similar tissue from non‐IBD patients.
Figure 4.
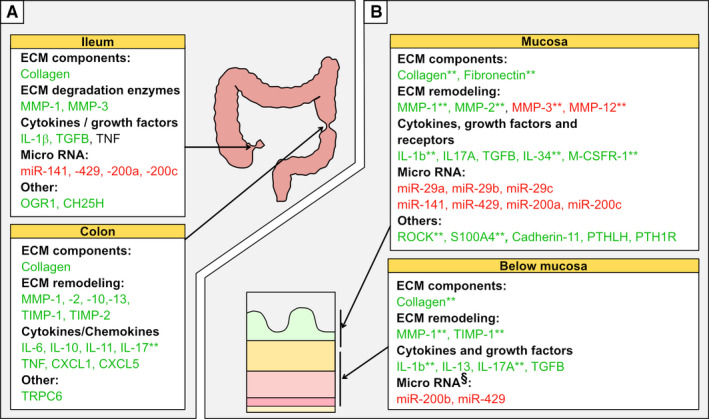
Genes and proteins expressed in strictured tissue compared to normal tissue. A, Shows differences in whole tissue analyses of mRNA and/or protein in strictured ileum (upper panel) and colon (lower panel) compared to non‐strictured tissue. B, Shows differences in intestinal wall layer expression of relevant proteins, mRNAs and miRNAs in the mucosa (upper panel) or below the mucosa (lower panel) of strictured tissue compared non‐strictured tissue. Green indicates expression higher than, and red indicates expression lower than, non‐strictured tissue. Standard text indicates mRNA level; * indicates protein level; ** indicates changes detected at both the protein and mRNA levels. § indicates submucosa only. Abbreviations; miR = micro‐RNA
3.4.2. Extracellular matrix components and tissue remodelling
The primary hallmark for strictures, besides smooth muscle cell expansion, is fibrosis. Fibrosis, described in its simplest form, is the result of a disturbed balance between deposition and degradation of extracellular matrix components, leading to a net accumulation of proteins such as collagen. It thus follows that the top genes or proteins altered in strictured tissue are coupled to this deposition‐degradation balance. For example, transcription of various collagen genes is increased in strictured colon, 12 , 32 ileum, 10 , 33 , 34 and mucosa 35 (Figure 4A and B). Strictured mucosa (Figure 4B) also contains more total fibronectin and disproportional increases in its ED‐A and ED‐B isoforms. 36 This may represent the presence of myofibroblasts, whose migration depends largely upon these molecules. 37 Moreover, strictured tissue shows alteration in several types of matrix metalloproteinases (MMPs), 10 , 11 , 12 , 32 , 34 , 38 at both the whole tissue and wall layer levels (Figure 4), which are involved in the degradation and remodelling of the extracellular matrix. Matrix degradation can also release stored growth factors, adding yet another aspect when considering the resultant effect in vivo. In general, MMP‐1 enrichment seems widespread in strictured tissue 11 , 32 , 34 while changes in other MMPs are more compartmentalized (Figure 4A and B). 10 , 11 , 12 , 32 , 34 , 38 Alterations in tissue inhibitors of metalloproteinases (TIMPs), which act by counteracting MMPs, are also present in strictured tissue. In particular, increased TIMPs are primarily seen in the colon 32 rather than the ileum 34 (Figure 4A), and in muscle layers 11 rather than the mucosa (Figure 4B). Thus, different compartments display different types of disturbances and may relate to the enrichment of different cell types in each layer. A detailed characterization of MMPs and TIMPs in inflammatory bowel disease has recently been reviewed. 39
3.4.3. Cytokines and growth factors that affect collagen deposition
Cells in the intestinal wall communicate through cytokines, and several cytokines show increased expression in strictures. Indeed, many cytokines, as discussed below, affect the expansion and behaviour of mesenchymal cells and their production of collagen, MMPs and TIMPs in favour of matrix degradation. For example, strictured colon has increased expression of IL‐10, which has anti‐inflammatory properties. There is also increased transcription of pro‐inflammatory cytokines like IL‐17, 40 IL‐6 and TNF 12 as well as several chemokines that recruit immune cells (Figure 4A: colon). Strictured ileum, on the other hand, has increased IL‐1β transcripts 10 but not TNF transcripts 34 (Figure 4A: ileum). Another interesting cytokine that is upregulated in colonic strictures is IL‐11. 32 The role of IL‐11 in fibrosis and stromal inflammation has recently been reviewed. 41
At the intestinal wall level, strictured mucosa (Figure 4B: top) displays increased IL‐1β 11 and IL17A. 40 Similar to the mucosa, deeper layers (defined as below muscularis mucosa) (Figure 4B: bottom) also have increased IL‐1β and IL17A, among others, 11 , 42 and secrete more collagen. 11 , 42 , 43 IL‐17 may have a role in stricture development, and a recent review summarized the potential for Th17/IL‐17 involvement in intestinal fibrosis. 44 Beyond the classical cytokines, ileal mucosa has upregulation of IL‐34 and its receptor M‐CSFR‐1. 45
TGF‐β, one of the main pro‐fibrotic factors that also has immunoregulatory properties, shows upregulation in the mucosa, 38 submucosa and muscle 42 , 43 (Figure 4B). Based on histopathological studies, TGF‐β shows widespread staining in stricture sections (in all wall layers), and TGF‐β co‐localizes with aggregates of mononuclear or lymphoid cells. 13 , 16 , 20 , 21 Thus, these immune cell aggregates seem to be the major source of TGF‐β.
Overall, data suggest widespread upregulation of IL‐1β, IL‐17A and TGF‐β, while several other cytokines are reported to be increased although their significance remains to be determined.
3.4.4. Micro‐RNAs and environment‐sensing receptors in stricture formation
Additional reports have shown alterations in several other molecules, including micro‐RNAs (miRNA), in strictures compared to normal margin or control tissue. miRs are non‐coding RNAs that exert post‐transcriptional silencing of gene expression, and if dysregulated, can potentially influence critical pathogenic pathways. In particular, strictures show downregulation of the miRNA‐200 family 25 (including miR‐141, miR‐200, and miR‐429), as well as the miRNA‐29 family 35 (Figure 4A and B). Downregulation of these gene silencers is interesting, especially as both miRNA families are suggested to interfere with TGF‐β1 signalling, which, as mentioned above, is an important pro‐fibrotic factor. Moreover, transcripts of the pH‐sensing receptor OGR1, shown to be expressed by lamina propria cells, 33 and the oxysterol‐synthesizing enzyme CH25H 46 are increased in strictured ileum (Figure 4A: ileum). Both of these are strongly correlated to transcripts of fibrosis‐related genes such as COL1A1, COL3A1, ACTA2 and TGF‐β1. Although these observations are interesting, their significance is unclear; further studies are needed to determine if they influence the fibrotic process.
3.4.5. Can tissue changes be used to predict future development of strictures?
So far we have discussed tissue changes in strictures that have already developed in patients. The question arises if a gene expression pattern exists in (inflamed) intestinal tissue before a stricture develops. If so, this could be a means to predict patients that will develop strictures with time. A recent paper addressed this intriguing question by analysing gene expression in biopsies from a large cohort of newly diagnosed paediatric Crohn's patients. 47 Gene expression patterns in those who developed strictures at their 5‐year follow‐up were compared to those who maintained a purely inflammatory disease behaviour. Over 500 differentially expressed genes were identified and revealed an inflammatory signature associated with extracellular matrix genes and future stricture development. 47 This suggests that gene expression changes occur in the intestinal tissue of patients prone to develop strictures early in the process. It also identifies genes associated with stricture development to aid in classification of patients and facilitate research to identify targets to prevent stricture development.
4. WHEN HEALING GOES WRONG: THE ROLE OF MESENCHYMAL CELLS IN STRICTURE FORMATION
As discussed above, the thickened bowel wall in strictures is associated with the accumulation of mesenchymal cells, particularly fibroblasts, myofibroblasts and SMCs. Fibroblasts (among other cell types as discussed below) are precursors to the cell type considered to be the main effector cell responsible for making up the fibrosis matrix, namely myofibroblasts. In addition to the fibrosis per se, which perhaps makes the intestine unique compared to other fibrotic diseases, is the concomitant expansion of the smooth muscle layers, which contributes to organ dysfunction.
Understanding the behaviour of mesenchymal cells, how they are influenced by the surrounding microenvironment, and how they, in turn, affect other cells, is crucial for understanding stricture pathogenesis. Thus, the next sections focus on these aspects of the three major cell types (fibroblasts, myofibroblasts, and SMCs) that are thought to explain, directly or indirectly, the majority of changes seen at the tissue level in strictures. Discussion regards differences between mesenchymal cells isolated from strictured versus non‐strictured tissue, and Figure 5 puts the discussed topics into a context illustrated using a general wound healing model.
Figure 5.
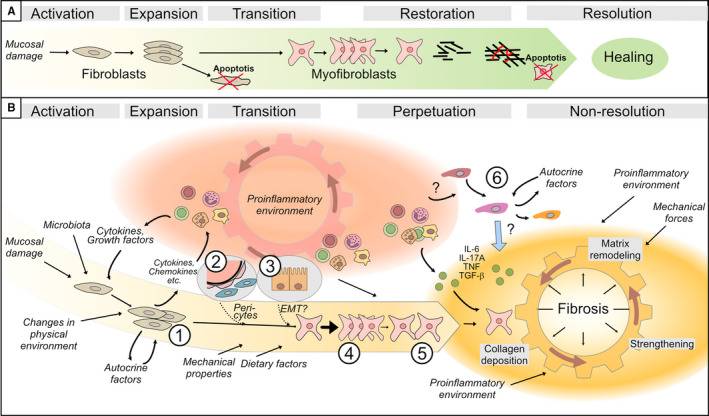
Schematic illustration of the cells and factors involved in the indicated phases of the stricturing process in intestinal tissue. A, The sequence of events during a simplified and idealized normal healing process. This involves activation and expansion of fibroblasts and subsequent transition of fibroblasts into myofibroblasts, which finalize the restoration of the extracellular matrix. The process occurs during a limited time period, and when the repair process is complete, redundant cells undergo apoptosis (indicated by cells with a red ‘x’) and healing is achieved. B, Under the prevailing conditions in the intestinal wall of a subset of Crohn's patients, the ‘normal’ healing process is aberrant. The pro‐inflammatory environment (red cogwheel) activates and fuels the activation and recruitment of myofibroblast precursors, which include ①matrix‐resident fibroblasts, ② pericytes and ③ epithelial cells. The transition to myofibroblasts can be further facilitated in the presence of certain environmental cues. Released products from activated fibroblasts, as well as from immune cells, create a positive feedback loop so the cogwheel keeps getting fuelled. This results in a shift from an initial inflammation‐driven process to an inflammation‐facilitated process, as the activated mesenchymal cells successively show independence from stimulatory signals. Subsequently, myofibroblasts ④ expand, and ⑤ migrate into the ‘injured’ site where they secrete collagen. Newly formed matrix is strengthened by contraction and cross‐linking, as indicated in the Fibrosis area (yellow cogwheel). However, in a setting where pro‐inflammatory mediators are additionally supplied by immune cells, and perhaps also by ⑥ infiltrating smooth muscle cells, matrix degradation is also stimulated. This leads to a vicious cycle with alternating deposition and degradation of matrix, leading to non‐resolution and fibrosis. Cell symbols are as defined in the Figure 1 and 3 legends
4.1. Fibroblasts
4.1.1. Short introduction
Function
Fibroblasts populate connective tissue and are responsible for its maintenance and as such, secret extracellular matrix components. Collagen is one of the main constituents of the extracellular matrix, particularly in fibrotic areas in strictures.
Wound healing
During physiological tissue repair, collagen replaces the temporary fibrin plug to create a permanent sealant of damaged tissue. In response to signals from injury, which normally initiate a healing process, fibroblasts become activated, proliferate and expand (Figure 5A: activation and expansion). As illustrated in the transition step in Figure 5A, expanded fibroblasts transform into myofibroblasts, a cell type which has several qualities needed during tissue repair. Subsequently, after performing the healing task, fibroblasts and myofibroblasts undergo apoptosis (Figure 5A: Restoring and Resolution).
Dysregulated wound healing
However, the sequence of events in the normal healing process (Figure 5A) may not apply to strictured tissue where several factors are present that maintain and misdirect the healing process (Figure 5B). In fact, factors in strictured tissue might themselves serve as damage signals. This is supported by the apparent lack of previous insults and may rather reflect persistent injury‐like stimuli resulting from the chronic inflammatory environment (Figure 5B: inflammation ‘cogwheel’), microbiota, food, etc. This scenario would lead to perpetuation and non‐resolution (Figure 5B, right), rather than restoration and resolution of the healing process (Figure 5A, right), and ultimately to fibrosis (Figure 5B: fibrosis ‘cogwheel’). Therefore, fibrosis is often described as ‘a healing process that went wrong’ (Figure 5B).
4.2. Cells and factors affecting fibroblast collagen production
To understand the stimuli that activate fibroblasts, we will start off by discussing factors affecting fibroblast proliferation and collagen production, and the sources of such molecules. These stimuli can be physical, such as mechanical forces, or occur after contact with matrix components, or loss thereof; stimuli can also come from autocrine or paracrine sources. The impact of the surroundings can be exemplified by ileal stricture fibroblasts, which produce twice as much collagen as colon stricture fibroblasts. 14 This is also evident from in vitro findings where stricture fibroblasts show increased proliferation compared to margin and control cells. Moreover, stricture fibroblasts display abnormally high collagen production (quantitatively, up to 10‐fold higher production 14 ) in the basal state when cultured in the same conditions as normal fibroblasts. 14 , 20
4.2.1. TGF‐β
TGF‐β is perhaps the most well‐known secreted factor that is enriched in stricture tissue that, among other functions, increases collagen production in fibroblasts. As discussed above, aggregates of mononuclear or lymphoid cells are most likely the source of TGF‐β. 13 , 16 , 20 , 21 Interestingly, stricture fibroblasts demonstrate an excessive response to TGF‐β when compared to normal cells. 14 , 20 However, following chronic activation with TGF‐β, fibroblasts might display functional changes, so they respond to stimuli in a different way than normal fibroblasts. For example, diminished responses of stricture fibroblasts to TGF‐β have also been reported, 48 and endoglin (an auxiliary receptor for TGF‐β1 and TGF‐β3) is suggested to at least partially explain this desensitization. Endoglin expression is increased in stricture fibroblasts, and, although endoglin itself is not capable of transducing signals, it can influence the activation of targets downstream of the TGF‐β receptor in fibroblasts. Endoglin could thus counteract or manipulate TGF‐β‐mediated effects. 48 It could thus be a compensatory mechanism to impede chronic, over‐activated TGF‐β signalling in stricture fibroblasts.
The stricture environment may also contain other factors that interfere with signals that activate fibroblasts. One example is miRNAs. These are non‐coding, single‐stranded RNA molecules that bind transcripts and lead to their destruction. Nijhuis et al investigated the effect of miR‐29b in isolated fibroblasts. 35 They showed that increasing gene silencing by transfecting fibroblasts with miR‐29b reduced collagen expression and normalized TGF‐β‐induced collagen expression. They also showed that miR‐29b, in turn, is negatively regulated by TGF‐β. Hence, there seems to be a reciprocal regulatory relationship between miR29‐b and TGF‐β. Indeed, stricture mucosa showed lower expression of miR29b (loss of negative regulation of TGF‐β), which is consistent with increased collagen and fibrosis as expected in strictured tissue.
4.2.2. PDGF
Another well‐known factor that influences collagen production is platelet‐derived growth factor (PDGF). PDGF normally shows low expression restricted to the submucosa but it is widely distributed in strictured tissue. 13 PDGF certainly affects fibroblasts, but its effect on stricture fibroblasts is unclear. That is, while some report a dose‐dependent decrease in fibroblast production of the collagen propeptide PIIIP in the presence of PDGF, 14 others showed enhanced collagen secretion with PDGF. 20 These differences may be related to selective activation or inhibition of different collagen genes that affect the production of different collagen subtypes. Indeed, strictures are associated with relative changes in collagen subtypes, 20 which might reflect the tissue balance of different pro‐fibrotic stimuli. However, a clinical significance of this observation remains to be determined.
4.2.3. IGF‐1 and bFGF
Growth factors like IGF‐1 and bFGF 49 increase collagen production and proliferation. IGF‐1 positive cells in strictures are mononuclear cells inside inflammatory infiltrates and are found in all wall layers, but predominantly in the mucosa and submucosa. 20 This is in contrast to healthy intestine where IGF‐1 positive cells are mainly in the mucosa, restricted to the lamina propria. It thus seems that immune cells, similar to the case with TGF‐β, provide IGF‐1 to fibroblasts to increase proliferation and collagen production. Interestingly, IGF‐1 was also found among the top upregulated genes in the stricture fibroblast transcriptome, 50 indicating that following activation, stricture fibroblasts could themselves become sources of IGF‐1. In a broad sense, it is possible that factors such as IGF‐1 initially produced by the inflammatory infiltrate can subsequently be produced by fibroblasts following sufficient activation.
4.2.4. Cytokines
The pro‐inflammatory cytokines IL‐1β 49 and TNF 51 also influence collagen production. The increased tissue expression of TGF‐β, 33 , 38 IL‐1β 10 , 11 and TNF 12 (Figure 4) reflects the microenvironment these cells reside in and the presence of immune cells. Moreover, it has recently been shown that IL‐34 and its receptor M‐CSFR‐1 are upregulated in ileal mucosal strictures. Knocking down IL‐34 in fibroblasts isolated from these specimens reduced collagen production. 45 Thus, several cytokines in the stricture microenvironment influence fibroblast behaviour including collagen production.
4.3. Summary: cells and factors
This section summarized growth factors and cytokines that influence fibroblast proliferation and stimulate collagen production, and are thus involved in the cogwheel driving fibrosis (Figure 5B). Immune cells seem to be a major source of many of the molecules that influence fibroblasts. However, some stricture fibroblasts themselves show an altered phenotype following chronic stimulation from the microenvironment that seems to persist, as demonstrated experimentally using ex vivo cell cultures. Fibrosis could thus be depicted as a positive feedback loop, driven by the inflammatory environment, that successively yields fibroblasts self‐sufficient in stimulatory signals that drive the fibrotic process (Figure 5B).
4.4. Factors in the microenvironment that affect fibroblast function
4.4.1. Survival of fibroblasts in strictures
In the normal healing process, after fibroblasts are activated, expand and perform their tasks, they undergo apoptosis as their role in healing decreases (Figure 5A). Failure of fibroblasts to undergo apoptosis would contribute to their accumulation and potentially perpetuate fibrosis and promote non‐resolution (Figure 5B). This section discusses factors that influence fibroblast survival.
4.4.2. Pathogen recognition receptor activation and IL‐8 are anti‐apoptotic and upregulated in stricture fibroblasts
The chemoattractant IL‐8 has anti‐apoptotic effects in fibroblasts. 52 Stricture fibroblasts themselves secrete higher amounts of IL‐8 that increases upon LPS stimulation. 53 Indeed, in addition to IL‐8, 54 several proteins involved in LPS signalling were among the top upregulated genes found when mapping the stricture fibroblast transcriptome. These include TLR4, 50 which binds LPS, as well as LITAF, 54 a TLR4 downstream protein. The microbial receptor Chitinase 3‐like‐1 also shows elevated expression in stricture fibroblasts, but its significance in fibrosis is currently unclear. 54 Thus, a weakened mucosal barrier, as is commonly seen in IBD, might increase bacterial exposure leading to increased fibroblast survival.
4.4.3. The adipokine CTRP‐3 antagonizes TLR signalling and IL‐8 secretion
C1q/TNF‐related protein‐3 (CTRP‐3) is an anti‐inflammatory adipokine produced by mesenteric adipose tissue. Importantly, it is thought to be a natural antagonist of TLR4. Hofmann et al explored how CTPR‐3 affected isolated fibroblasts. 53 They showed that incubation of stricture fibroblasts with CTPR‐3 normalized their IL‐8 secretion, both their upregulated basal IL‐8 and LPS‐induced IL‐8 levels. They also showed that CTPR‐3 reduced stricture fibroblast expression of pro‐fibrotic genes. However, stricture fibroblasts required a higher concentration of CTPR‐3 to achieve these effects, suggesting they are less responsive to this adipokine compared to normal fibroblasts. Indeed, as presented above, TLR signalling is upregulated in stricture fibroblasts and may be related to impaired mucosal barrier. However, a better understanding of factors released from the mesenteric fat, like CTPR‐3, might open up possibilities for finding new ways to treat fibrosis and stricture development.
4.4.4. miR29b is linked to IL‐6 and IL‐8 secretion and fibroblast survival
micro‐RNAs may also influence fibroblast survival. For instance, silencing genes in cultured stricture fibroblasts by transfection with miR‐29b leads to increased IL‐6 and IL‐8 secretion as well as to increased expression anti‐apoptotic proteins. 52 Although a bit counterintuitive, the gene silencing mediated by miR‐29b seems to release inhibition of as yet unidentified IL‐6/IL‐8 suppressive mediators. However, downregulation, rather than upregulation, of miR‐29b expression has been reported in stricture ileal mucosa. 35 Thus, whether mir‐29b has any implication for stricture pathogenesis is uncertain.
4.4.5. Fibroblasts show increased susceptibility to apoptosis in vitro
Stricture fibroblasts could be more susceptible to apoptosis, but whether apoptosis occurs or not is determined by the balance of pro‐ versus anti‐apoptotic factors in the stricture environment. This is illustrated by the example of TNF‐related apoptosis‐inducing ligand (TRAIL). TRAIL is secreted from various cells and, on the one hand, induces apoptosis after death‐receptor binding. Alternatively, TRAIL can also bind the soluble decoy receptor osteoprotegerin (OPG) that neutralizes TRAIL resulting in an anti‐apoptotic effect. Indeed, with regard to stricture fibroblasts, Reenaers et al demonstrated that they are more susceptible to TRAIL stimulation, leading to apoptosis even with low levels of TRAIL. 55 Stricture fibroblasts were further shown to be more susceptible to apoptosis in additional experiments. That is, while several cytokines decreased apoptosis of TRAIL‐stimulated fibroblasts bordering strictures (so‐called margin cells), only one cytokine (IL‐6 and its soluble receptor) decreased apoptosis of stricture fibroblasts. Fibroblasts in strictures also needed higher concentrations of OPG to neutralize TRAIL and achieve an anti‐apoptotic effect. Overall, in vitro experiments suggest that stricture fibroblasts have increased susceptibility to apoptosis, which might result from accumulated damage following long‐standing excessive proliferation the cells undergo in vivo. It follows that the pro‐survival state of stricture fibroblasts in vivo likely depends on the factors present in strictures and their concentration. A better understanding of apoptosis mechanisms in (myo)‐fibroblasts might open up possibilities to stimulate apoptosis to reduce fibroblasts accumulated in strictures as a means to treat fibrosis.
4.5. The impact of fibroblasts on the stricture microenvironment
The previous section summarized factors in the stricture environment that influence fibroblasts. This section focuses on the opposite side of the coin, that is, molecules in stricture fibroblasts that influence the microenvironment. Indeed, several differentially expressed molecules are observed in stricture fibroblasts. These molecules, in turn, can influence cells in the vicinity and might be involved in the cogwheel driving fibrosis (Figure 5B).
4.5.1. Fibroblast activating protein
As discussed above, despite that FAP is present in strictures and is expressed by stricture fibroblasts and myofibroblasts, its role is unclear. However, considering that FAP is exclusively expressed on fibroblasts or myofibroblasts, data from FAP inhibition experiments suggest FAP expression on these cells indirectly influences surrounding cells. For example, FAP inhibition in mucosal explants led to reduced activation of important transcription factors such as STAT3 and T‐bet. 29 The effect on T‐bet is especially interesting since it is only expressed by T cells; in other words, this indirect effect of FAP inhibition might block communication between (myo)‐fibroblasts and other cells.
4.5.2. Prostaglandins
Prostaglandins have numerous functions in both health and disease including, among others, vasodialation and inflammation. Prostaglandins have long been targeted by classical IBD drugs, underscoring their importance in IBD. Indeed, a disturbed balance between syntheses of different prostaglandins can have a significant impact on its surroundings. Furthermore, the expression of prostaglandin D2 synthetase 50 is upregulated with concomitant downregulation of prostaglandin I2 (prostacyclin) synthase in stricture fibroblasts. 54 Thus, fibroblast production of prostaglandins may influence the stricture environment.
4.5.3. Angiogenesis
Stricture fibroblasts also promote angiogenesis by, for example, secreting higher amounts of connective tissue growth factor 56 , 57 and vascular endothelial growth factor (VEGF). 58 They also show reduced expression of the serpin family F member 1, which has inhibiting effects on VEGF. 59 Thus, angiogenesis might be a mechanism to overcome the hypoxic environment in fibrotic areas.
4.5.4. Other
Additionally, stricture fibroblasts show upregulated IL‐33 expression. IL‐33 is implicated in allergic diseases where it activates mast cells, among other cell types. 60 Whether IL‐33 has any role in the enrichment of mast cells seen in strictured tissue is unknown. Moreover, reduced levels of the wnt family member 2B in stricture fibroblasts compared to control fibroblasts have also been reported in two transcriptomic screens. 50 , 59 However, given the complex regulatory mechanisms steered by WNT family factors, the significance of this finding needs to be determined.
4.6. Summary: Fibroblast effects on the microenvironment
To summarize, there are several examples of potential bidirectional communication, either direct or indirect, between fibroblasts and immune cells (Figure 5B: interphase between fibroblasts and the red oval area) or other cells in the vicinity.
4.7. Overall summary of the fibroblast section
The replenishment of the cells in the fibrosis‐forming core can be depicted as an initially benign reaction to mucosal injury, which in the presence of certain pro‐inflammatory signals and microenvironmental cues, takes an alternative path (Figure 5B). This path involves a positive feedback loop that, once initiated by immune cells, persists due to feedback signals from the expanded pool of activated fibroblasts. Thus, there is a shift from an inflammation‐driven to an inflammation‐facilitated process.
4.8. Myofibroblasts
4.8.1. Introduction to myofibroblasts
In parallel to fibroblast activation and expansion during healing, fibroblasts (and presumably other cells such as pericytes) undergo a transition to myofibroblasts, the main effector cells in fibrosis. As outlined below, the cellular origin of myofibroblasts in strictured tissue, as well as the relative contribution of different precursors to the stricture myofibroblast pool, is not fully elucidated.
4.8.2. Function and Context
Regardless of origin, the behaviour of myofibroblasts is complicated and differs from fibroblasts. Although the precise chronological order of events may be different, Figure 5B gives a simplified view of the concepts ‘Perpetuation’ and ‘Non‐resolution’. In this view, newly formed myofibroblasts migrate into damaged tissue and anchor themselves to the surrounding extracellular matrix, and to adjacent myofibroblasts, through integrins and cadherins, respectively. These integrins and cadherins are, in turn, connected to the actin cytoskeleton, which contracts and exerts tension on the matrix. Thus, myofibroblasts create a mesh covering the damaged area and take over the mechanical load until it becomes stabilized and sealed by secreted and cross‐linked collagen. However, matrix degradation and remodelling also take place, and if continuous, creates a vicious cycle that prevents the final resolution of tissue repair (Figure 5B: fibrosis). The section below describes experimental observations from stricture myofibroblasts and compares them to ‘normal’ myofibroblasts. We will discuss the cellular origin of, and the process of transition to, myofibroblasts. Factors that affect the key features of myofibroblasts, namely migration, contraction and matrix synthesis and degradation, will also be discussed.
4.8.3. The cellular origin of the myofibroblast pool
Understanding the origin of the myofibroblasts, and the molecular mechanism behind their transition, is crucial to understanding how it can be manipulated and potentially exploited to treat fibrosis.
4.8.4. Matrix‐resident fibroblasts
In the general healing model presented above, fibroblasts are referred to as the matrix‐resident fibroblasts, which in case of injury, differentiate into myofibroblasts as a part of the normal healing process (Figure 5A: transition). These matrix‐resident fibroblasts likely also contribute largely to the myofibroblast pool in strictured tissue (Figure 5B: ①). The transition is induced by cues from the microenvironment. For example, both TGF‐β and TNF are capable of inducing transition in cultured fibroblasts, 51 but the transition also depends on changes in the physical properties of the surrounding matrix. 10 Irrespective of initial cues, these signals converge into intracellular pathways that lead to the expression of contractile proteins, which is the hallmark of myofibroblast differentiation. Although still debated, sources of myofibroblasts other than matrix‐resident fibroblasts may also contribute to the myofibroblast population. Both pericytes from blood vessel walls and epithelial cells, through epithelial‐to‐mesenchymal transition, among others, have been proposed.
4.8.5. Pericytes
Fibroblasts found around blood vessels are named pericytes. Fate‐mapping studies in a murine model of kidney fibrosis showed pericytes as a major myofibroblast precursor. 61 This has led to raised interest in pericytes during the past decade. Indeed, there is increasing evidence for pericytes as a major contributor to myofibroblasts in several other fibrotic diseases that share similarities with stricture fibrosis. 30 , 62 , 63 Recently, pericytes (as well as matrix‐resident fibroblasts) were shown to be absent in sections of strictured human intestine relative to normal intestine where pericytes were abundant around blood vessels. 16 The authors concluded that this is at least indirect evidence that pericytes can, in addition to matrix‐resident fibroblasts, undergo transformation to myofibroblasts (Figure 5B: ②).
4.8.6. Epithelial‐to‐mesenchymal transition
In addition to fibroblasts and pericytes, epithelial cells, through an epithelial‐to‐mesenchymal transition (EMT), have also been suggested to aid in the generation of myofibroblasts in strictures (Figure 3: ⑧, and Figure 5B: ③). This is based on observations that strictures show alterations in EMT‐associated markers. 21 , 25 , 40 , 64 For example, several papers describe lower expression of epithelial cell markers (and/or increased extra‐epithelial staining of such a marker) in stricture mucosa concomitant with increases in markers for mesenchymal cells or EMT onset. However, the contribution of EMT to the population of myofibroblasts in other fibrotic diseases has been questioned. 30 Although an interesting possibility, further investigation is needed to determine the importance of EMT in the setting of intestinal strictures.
4.9. The mechanism behind the myofibroblast transition
As mentioned above, the transition of precursors to myofibroblasts is induced by cues from the microenvironment, including secreted factors and changes in the physical properties of the surrounding matrix. These cues converge into intracellular pathways that ultimately lead to the expression of contractile proteins which, regardless of precursor cell, is the final common pathway that defines the transition of a cell into a myofibroblast. Here, we discuss some factors, investigated in strictured tissue or isolated cells that are important in the myofibroblast transition.
4.9.1. ROCK relays transition‐inducing signals and can be targeted
One protein suggested to be crucial in relaying signals leading to the expression of contractile proteins is Rho‐associated protein kinase (ROCK), which is overexpressed in strictured mucosa. Inhibition of ROCK in isolated fibroblasts (using the investigational drug AMA0825) blocked the transition to myofibroblasts. 65 Moreover, ROCK blockade had multiple effects on cultured mucosal tissue explants, with decreased secretion of several factors involved in fibrosis including MMPs, TGF‐β and IL6. 65 Thus, ROCK is implicated in fibrosis and the myofibroblast transition. However, further studies are needed to elucidate the role of ROCK‐associated factors in stricture pathogenesis.
4.9.2. AhR‐signalling interferes with the transition
Another factor that can potentially influence fibroblast to myofibroblast transition is signalling through the Aryl Hydrocarbon Receptor (AhR). Ahr is a widely expressed transcription factor that binds to a broad arsenal of environmental stimuli, including dietary components and contaminants in food. Monteleone et al showed that AhR activation reduces TGF‐β and TNF‐induced transcription of collagen genes (COL1A1, COL3A1) and α‐SMA, by selectively inhibiting MAPK activation in fibroblasts. In contrast, inhibiting AhR had the opposite effect and induced collagen secretion, even in unstimulated conditions. 51 Whether Ahr‐signalling following food ingestion is sufficient to replicate these effects (either pro‐ or anti‐fibrotic) in vivo is unknown but an interesting possibility. For example, if looking beyond AhR, diet can have powerful effects on intestinal inflammation, as exemplified by the relief given to paediatric Crohn's patients on exclusive enteral nutrition therapy that is used clinically. 66 , 67 , 68 It is thus tempting to think that diet might modify the development of fibrosis.
4.9.3. Cadherins are involved in creating necessary cell‐cell contacts
Cadherins are involved in creating cell‐cell contacts between adjacent myofibroblasts, and consequently, these proteins may be important for both initiating and maintaining myofibroblast function. For example, N‐cadherin 69 and the downstream calcium channels TRPC6 and TRPC4 32 show increased expression in strictures and are thought to be necessary to develop contractile proteins. Moreover, cadherin‐11 co‐localizes with α‐SMA and is upregulated in stricture mucosa (Figure 4B) as well as in isolated stricture fibroblasts. Cadherin‐11 also increases with several pro‐inflammatory stimuli. 70 However, knock‐down of cadherin‐11 led to decreased collagen production without affecting α‐SMA. Thus, targeting cadherin‐mediated signals might represent a way to modulate the behaviour of fibroblasts and myofibroblasts, but further investigation is needed to define the role and significance of different cadherins.
4.9.4. FAP induction, polarity and effects on tissue
Fibroblast Activating Protein is absent from healthy intestine 16 , 28 but increases in strictured tissue, predominately in areas affected by fibrosis (Figure 3: ① and ③). FAP co‐localizes with α‐SMA, 29 and the majority of myofibroblasts isolated from strictured tissue are FAP+. 28 , 29 Thus, FAP seems to be expressed primarily on myofibroblasts, and data suggest it is induced by cytokines in the environment. For example, TGF‐β and TNF induce FAP expression in cultured fibroblasts. However, these cytokines fail to do so in ‘normal’ fibroblasts. 28 Indeed, inflammation per se might not be sufficient for FAP induction since FAP is absent from purely inflamed (non‐strictured) tissue. Moreover, blocking FAP does not seem to affect α‐SMA expression, suggesting that α‐SMA and FAP may be co‐expressed in cells independently of each other. However, blocking FAP decreased TIMP production and increased migration in isolated stricture myofibroblasts. Blocking FAP also decreased collagen secretion and TIMP‐1 in strictured mucosal explants. 29 Thus, FAP is interesting since it is restricted to strictured tissue and blocking FAP has anti‐fibrotic effects without any apparent effect on ‘normal’ tissue. However, based on available data, it cannot be ruled out that there could also be FAP− myofibroblasts, as well as FAP+ fibroblasts, in strictures. It could thus be speculated that perhaps the FAP status may define different phenotypes of both fibroblasts and myofibroblasts. However, the specific function performed by such FAP+/FAP− subpopulations remains to be established.
4.10. Proliferation, migration and matrix production of myofibroblasts in strictures
4.10.1. Proliferation
Similar to fibroblasts, the expansion (Figure 5B: ④) of newly differentiated myofibroblasts is driven by bFGF. 71 Although the proliferation of myofibroblasts in strictures is increased compared to controls, they proliferate less than myofibroblasts from inflamed tissue. 72 This highlights the importance of inflammatory signals in driving the generation of myofibroblasts. In contrast, substances in the diet can negatively impact myofibroblast proliferation. For example, Luna et al investigated the effects of tocotrienols, a group of substances with Vitamin E activity typically found in food. 71 Tocotrienols dose‐dependently decreased proliferation and diminished the bFGF‐induced proliferation of myofibroblasts. It also led to reduced production of matrix components and increased apoptosis. Whether the concentrations used in the experiments are realistic to achieve through a regular diet is uncertain, but the data suggest that diet can potentially have protective effects under the right conditions. However, a deficiency of these substances might facilitate pro‐fibrotic processes, which is currently not known.
4.10.2. Migration
Migration (Figure 5B: ⑤) enables myofibroblasts to reach and spread out in the damaged area. This is typically tested experimentally through a scratch assay. Using this assay, Meier et al showed that stricture myofibroblast migration, both in the basal state and in the fibronectin‐stimulated state, is higher compared to myofibroblasts isolated from inflamed or fistulae tissue. 37 This correlated directly to the higher activation of Focal adhesion kinase (FAK), a kinase involved in transducing integrin‐mediated signals to the contractile elements of the cells. However, somewhat contradictory to the above findings, Di Sabatino et al reported that stricture fibroblasts have less migratory ability relative to fibroblasts from non‐stricture tissue and were more senescent. 38 They also showed that blocking TGF‐β did not affect the migration of stricture myofibroblasts but led to increased mobility of margin cells and fibroblasts from control (healthy) subjects. Where in the signalling pathway this interaction leading to non‐responsiveness takes place is uncertain. Furthermore, it may, for example, depend on the duration and concomitant stimulation from other factors that interact with FAK.
4.11. Collagen deposition is a prerequisite for wound healing but becomes excessive in fibrosis
Collagen is secreted in a soluble form that self‐assembles into fibrils outside the cell and, upon cross‐linking, the newly formed matrix is strengthened, and the tissue returns to homeostasis. Thus, when produced in normal amounts, collagen is necessary for wound healing. However, in the case of strictures, this becomes exaggerated and leads to fibrosis. Indeed, myofibroblasts isolated from strictures are characterized by excessive production of collagen. 38 , 42 , 72
4.11.1. The net effect on the matrix is dictated by the balance between enzymes
What dictates whether or not excessive collagen production leads to a net accumulation of matrix is the balance between matrix degradation enzymes, their inhibitors, and cross‐linking enzymes. Below we discuss some of the factors that can disturb this balance.
4.11.2. TGF‐β tips the balance towards fibrosis
Stricture myofibroblasts produce less MMP‐12 38 , 42 in combination with increased TIMP‐1, 38 thus favouring matrix deposition and fibrosis. This pattern has been demonstrated in vitro following incubation with TGF‐β. 38 Thus, stricture myofibroblasts show activated TGF‐β signalling. As discussed earlier, immune cell infiltrates might serve as the principal source of TGF‐β. Interestingly, stricture myofibroblasts show upregulation of ER stress‐related proteins (GRP78, ATF6a, XBP1‐S/U) compared to normal margin or other non‐stricturing Crohn's disease phenotypes. It has recently been shown that endoplasmic reticulum (ER) stress, which occurs following the accumulation of misfolded proteins, induces TGF‐β1 gene expression. 73
4.11.3. Pro‐inflammatory cytokines favour matrix remodelling and decrease wound healing
Stricture myofibroblasts exposed experimentally to the pro‐inflammatory cytokines IL17A and TNF release MMP‐3 and MMP‐12, in addition to increasing TIMP‐1 and collagen production. 42 Moreover, IL‐17E increased MMP‐3 and MMP‐12 without concomitant stimulation of TIMP and collagen. Pro‐inflammatory cytokines like IFN‐γ and TNF, 37 as well as IL17A, 42 have been shown to reduce the migration of stricture fibroblasts. Thus, the effect of having pro‐inflammatory cytokines present impairs wound healing in several ways; it reduces migration and favours both matrix deposition and degradation. The net effect in vivo, where TGF‐β and other molecules are present simultaneously, is hard to predict. Regardless, the general conclusion is that inflammation delays resolution.
4.11.4. Inhibiting the cross‐linking enzyme LOX decreases matrix contraction and increases MMPs
Myofibroblasts that have migrated into the affected area exert tension (contract) the matrix and aid in enzymatic cross‐linking of collagen. 74 Cross‐linking of newly secreted collagen is necessary to make a strong matrix but also makes it harder to degrade. This, indeed, is an unfavourable output in the setting of fibrosis. Stricture myofibroblasts display concomitantly higher degrees of matrix contraction and enhanced expression of the cross‐linking enzymes LOX and LOXL2, 72 which further contributes to fibrosis. Matrix stiffness per se modulates myofibroblast behaviour. However, stricture myofibroblasts behave aberrantly in this regard. For example, an increase in matrix stiffness is normally associated with increased MMP activity, but in stricture myofibroblasts, this leads to inhibited activity. Interestingly, inhibiting the ability of stricture myofibroblasts to cross‐link collagen (with a LOX inhibitor) had dual effects: it reduces contraction to normal levels and increases MMP‐3 activity. 72 Even though the significance of these effects needs to be determined, the results suggest that changing myofibroblasts' perception of the physical environment might have beneficial effects resulting from changed cell‐matrix signalling.
4.12. Summary for myofibroblast section
Myofibroblasts are the primary effector cells in normal wound healing where they contract, produce collagen, and reinforce the newly made matrix. While the matrix heals, the physical environment changes, which is sensed by the myofibroblasts, ultimately leading to their apoptosis. In the setting of chronic inflammation, as is the case in the intestine of CD patients, myofibroblast expansion is facilitated, and their normal behaviour is exaggerated. However, as inflammatory stimuli also stimulate degradation enzymes, it might create a vicious cycle of matrix production and degradation, leading to non‐resolution and, ultimately, fibrosis.
5. SMOOTH MUSCLE CELLS
5.1. Introduction
The discussion thus far has focused on the role of fibroblasts and myofibroblasts in fibrosis and strictured tissue. However, the fact that SMCs constitute the majority of the increased stricture bowel wall thickness makes these cells a highly relevant topic for further studies. This section focuses on SMCs in the context of strictures, about which less is known. Interestingly, factors that influence SMCs can be produced by SMCs themselves. In addition, factors effecting SMCs are also released by inflammatory infiltrates, which is consistent with inflammation preceding fibrosis.
5.2. Factors affecting hyperplasia and hypertrophy
Smooth muscle hyperplasia (increased number) and hypertrophy (increased size) are hallmarks of strictures. An understanding of the factors driving these responses in SMCs might facilitate deciphering the underlying causes.
5.2.1. Hyperplasia
Several factors increase the proliferation of SMCs, including stimulation via αVβ3 integrin and IGF‐1 receptor. 75 TGF‐β stimulation of SMCs, for example, leads to increased expression of IGF‐1 mRNA. Given the TGF‐β‐rich environment in strictures, this factor could thus indirectly facilitate SMC hyperplasia. Indeed, stricture SMCs show increased activation of targets downstream of the IGF‐1 receptor. Similarly, stricture SMCs show increased activation of the transcription factor STAT3, a second messenger relaying signals from several cytokines, compared to normal SMCs (from non‐strictured tissue). Indeed, STAT3 activation has proliferative effects. 76
5.2.2. Hypertrophy
In addition to producing IGF‐1 protein, transcription of IGF‐1 may also result in the production of Mechano Growth Factor (MGF), instead of IGF‐1, through alternative splicing. 77 Mechano Growth Factor acts hypertrophically on SMCs and might contribute to enlargement of the muscle layers. Indeed, Mechano Growth Factor is increased on both the transcript and protein levels in SMCs, and stricture SMCs show higher activation of effectors downstream of the MGF receptor. Interestingly, incubation of healthy margin SMCs with conditioned media from stricture cells led to SMC hypertrophy, which was blocked using an anti‐MGF antibody. 77
5.3. Different smooth muscle layers undergo distinct changes during stricture formation
The hyperplasia and hypertrophy of stricture SMCs are associated with other changes in the surrounding extracellular matrix. Interesting in this regard is that each of the three intestinal wall layers that contain smooth muscle (muscularis mucosae, muscularis propria interna and muscularis propria externa; Figure 3) are affected in different ways in a polarized fashion. 9
5.3.1. Muscularis mucosae
As discussed earlier, SMC bundles in stricture muscularis mucosae have a distorted architecture because of abundant extracellular fibrosis. Interestingly, the type of collagen in stricture muscularis mucosae (presence of type V, absence of type I and III) is different from collagen in the submucosa (contains type I and III). 9
5.3.2. Muscularis propria interna
In contrast, the internal layer of muscularis propria is thickened but displays a well‐preserved architecture with no extracellular collagen (Figure 3). Fibrosis in this layer is instead situated in the expanded septa and contains similar collagen types as the submucosa (type 1 and III, no type V). 9
5.3.3. Muscularis propria externa
The layer reported to be least affected, with no apparent thickening or fibrosis, is the mucularis propria externa.
The reasons for the polarized differences seen in smooth muscle layers are not understood. In muscularis mucosae, it might relate to its proximity to immune cells in the mucosa. In contrast, the deeper layers are far away from the mucosa but are surrounded by the fibrotic submucosa and subserosa (including chronic inflammatory infiltrates) and lie closer to the mesenteric fat. Further investigations are needed to understand the differences in changes, and whether SMCs themselves have any role in the changes described above.
5.3.4. Phenotypic changes in SMCs
As discussed earlier, in vascular biology research, it is proposed that SMCs can switch from a highly mature, contractile phenotype to a secretory phenotype with the ability to secrete extracellular matrix. 31 If SMCs are involved in the changes discussed above, they may have undergone this type of switch. Experimentally, stricture SMCs show less contractile response to acetylcholine. An altered phenotype in SMCs, after protracted growth, has been suggested by Bonafiglia et al, who demonstrated lower expression of the markers α‐SMA and SM‐22 in strictures, in combination with alterations in epigenetic modification enzymes. 78 Phenotypically changed SMCs have been observed histologically along the submucosal borders of muscularis mucosae and muscularis propria (Figure 3: ⑥), respectively. Altered SMCs have also been reported in the fibromuscular bundles infiltrating the submucosa (Figure 3: ⑦). Below is a discussion of factors released by SMCs cultured ex vivo that, in many cases, are related to fibrosis.
5.4. SMC—beyond the contraction
5.4.1. SMCs produce collagen
It has been shown experimentally that SMCs express collagen. Similar to stricture fibroblasts and myofibroblasts, basal collagen production by stricture SMCs is higher compared to SMCs in margin or control tissue. 79 Among the factors shown to induce collagen in SMCs are IL‐6 and TGF‐β. 79 IL‐6‐induced STAT3 activation that, in turn, led to transcription of the pro‐fibrotic factors TGF‐β and CTGF. 76 Neutralization of TGF‐β in cultured stricture SMCs resulted in the downregulation of COL1A1‐transcription, indicating autocrine signalling of TGF‐β. 79 However, based on the data, the role of TGF‐β signalling in stricture SMCs, both as responders and producers of this factor, is not clear. While some studies show increased TGF‐β expression and activated signalling, 79 , 80 another showed less TGF‐β expression and diminished activation of the TGF‐β‐signalling pathway. 13 Although not fully elucidated, it has been suggested that PDGF is involved in the SMC phenotypic switch discussed above by downregulating TGF‐β signalling. Consistent with this, reduced TGF‐β activation was associated with reduced COL1A1 (which is induced by STAT3 and TGF‐β) together with upregulated PDGF‐B and COL3A1. 13 The significance of these findings in strictures needs further investigation. However, deciphering these mechanisms might open up possibilities to find key molecules that can be targeted to interfere with the fibrosis process.
5.4.2. SMCs also produce cytokines
It is not clear whether the switch of SMCs to a secretory phenotype actually occurs in vivo. Moreover, it is unclear whether the highly differentiated, contractile SMCs from strictures investigated ex vivo maintain their in vivo phenotype, or if a switch to a secretory phenotype occurs. Regardless, SMCs isolated from strictures and studied ex vivo are indeed secretors. For example, IL‐6 and IL‐17A levels were markedly increased in comparison to SMCs from inflamed (non‐strictured) and fistulae tissue. 76 Other cytokines produced in higher quantities by cultured, stricture SMCs include IL‐10, IL‐12 and IL‐13. Of note, all of the cytokines mentioned here can, in an autocrine fashion via the cytokine receptor or the gp130 receptor, activate STAT3. 76 As discussed previously, STAT3 activation is increased in stricture SMCs and increases proliferation. It also increases expression of pro‐fibrotic genes. Thus, STAT3 activation in stricture SMCs contributes to both hyperplasia and fibrosis.
Importantly, the contribution of cytokines produced by SMCs on SMC function, relative to other cellular sources like immune cells, remains to be established. What is established is that SMCs show autocrine production of proliferation signals. However, these factors can also be secreted by, and/or have an impact on, other cells in the proximity.
5.5. Interaction between SMCs and fibroblasts/myofibroblasts
We have thus far discussed SMCs as responders to autocrine or paracrine signals that influence their function. However, several of the autocrine factors made by SMCs have pro‐fibrotic effects on myofibroblasts. Thus, bidirectional paracrine signalling between SMCs and myofibroblasts could occur in appropriate microenvironments. For example, the αVβ3 ligand fibronectin stimulates SMC proliferation 75 and myofibroblast migration. 37 In strictures, both of these cell types have increased fibronectin expression. Although the cause and effect of these changes are unknown, it can be viewed from two perspectives. On the one hand, myofibroblasts are attracted by fibronectin‐producing SMCs. On the other hand, SMCs expand in regions enriched in active myofibroblasts. An analogous discussion could also be done with other factors like IGF‐1, IL‐6 and IL‐17.
Thus, in microenvironments where SMCs, fibroblasts and myofibroblasts are in close proximity, factors produced by these cells could influence the behaviour of the other cell types in a paracrine fashion. Indeed, paracrine effects could occur in bidirectional manner, and autocrine effects are also likely. (Figure 5B: ⑥).
5.6. Summary
Expanded smooth muscle layers in strictured intestinal wall are a major feature of CD strictures. The characteristic changes in these layers are different in terms of SMC arrangement and fibrosis and have a polarized pattern. Phenotypically changed SMCs with secretory abilities have been described in, for instance, histological studies. Data show that ex vivo stricture SMCs produce collagen and several cytokines, but whether a phenotypic switch lies behind this behaviour of ex vivo SMCs is not known. Regardless, the production of both collagen and cytokines by stricture SMCs is magnitudes higher than that of SMCs isolated from non‐strictured muscle. In areas where SMCs and other mesenchymal cells are juxtaposed, there is likely cross‐talk between these cells. Although far from elucidated, SMCs may not necessarily be solely regarded as by‐standers, that is, a victim of the fibrotic process, but may have an active role in contributing to the pathogenesis of CD strictures. Further studies are warranted to explore this intriguing possibility.
6. SIMILARITIES WITH FIBROSIS IN OTHER ORGANS
The fibrosis process in intestinal strictures shares many similarities with fibrosis in other organs, including, but not limited to, the kidney, heart, liver and skin. 30 , 62 , 63 , 74 , 81 , 82 A common theme is that there is dysregulated healing following persistent or repetitive insults to the organ. Like intestinal strictures, fibrosis in other organs is associated with concomitant chronic inflammation and accumulation of myofibroblasts and ECM matrix in the interstitial space of the organ. Despite diverse aetiologies (infectious, metabolic, toxic, ischaemic, mechanical injury, etc), fibrosis is the common endpoint that ultimately leads to organ failure. The convergence on a common fibrotic pathway provides hope that understanding the fibrotic process will lead to therapeutic targets with beneficial effects in several diseases. Additional hope comes from examples where regression of fibrosis has occurred, for example in the liver, after eradicating the aetiologic factor. 83 , 84 In IBD, this could be improving remission rate and sustaining remission length. That is, ultimately alleviating the chronic inflammation that underlies IBD.
7. DIFFERENCES WITH FIBROSIS IN OTHER ORGANS
Despite optimism to develop broadly applicable treatments based on common fibrotic mechanisms, the nature of the gastrointestinal tract makes it unique from other internal organs. That is, while other organs are sterile, the gastrointestinal tract is continually exposed to everything we ingest including nutrients, microbes (both commensals and pathogens), toxins and drugs. In contrast, other organs, except the liver, are relatively protected. Moreover, other than the heart, the intestine is also unique in being a contractile organ. In particular, the muscular hypertrophy that evolves in association with intestinal fibrosis contributes substantially to the wall thickness, as discussed above. Moreover, fibroblasts from the gastrointestinal tract seem to be different from those in other tissues, highlighting again the unique intestinal environment. 85 Thus, despite optimism to develop broadly applicable anti‐fibrotic drugs based on common fibrotic mechanisms, affected internal organs are obviously distinct and challenges remain. For example, considering the intestine's unique features and constant exposure to external insults, drugs that halt fibrosis in other organs may not be effective in the intestine.
8. CONCLUSIONS AND FUTURE PERSPECTIVES
In a subgroup of CD patients, the normal process of healing the inflammation‐driven intestinal damage becomes exaggerated, dysregulated and self‐perpetuating. This results in the establishment of cell populations not seen in healthy intestinal tissue that drive an aberrant cycle of continuous matrix deposition and degradation. Simultaneous to this successive development of fibrosis, the muscle layers expand and infiltrate the fibrotic area. This culminates in the serious complication of stricture formation and ultimately gastrointestinal obstruction.
While modern therapies might benefit symptomatic strictures in a shorter perspective, 6 significant numbers of patients still need stricture‐associated surgery. As not all CD patients with pronounced intestinal inflammation develop strictures, other as yet unknown factors, present together with the chronic inflammation, seem necessary for the initiation and perpetuation of fibrosis. Thus, despite being an initial prerequisite for stricture development, immune cells and inflammatory cytokines per se seem to be only one piece of the puzzle. Indeed, the stricture environment is not highly inflammatory yet fibrosis continues.
The complex interplay of cytokines with diverse cell types and growth factors in fibrotic tissue underscores the elaborate environment that drives fibrosis and therein the difficulty of treating. Carefulness in treatment must be exercised as it is possible treatment could exaggerate pathogenesis by, for instance, blocking beneficial effects of the same targeted cytokine. Examples of cytokines with this Janus‐faced nature include TGF‐β and IL‐17, where inhibiting their pleiotropic effects often has undesired consequences. Where blocking a single cytokine might be insufficient, a multi‐target approach may offer advantages. For instance, two drugs recently approved for idiopathic pulmonary fibrosis, 84 target several cytokines/growth factors. While one (Nintedanib) is thought to inhibit PDGF, FGF and VEGF receptor signalling, another (Pirfenidone) inhibits TGF‐β and TNFα. Whether these could be used for intestinal strictures is currently not known.
However, increases in non‐immune cell types of mesenchymal origin, particularly myofibroblasts and SMCs, are substantial. Both of these cell types display unique behaviour in strictures and clearly impact the microenvironment. Thus, effective treatments must consider not only inflammation and immune cells, but also the fibrotic process and muscular aspects.
8.1. Targeting accumulation of myofibroblasts
Indeed, the terminal effector cell creating fibrosis is the myofibroblast. Selectively targeting these cells, or ideally hindering myofibroblast precursor cell activation, raises possibilities for anti‐fibrotic therapy. Thus, strategies that prevent their accumulation, for example, by blocking their transition into myofibroblasts would be beneficial. An example of this is the ROCK inhibitor AMA0825, 65 which shows promise. Moreover, a better understanding of mechanisms driving (myo)‐fibroblast apoptosis might open other possibilities. For example, identifying and eradicating signals that facilitate myofibroblast escape from apoptosis could reduce myofibroblast burden. However, specificity in such an approach would be a challenge.
8.2. Targeting pathological myofibroblasts
Identifying targets unique to pathological myofibroblasts, leaving ‘normal’ myofibroblasts untouched, would increase treatment specificity and minimize side effects. In this regard, one such target could be FAP, as FAP+ myofibroblasts are restricted to fibrotic areas. Experimentally, FAP inhibition has shown interesting anti‐fibrotic effects as discussed above. 29 However, understanding the mechanism behind this effect is necessary to develop specific and safe FAP‐based treatments.
8.3. Targeting mechanical aspects
There are reports of stricture patients that have undergone strictureplasty (reconstruction of the strictured segment without resection) that show signs of fibrosis regression. 81 , 84 It could be speculated that changing the physical environment in this way affects myofibroblast behaviour and survival, as myofibroblast functions rely heavily on mechano‐sensing. This is also illustrated by blocking collagen cross‐linking with LOX inhibition, 72 which normalizes stricture myofibroblast responses to mechanical stimuli and increases matrix degradation enzymes. Given the importance of tissue remodelling in general, directly targeting the balance between matrix deposition and degradation might be risky. However, a tempting possibility may be to modulate the deposition enzymes, or their inhibitors, to tip the balance towards resolution.
8.4. Targeting smooth muscle cells
Another possible cell type to target is SMCs. This is indeed appealing, as these cells constitute the majority of the increased bowel wall thickness in strictures. Therapies directed against pathways critical to driving SMC expansion may increase lumen size and wall compliance sufficiently to avoid or postpone the need for surgery, regardless of fibrosis. Changing the mechanical properties of the intestinal wall by targeting SMCs could also have secondary beneficial effects on myofibroblast behaviour as discussed above.
8.5. Other targets
Other molecules that have shown anti‐fibrotic effects experimentally might be exploited. These include adipokines from mesenteric fat, micro‐RNA, and signalling pathways evoked by substances in food, as discussed earlier. Additional research is warranted to explore these possibilities.
8.6. Treatment challenges
Given the inherent complexity of biological systems, targets with potential as anti‐fibrotic therapies must be considered together with undesirable side effects on processes involved in maintaining health, especially in extraintestinal organs. One way to avoid undesired systemic effects may involve choosing the route of administration. For example, oral, rectal (with colonic involvement) or endoscopic administration of depot formulations with high first passage metabolism would be advantageous. Indeed, pharmacokinetic aspects must be taken into account when developing drugs to minimize adverse systemic effects.
In summary, strictures are a severe complication of CD where mechanism(s) underlying fibrosis are complex and poorly understood. Finding novel drug targets requires better understanding of the interplay between immune cells, cytokines, mechanics and the unique and temporal behaviour of the mesenchymal cells.
CONFLICT OF INTEREST
Both authors declare no conflict of interest.
AUTHOR CONTRIBUTIONS
Both authors contributed equally to writing the review; JA evaluated the literature and created the figures.
Alfredsson J, Wick MJ. Mechanism of fibrosis and stricture formation in Crohn's disease. Scand J Immunol. 2020;92:e12990 10.1111/sji.12990
Funding information
The author's research is supported by the Swedish Cancer Foundation.
DATA AVAILABILITY STATEMENT
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
REFERENCES
- 1. Molodecky NA, Soon IS, Rabi DM, et al. Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology. 2012;142(1):46‐54.e42; quiz e30. [DOI] [PubMed] [Google Scholar]
- 2. Torres J, Mehandru S, Colombel J‐F, Peyrin‐Biroulet L. Crohn's disease. Lancet. 2017;389(10080):1741‐1755. [DOI] [PubMed] [Google Scholar]
- 3. Cosnes J, Cattan S, Blain A, et al. Long‐term evolution of disease behavior of Crohn's disease. Inflamm Bowel Dis. 2002;8(4):244‐250. [DOI] [PubMed] [Google Scholar]
- 4. Satsangi J, Silverberg MS, Vermeire S, Colombel JF. The Montreal classification of inflammatory bowel disease: controversies, consensus, and implications. Gut. 2006;55(6):749‐753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rieder F, Latella G, Magro F, et al. European Crohn's and colitis organisation topical review on prediction, diagnosis and management of fibrostenosing Crohn's disease. J Crohns Colitis. 2016;10(8):873‐885. [DOI] [PubMed] [Google Scholar]
- 6. Bouhnik Y, Carbonnel F, Laharie D, et al. Efficacy of adalimumab in patients with Crohn's disease and symptomatic small bowel stricture: a multicentre, prospective, observational cohort (CREOLE) study. Gut. 2018;67(1):53‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rimola J, Capozzi N. Differentiation of fibrotic and inflammatory component of Crohn's disease‐associated strictures. Intest Res. 2020;18(2):144‐150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mohan HM, Coffey JC. Surgical treatment of intestinal stricture in inflammatory bowel disease. J Dig Dis. 2020;21(6):355‐359. [DOI] [PubMed] [Google Scholar]
- 9. Zhang X, Ko HM, Torres J, et al. Luminally polarized mural and vascular remodeling in ileal strictures of Crohn's disease. Hum Pathol. 2018;79:42‐49. [DOI] [PubMed] [Google Scholar]
- 10. Johnson LA, Rodansky ES, Sauder KL, et al. Matrix stiffness corresponding to strictured bowel induces a fibrogenic response in human colonic fibroblasts. Inflamm Bowel Dis. 2013;19(5):891‐903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bailey JR, Bland PW, Tarlton JF, et al. IL‐13 promotes collagen accumulation in Crohn's disease fibrosis by down‐regulation of fibroblast MMP synthesis: a role for innate lymphoid cells? PLoS One. 2012;7(12):e52332‐e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Scheibe K, Kersten C, Schmied A, et al. Inhibiting Interleukin 36 Receptor Signaling Reduces Fibrosis in Mice With Chronic Intestinal Inflammation. Gastroenterology. 2019;156(4):1082‐1097.e11. [DOI] [PubMed] [Google Scholar]
- 13. Severi C, Sferra R, Scirocco A, et al. Contribution of intestinal smooth muscle to Crohn's disease fibrogenesis. European journal of histochemistry : EJH. 2014;58(4):2457‐. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Stallmach A, Schuppan D, Riese HH, Matthes H, Riecken EO. Increased collagen type III synthesis by fibroblasts isolated from strictures of patients with Crohn's disease. Gastroenterology. 1992;102(6):1920‐1929. [DOI] [PubMed] [Google Scholar]
- 15. Chen W, Lu C, Hirota C, Iacucci M, Ghosh S, Gui X. Smooth muscle hyperplasia/hypertrophy is the most prominent histological change in Crohn's fibrostenosing bowel strictures: a semiquantitative analysis by using a novel histological grading scheme. J Crohns Colitis. 2017;11(1):92‐104. [DOI] [PubMed] [Google Scholar]
- 16. Zidar N, Langner C, Jerala M, Bostjancic E, Drobne D, Tomazic A. Pathology of fibrosis in Crohn's disease‐contribution to understanding its pathogenesis. Front Med (Lausanne). 2020;7:167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mao R, Kurada S, Gordon IO, et al. The mesenteric fat and intestinal muscle interface: creeping fat influencing stricture formation in Crohn's disease. Inflamm Bowel Dis. 2019;25(3):421‐426. [DOI] [PubMed] [Google Scholar]
- 18. Masterson JC, Capocelli KE, Hosford L, et al. Eosinophils and IL‐33 perpetuate chronic inflammation and fibrosis in a pediatric population with stricturing Crohn's ileitis. Inflamm Bowel Dis. 2015;21(10):2429‐2440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gordon IO, Bettenworth D, Bokemeyer A, et al. Histopathology scoring systems of stenosis associated with small bowel Crohn's disease: a systematic review. Gastroenterology. 2020;158(1):137‐150.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lawrance IC, Maxwell L, Doe W. Inflammation location, but not type, determines the increase in TGF‐beta1 and IGF‐1 expression and collagen deposition in IBD intestine. Inflamm Bowel Dis. 2001;7(1):16‐26. [DOI] [PubMed] [Google Scholar]
- 21. Scharl M, Huber N, Lang S, Fürst A, Jehle E, Rogler G. Hallmarks of epithelial to mesenchymal transition are detectable in Crohn's disease associated intestinal fibrosis. Clin Transl Med. 2015;4:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gelbmann CM, Mestermann S, Gross V, Köllinger M, Schölmerich J, Falk W. Strictures in Crohn's disease are characterised by an accumulation of mast cells colocalised with laminin but not with fibronectin or vitronectin. Gut. 1999;45(2):210‐217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Suekane T, Ikura Y, Watanabe K, et al. Phenotypic change and accumulation of smooth muscle cells in strictures in Crohn's disease: relevance to local angiotensin II system. J Gastroenterol. 2010;45(8):821‐830. [DOI] [PubMed] [Google Scholar]
- 24. Powell DW, Pinchuk IV, Saada JI, Chen X, Mifflin RC. Mesenchymal cells of the intestinal lamina propria. Annu Rev Physiol. 2011;73:213‐237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mehta SJ, Lewis A, Nijhuis A, et al. Epithelial down‐regulation of the miR‐200 family in fibrostenosing Crohn's disease is associated with features of epithelial to mesenchymal transition. J Cell Mol Med. 2018;22(11):5617‐5628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Busek P, Mateu R, Zubal M, Kotackova L, Sedo A. Targeting fibroblast activation protein in cancer ‐ Prospects and caveats. Front Biosci (Landmark Ed). 2018;23:1933‐1968. [DOI] [PubMed] [Google Scholar]
- 27. Šimková A, Bušek P, Šedo A, Konvalinka J. Molecular recognition of fibroblast activation protein for diagnostic and therapeutic applications. Biochim Biophys Acta Proteins Proteomics. 2020;1868(7):140409. [DOI] [PubMed] [Google Scholar]
- 28. Rovedatti L, Di Sabatino A, Knowles CH, et al. Fibroblast activation protein expression in Crohn's disease strictures. Inflamm Bowel Dis. 2011;17(5):1251‐1253. [DOI] [PubMed] [Google Scholar]
- 29. Truffi M, Sorrentino L, Monieri M, et al. Inhibition of fibroblast activation protein restores a balanced extracellular matrix and reduces fibrosis in Crohn's disease strictures ex vivo. Inflamm Bowel Dis. 2018;24(2):332‐345. [DOI] [PubMed] [Google Scholar]
- 30. Greenhalgh SN, Conroy KP, Henderson NC. Healing scars: targeting pericytes to treat fibrosis. QJM. 2015;108(1):3‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Liu M, Gomez D. Smooth muscle cell phenotypic diversity. Arterioscler Thromb Vasc Biol. 2019;39(9):1715‐1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kurahara LH, Sumiyoshi M, Aoyagi K, et al. Intestinal myofibroblast TRPC6 channel may contribute to stenotic fibrosis in Crohn's disease. Inflamm Bowel Dis. 2015;21(3):496‐506. [DOI] [PubMed] [Google Scholar]
- 33. Hutter S, van Haaften WT, Hünerwadel A, et al. Intestinal activation of pH‐sensing receptor OGR1 [GPR68] contributes to fibrogenesis. J Crohns Colitis. 2018;12(11):1348‐1358. [DOI] [PubMed] [Google Scholar]
- 34. Warnaar N, Hofker HS, Maathuis MHJ, et al. Matrix metalloproteinases as profibrotic factors in terminal ileum in Crohn's disease. Inflamm Bowel Dis. 2006;12(9):863‐869. [DOI] [PubMed] [Google Scholar]
- 35. Nijhuis A, Biancheri P, Lewis A, et al. In Crohn's disease fibrosis‐reduced expression of the miR‐29 family enhances collagen expression in intestinal fibroblasts. Clin Sci (Lond). 2014;127(5):341‐350. [DOI] [PubMed] [Google Scholar]
- 36. Brenmoehl J, Lang M, Hausmann M, et al. Evidence for a differential expression of fibronectin splice forms ED‐A and ED‐B in Crohn's disease (CD) mucosa. Int J Colorectal Dis. 2007;22(6):611‐623. [DOI] [PubMed] [Google Scholar]
- 37. Meier JKH, Scharl M, Miller SN, et al. Specific differences in migratory function of myofibroblasts isolated from Crohn's disease fistulae and strictures. Inflamm Bowel Dis. 2011;17(1):202‐212. [DOI] [PubMed] [Google Scholar]
- 38. Di Sabatino A, Jackson CL, Pickard KM, et al. Transforming growth factor beta signalling and matrix metalloproteinases in the mucosa overlying Crohn's disease strictures. Gut. 2009;58(6):777‐789. [DOI] [PubMed] [Google Scholar]
- 39. de Bruyn M, Vandooren J, Ugarte‐Berzal E, Arijs I, Vermeire S, Opdenakker G. The molecular biology of matrix metalloproteinases and tissue inhibitors of metalloproteinases in inflammatory bowel diseases. Crit Rev Biochem Mol Biol. 2016;51(5):295‐358. [DOI] [PubMed] [Google Scholar]
- 40. Zhang H‐J, Zhang Y‐N, Zhou H, Guan L, Li Y, Sun M‐J. IL‐17A promotes initiation and development of intestinal fibrosis through EMT. Dig Dis Sci. 2018;63(11):2898‐2909. [DOI] [PubMed] [Google Scholar]
- 41. Cook SA, Schafer S. Hiding in plain sight: interleukin‐11 emerges as a master regulator of fibrosis, tissue integrity, and stromal inflammation. Annu Rev Med. 2020;71:263‐276. [DOI] [PubMed] [Google Scholar]
- 42. Biancheri P, Pender SL, Ammoscato F, et al. The role of interleukin 17 in Crohn's disease‐associated intestinal fibrosis. Fibrogenesis Tissue Repair. 2013;6(1):13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Biancheri P, Di Sabatino A, Ammoscato F, et al. Absence of a role for interleukin‐13 in inflammatory bowel disease. Eur J Immunol. 2014;44(2):370‐385. [DOI] [PubMed] [Google Scholar]
- 44. Latella G, Viscido A. Controversial contribution of Th17/IL‐17 toward the immune response in intestinal fibrosis. Dig Dis Sci. 2020;65(5):1299‐1306. [DOI] [PubMed] [Google Scholar]
- 45. Franze E, Dinallo V, Laudisi F, et al. Interleukin‐34 stimulates gut fibroblasts to produce collagen synthesis. J Crohns Colitis. 2020;14(10):1436‐1445. [DOI] [PubMed] [Google Scholar]
- 46. Raselli T, Wyss A, Gonzalez Alvarado MN, et al. The oxysterol synthesising enzyme CH25H contributes to the development of intestinal fibrosis. J Crohns Colitis. 2019;13(9):1186‐1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Haberman Y, Minar P, Karns R, et al. Mucosal inflammatory and wound healing gene programs reveal targets for stricturing behavior in pediatric Crohn's disease. J Crohns Colitis. 2020:jjaa166. 10.1093/ecco-jcc/jjaa166. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Burke JP, Watson RWG, Mulsow JJ, Docherty NG, Coffey JC, O'Connell PR. Endoglin negatively regulates transforming growth factor beta1‐induced profibrotic responses in intestinal fibroblasts. Br J Surg. 2010;97(6):892‐901. [DOI] [PubMed] [Google Scholar]
- 49. Lawrance IC, Maxwell L, Doe W. Altered response of intestinal mucosal fibroblasts to profibrogenic cytokines in inflammatory bowel disease. Inflamm Bowel Dis. 2001;7(3):226‐236. [DOI] [PubMed] [Google Scholar]
- 50. Sadler T, Bhasin JM, Xu Y, et al. Genome‐wide analysis of DNA methylation and gene expression defines molecular characteristics of Crohn's disease‐associated fibrosis. Clin Epigenet. 2016;8:30‐. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Monteleone I, Zorzi F, Marafini I, et al. Aryl hydrocarbon receptor‐driven signals inhibit collagen synthesis in the gut. Eur J Immunol. 2016;46(4):1047‐1057. [DOI] [PubMed] [Google Scholar]
- 52. Nijhuis A, Curciarello R, Mehta S, et al. MCL‐1 is modulated in Crohn's disease fibrosis by miR‐29b via IL‐6 and IL‐8. Cell Tissue Res. 2017;368(2):325‐335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hofmann C, Chen N, Obermeier F, et al. C1q/TNF‐related protein‐3 (CTRP‐3) is secreted by visceral adipose tissue and exerts antiinflammatory and antifibrotic effects in primary human colonic fibroblasts. Inflamm Bowel Dis. 2011;17(12):2462‐2471. [DOI] [PubMed] [Google Scholar]
- 54. Lang M, Schlechtweg M, Kellermeier S, et al. Gene expression profiles of mucosal fibroblasts from strictured and nonstrictured areas of patients with Crohn's disease. Inflamm Bowel Dis. 2009;15(2):212‐223. [DOI] [PubMed] [Google Scholar]
- 55. Reenaers C, Franchimont N, Oury C, et al. Sensitivity of intestinal fibroblasts to TNF‐related apoptosis‐inducing ligand‐mediated apoptosis in Crohn's disease. Scand J Gastroenterol. 2008;43(11):1334‐1345. [DOI] [PubMed] [Google Scholar]
- 56. Beddy D, Mulsow J, Watson RWG, Fitzpatrick JM, O'Connell PR. Expression and regulation of connective tissue growth factor by transforming growth factor beta and tumour necrosis factor alpha in fibroblasts isolated from strictures in patients with Crohn's disease. Br J Surg. 2006;93(10):1290‐1296. [DOI] [PubMed] [Google Scholar]
- 57. Mulsow JJW, Watson RWG, Fitzpatrick JM, O'Connell PR. Transforming growth factor‐beta promotes pro‐fibrotic behavior by serosal fibroblasts via PKC and ERK1/2 mitogen activated protein kinase cell signaling. Ann Surg. 2005;242(6):880‐889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Beddy D, Watson RWG, Fitzpatrick JM, O'Connell PR. Increased vascular endothelial growth factor production in fibroblasts isolated from strictures in patients with Crohn's disease. Br J Surg. 2004;91(1):72‐77. [DOI] [PubMed] [Google Scholar]
- 59. Li Yim AYF, de Bruyn JR, Duijvis NW, et al. A distinct epigenetic profile distinguishes stenotic from non‐inflamed fibroblasts in the ileal mucosa of Crohn's disease patients. PLoS One. 2018;13(12):e0209656‐e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Saluja R, Ketelaar ME, Hawro T, Church MK, Maurer M, Nawijn MC. The role of the IL‐33/IL‐1RL1 axis in mast cell and basophil activation in allergic disorders. Mol Immunol. 2015;63(1):80‐85. [DOI] [PubMed] [Google Scholar]
- 61. Humphreys BD, Lin SL, Kobayashi A, et al. Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am J Pathol. 2010;176(1):85‐97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Campanholle G, Ligresti G, Gharib SA, Duffield JS. Cellular mechanisms of tissue fibrosis. 3. Novel mechanisms of kidney fibrosis. Am J Physiol Cell Physiol. 2013;304(7):C591‐603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Rockey DC, Bell PD, Hill JA. Fibrosis–a common pathway to organ injury and failure. N Engl J Med. 2015;372(12):1138‐1149. [DOI] [PubMed] [Google Scholar]
- 64. He S, Xue M, Liu C, Xie F, Bai L. Parathyroid hormone‐like hormone induces epithelial‐to‐mesenchymal transition of intestinal epithelial cells by activating the runt‐related transcription factor 2. Am J Pathol. 2018;188(6):1374‐1388. [DOI] [PubMed] [Google Scholar]
- 65. Holvoet T, Devriese S, Castermans K, et al. Treatment of intestinal fibrosis in experimental inflammatory bowel disease by the pleiotropic actions of a local Rho kinase inhibitor. Gastroenterology. 2017;153(4):1054‐1067. [DOI] [PubMed] [Google Scholar]
- 66. Narula N, Dhillon A, Zhang D, Sherlock ME, Tondeur M, Zachos M. Enteral nutritional therapy for induction of remission in Crohn's disease. Cochrane Database Syst Rev. 2018;4:CD000542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Swaminath A, Feathers A, Ananthakrishnan AN, Falzon L, Li FS. Systematic review with meta‐analysis: enteral nutrition therapy for the induction of remission in paediatric Crohn's disease. Aliment Pharmacol Ther. 2017;46(7):645‐656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Yu Y, Chen KC, Chen J. Exclusive enteral nutrition versus corticosteroids for treatment of pediatric Crohn's disease: a meta‐analysis. World J Pediatr. 2019;15(1):26‐36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Burke JP, Cunningham MF, Sweeney C, Docherty NG, O'Connell PR. N‐cadherin is overexpressed in Crohn's stricture fibroblasts and promotes intestinal fibroblast migration. Inflamm Bowel Dis. 2011;17(8):1665‐1673. [DOI] [PubMed] [Google Scholar]
- 70. Franze E, Monteleone I, Laudisi F, et al. Cadherin‐11 Is a Regulator of Intestinal Fibrosis. J Crohns Colitis. 2020;14(3):406‐417. [DOI] [PubMed] [Google Scholar]
- 71. Luna J, Masamunt MC, Rickmann M, et al. Tocotrienols have potent antifibrogenic effects in human intestinal fibroblasts. Inflamm Bowel Dis. 2011;17(3):732‐741. [DOI] [PubMed] [Google Scholar]
- 72. de Bruyn JR, van den Brink GR, Steenkamer J, et al. Fibrostenotic phenotype of myofibroblasts in Crohn's disease is dependent on tissue stiffness and reversed by LOX inhibition. J Crohns Colitis. 2018;12(7):849‐859. [DOI] [PubMed] [Google Scholar]
- 73. Li C, Grider JR, Murthy KS, et al. Endoplasmic reticulum stress in subepithelial myofibroblasts increases the TGF‐beta1 activity that regulates fibrosis in Crohn's disease. Inflamm Bowel Dis. 2020;26(6):809‐819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Hinz B, Phan SH, Thannickal VJ, et al. Recent developments in myofibroblast biology: paradigms for connective tissue remodeling. Am J Pathol. 2012;180(4):1340‐1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Flynn RS, Murthy KS, Grider JR, Kellum JM, Kuemmerle JF. Endogenous IGF‐I and alphaVbeta3 integrin ligands regulate increased smooth muscle hyperplasia in stricturing Crohn's disease. Gastroenterology. 2010;138(1):285‐293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Li C, Iness A, Yoon J, et al. Noncanonical STAT3 activation regulates excess TGF‐β1 and collagen I expression in muscle of stricturing Crohn's disease. J Immunol. 2015;194(7):3422‐3431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Li C, Vu K, Hazelgrove K, Kuemmerle JF. Increased IGF‐IEc expression and mechano‐growth factor production in intestinal muscle of fibrostenotic Crohn's disease and smooth muscle hypertrophy. Am J Physiol Gastrointest Liver Physiol. 2015;309(11):G888‐G899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Bonafiglia QA, Lourenssen SR, Hurlbut DJ, Blennerhassett MG. Epigenetic modification of intestinal smooth muscle cell phenotype during proliferation. Am J Physiol Cell Physiol. 2018;315(5):C722‐C733. [DOI] [PubMed] [Google Scholar]
- 79. Flynn RS, Mahavadi S, Murthy KS, et al. Endogenous IGFBP‐3 regulates excess collagen expression in intestinal smooth muscle cells of Crohn's disease strictures. Inflamm Bowel Dis. 2011;17(1):193‐201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Li C, Flynn RS, Grider JR, et al. Increased activation of latent TGF‐β1 by αVβ3 in human Crohn's disease and fibrosis in TNBS colitis can be prevented by cilengitide. Inflamm Bowel Dis. 2013;19(13):2829‐2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Rieder F, Bettenworth D, Imai J, Inagaki Y. Intestinal fibrosis and liver fibrosis: consequences of chronic inflammation or independent pathophysiology? Inflamm Intest Dis. 2016;1(1):41‐49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Zeisberg M, Kalluri R. Cellular mechanisms of tissue fibrosis. 1. Common and organ‐specific mechanisms associated with tissue fibrosis. Am J Physiol Cell Physiol. 2013;304(3):C216‐C225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Jun JI, Lau LF. Resolution of organ fibrosis. J Clin Invest. 2018;128(1):97‐107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Ma C, Jairath V, Click B, et al. Targeting anti‐fibrotic pathways in Crohn's disease ‐ The final frontier? Best Pract Res Clin Gastroenterol. 2019;38–39:101603. [DOI] [PubMed] [Google Scholar]
- 85. Higuchi Y, Kojima M, Ishii G, Aoyagi K, Sasaki H, Ochiai A. Gastrointestinal Fibroblasts Have Specialized, Diverse Transcriptional Phenotypes: A Comprehensive Gene Expression Analysis of Human Fibroblasts. PLoS One. 2015;10(6):e0129241. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.