

Summary
This protocol describes how to image fluorescently tagged proteins, RNA, or DNA inside living Saccharomyces cerevisiae cells at the single-molecule level. Imaging inside living cells, as opposed to fixed materials, gives access to real-time kinetic information. Although various single-molecule imaging applications are discussed, we focus on imaging of gene transcription at the single-RNA level. To obtain the best possible results, it is important that both imaging parameters and yeast culture conditions are optimized. Here, both aspects are described.
For complete details on the use and execution of this protocol, please refer to Lenstra et al. (2015) and Donovan et al. (2019).
Graphical Abstract
Highlights
-
•
Optimized protocol for single-molecule live-cell imaging in S. cerevisiae cells
-
•
Applicable for imaging gene transcription, DNA loci, or individual proteins
-
•
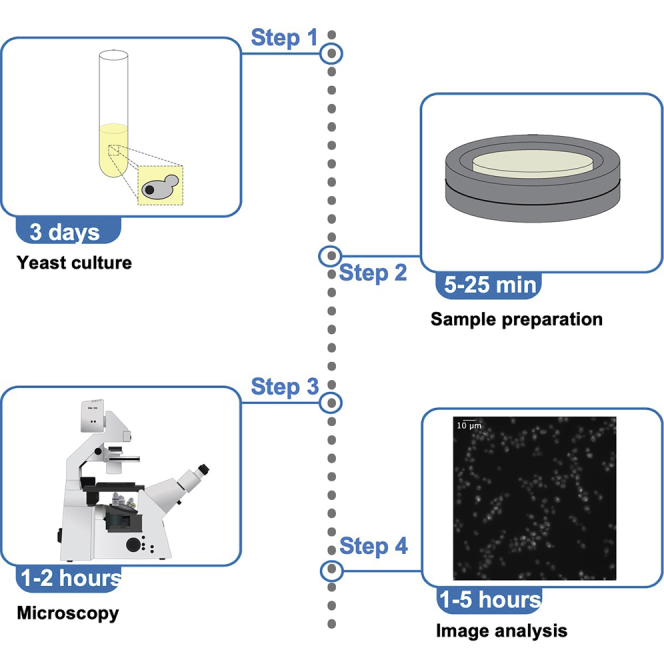
Includes yeast culture, sample preparation, microscopy settings, and image analysis
This protocol describes how to image fluorescently tagged proteins, RNA, or DNA inside living Saccharomyces cerevisiae cells at the single-molecule level. Imaging inside living cells, as opposed to fixed materials, gives access to real-time kinetic information. Although various single-molecule imaging applications are discussed, we focus on imaging of gene transcription at the single-RNA level. To obtain the best possible results, it is important that both imaging parameters and yeast culture conditions are optimized. Here, both aspects are described.
Before You Begin
Preparing Synthetic Complete (SC) Medium (500 mL)
Timing: approximately 2 h
For yeast imaging purposes, synthetic complete (SC) medium is used rather than yeast peptone dextrose (YPD) because YPD is opaque and contains autofluorescent components, leading to more background signal in fluorescence imaging.
-
1.In a 500 mL bottle, add
-
a.450 mL of Milli-Q water
-
b.3.35 gram Yeast Nitrogen Base Without Amino Acids and carbohydrate and with Ammonium Sulfate ( YNB w/o AA, carbohydrate & w/ AS)
-
c.1 gram Drop-out Mix Complete w/o YNB (alternatively, if the yeast strains used require this, a Drop-out Mix lacking specific amino acids should be used)
-
a.
-
2.
Mix until the powder is dissolved
-
3.
Sterilize by autoclaving 20 min at 121°C.
-
4.
If using the media immediately, wait for media to cool down to approximately 55°C–65°C before adding the carbon source. This can be done by placing the medium in a waterbath or incubator set within this temperature range.
-
5.
Alternatively, you can store medium at 18°C–22°C for future use, preferably in the dark and without carbon source added. It then remains stable for months.
Note: Before use, a filter sterilized solution of the appropriate carbon source should be added up to a final concentration of 2%. This can be done by adding 50 mL of 20% carbon source to 450 mL media. For most experiments, D-glucose should be used, but if the specific experiments require, other sugars such as D-raffinose or D-galactose can be used.
Note: When using Ade- strains, the medium should be supplemented with additional Adenine. Without this, the yeast cells will start to turn slightly red, leading to increased levels of background fluorescence during imaging.
Preparing Yeast Peptone Dextrose (YPD) Plates
-
6.
Put 15 gram yeast extract powder (YEP) in a 250 mL bottle.
-
7.
Put 10 gram bacto-agar in a 500 mL bottle.
-
8.
Divide 475 mL Milli-Q water over both bottles. The two bottles should contain roughly equal volumes, but they are combined in step 5 so the exact division over the bottles does not matter.
-
9.
Sterilize by autoclaving 20 min at 121°C.
-
10.
Add YEP medium to agar solution and mix by inversion.
-
11.
Wait for media to cool down to approximately 55°C–65°C. This can be done by placing the medium in a waterbath or incubator set within this temperature range.
-
12.
Add 50 mL 20% D-glucose and mix by inversion.
-
13.
Pour plates of roughly 25 mL each. Pour slowly to prevent the formation of bubbles.
-
14.
Leave the plates to dry for 3 days at 18°C–22°C.
-
15.
Put the plates upside down in a plastic bag and store at 4°C. Store plates upside down to prevent infections and to prevent waterdrops from the lid from falling on the plates. Plates can be stored for months.
Note: This is the standard plate that can be used for growing most yeast strains. However, if a selection marker is required, plates with additional selection should be used.
Preparing Agar Imaging Pad for Microscopy
Imaging samples can be prepared either by creating a layer of single cells by squeezing them between a coverslip and an agar pad, or by attaching the cells to a coverslip using concanavalin A (see protocol below). Select one of the two methods based on the experimental constraints. The advantage of using an agar pad is that the cells are pressed into a monolayer between the coverslip and the agar pad, such that they remain immobile during the experiment. The advantage of concanavalin A is that the cells remain in liquid medium, enabling media exchange or addition of drugs or compounds during the experiment. See Methods Video S1.
-
16.
Add 0.4 g of agarose (warning: do not use bacto-agar) to 18 mL of SC medium (see protocol above; use medium without added carbon source), yielding a final agarose concentration of 2%.
-
17.
Boil the mixture in the microwave. Make sure the agar is completely melted.
-
18.
Cool down to approximately 55°C–65°C, but do not let the mixture solidify.
-
19.
Add appropriate carbon source. For a 2% final concentration, add 2 mL of 20% D-glucose, D-raffinose, or D-galactose.
-
20.
Mix gently by inversion.
-
21.
Pour into a petri dish.
-
22.
Wait for pad to solidify (this takes at least 20 min).
-
23.
Keep at 30°C until use. Can be stored during one day, prepare fresh on the day of imaging.
Cleaning Coverslips for Microscopy
Generally, coverslips or glass-bottom dishes used for microscopy glass may have some dirt on the surface, which can lead to autofluorescent specles at the surface of the glass. This problem becomes especially apparent using high illumination conditions during single-molecule experiments. To remove this dirt, we advise the cleaning procedure outlined below. This cleaning procedure is only applicable to coverslips, and not to glass-bottom dishes. So, if he experiment requires cleaning, we recommend using cleaned coverslips. If cleaning is not necessary, both coverslips or glass-bottom dishes can be used.
-
24.
Place 25 mm round coverslips #1.5 (thickness 0.17 mm) in Wash-N-Dry coverslip racks. Depending on size of the beaker and sonicator, multiple racks can be cleaned simultaneously.
-
25.
Carefully place the filled racks in a beaker with 250 mL of 1M KOH. Make sure that they are fully submerged.
-
26.
Sonicate the beaker for 30 min at maximum intensity.
-
27.
Rinse the coverslips with Milli-Q water. This can be done by rinsing each rack; there is no need to remove the coverslips from the rack.
-
28.
Place the racks in beaker with 250 mL of 100% ethanol. Make sure that they are fully submerged.
-
29.
Sonicate the beaker for 30 min at maximum intensity.
Place the racks in a beaker with 250 mL of 70% ethanol for storage until use. Make sure that they are fully submerged. Cover the beaker with parafilm to prevent evaporation of the ethanol. Coverslips can be stored in this manner for months.
Coating Glass with Concanavilin-A for Yeast Cell Adhesion during Microscopy
-
30.
Dissolve concanavalin A in PBS at a 20 mg/mL.
-
31.
Divide this stock solution into aliquots of 0.5 mL and store at −20°C for up to 3 years.
-
32.
Dilute concanavalin A stock solution to 1 mg/mL using PBS or Milli-Q.
-
33.
Filter sterilize using a 0.22-μm filter.
-
34.
Pipet 300 μL of the dilution onto a (cleaned) coverslip (placed inside a 6-well culture dish) or glass-bottom dish.
-
35.
Cover the dishes with the lid.
-
36.
Wait 20 min at 18°C–22°C.
-
37.
Aspirate liquid.
-
38.
Air dry 20 min; leave the lids of the dishes open.
-
39.
Coated dishes and coverslips can be stored for several months at 18°C–22°C.
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, Peptides, and Recombinant Proteins | ||
| Yeast Nitrogen Base w/o AA, Carbohydrate & w/AS | US biological | Cat# Y2025 |
| Drop-out Mix Complete w/o Yeast Nitrogen Base | US biological | Cat# D9515 |
| D-glucose | Sigma Aldrich | Cat# 8270-10KG |
| D-raffinose | Bio-connect | Cat# OR06197_2kg |
| D-galactose | Sigma Aldrich | Cat# G0750-500G |
| Yeast extract powder (YEP) | VWR international | Cat# 212750 |
| Bacto-agar | Becton Dickinson | Cat# 214030 |
| Agarose MP | Sigma Aldrich | Cat# 11388991001 |
| Potassium Hydroxide | Sigma Aldrich | Cat# P5958-1KG |
| Ethanol | Honeywell | Cat# 32221-2.5L |
| Concanavalin A from Canavalia ensiformis (Jack bean) | Sigma Aldrich | Cat# L7647-250MG |
| PBS tablets | Gibco | Cat# 17912-014 |
| Other | ||
| Cover glasses (#1.5, thickness 0.17 mm round, 25 mm) | VWR | Cat# 631-0172 |
| Wash-N-Dry coverslip rack | Sigma Aldrich | Cat# Z688568-1EA |
| 400 mL tall glass beakers | Novodirect | Cat # 15439093 |
| Whatman® Puradisc syringe filters (FP 30/0.2, pore size 0.2 μm) | Sigma Aldrich | Cat# Z612529 |
| CELLView cell culture dish, PS, 35/10 mm, glass bottom | Greiner Bio-One B.V. | Cat# 627861 |
| Attofluor™ Cell Chamber, for microscopy | Thermofisher Scientific | Cat# A7816 |
Materials and Equipment
Microwave with at least 800 W power
Incubator at 30C with humidity control and/or air circulation
Shaker (at least 150 rpm) or rotating wheel (such as Brunswick Scientific TC-7 Tissue Culture Roller Drum Rotator M1053-4004)
Sonicator (Bioruptor Diagenode)
Milli-Q machine
Spectrophotometer with capability to measure OD at 600 nm in cuvettes with a light path of 10 mm
Inverted widefield microscope (e.g., Zeiss AxioObserver) with the following parts, see schematic in Figure 1:
-
•
Objective with at least 100× magnification and numerical aperture (NA) of at least 1.3; e.g., 100× (Zeiss 4-alpha Plan-Apochromat 100×/1.46 Oil, 420792-9800-000).
-
•Fluorescence excitation source:
-
○Widefield source (e.g., SPECTRA X light engine WL:360–680 nm, Lumencor) OR
-
○Excitation lasers in appropriate wavelengths
-
○
-
•
Excitation and emission filters optimized for the fluorophore(s) of interest; e.g., for GFP: excitation filter 470/24 (Lumencor) and emission filter FF01-515/30-25 (Semrock) or for mScarlet-i: excitation filter 550/15 (Lumencor) and emission filter FF01-600/52-25 (Semrock). For both colors a dichroic mirror 59012bs (Chroma) can be used.
-
•
Camera for fluorescence detection. A highly sensitive camera is needed; e.g., sCMOS (ORCA Flash 4.0 V3, 000000-1370-927) or EMCCD (iXon Ultra 888, DU-888U-CS0-#BV).
-
•
Optional (for experiments longer than approximately 10 min): focusing device; e.g., Zeiss definite focus.
-
•
Microscope stage incubator; e.g., stage-top incubator (OKO Labs).
Note: Although a broad range of imaging applications is available, the most straightforward modality is widefield fluorescence microscopy, which is available in many institutes. In general, this approach is very suitable for imaging inside living yeast cells when detection and localization of individual fluorophores is not required. Another modality that is widely used is confocal microscopy. However, confocal microscopy requires higher excitation intensities, which are often phototoxic for yeast cells, making confocal microscopy less suitable for imaging in living yeast cells. In experiments where localization of single molecules is necessary, another approach is to use HILO imaging (Tokunaga et al., 2008). Here, a highly inclined laser beam is used for fluorescence excitation, exciting a smaller number of out-of-focus molecules, thus resulting in an increased signal-to-noise ratio as compared to widefield microscopy. For HILO imaging, a high-NA TIRF objective and inclined illumination is essential. In this manuscript we focus on widefield imaging, but in the other modalities are also possible inside living yeast cells.
Note: For selecting an objective the main considerations are the magnification and light collection capabilities (specified by the NA). The optical resolution of the obtained microscopy image depends on the objective’s NA and the wavelength of light, and with high-NA objectives (>1.3) and GFP excitation is roughly around 200–250 nm. For optimal resolution, the total magnification should be set such that this 200–250 nm is collected by 2 camera pixels. The necessary magnification of the objective thus also depends on the pixel size of the camera chip; e.g., for the sCMOS mentioned above, with a chip pixel size of 6.5 μm, we recommend an objective that magnifies at least 63× in order to get a calibrated pixel size of around 100 nm in the resulting fluorescence image for optimal resolution. For typical fluorescence imaging purposes, we advise an NA of at least 1.3 in order to collect enough fluorescence emission light.
Note: For selecting appropriate excitation and emission filters, the following considerations can be kept in mind:
-
•
Excitation of the fluorophore should occur in a narrow band around its excitation peak. In that manner, fluorescence signal is maximized while photobleaching and phototoxicity are minimized. Using a laser as an excitation source results in a naturally narrow excitation peak and the use of excitation filters might is not needed. Using an LED source requires the use of narrow bandpass excitation filter. For GFP, we obtain good results with a 470/24 filter, and with mScarlet with a 550/15 filter.
-
•
The emission filter is ideally designed such that it transmits as much of the fluorophore’s emission light as possible but reflects light from the excitation peak.
Note: For imaging living yeast cells, they must be kept at a constant temperature (generally 30C for yeast). Even when using a sensitive stage incubator, we advise to monitor the temperature inside the incubator or inside the sample closely, for example by using a thermocouple.
Figure 1.

Schematic of Microscope Setup Used
Main components are indicated in the figure.
Step-By-Step Method Details
Culturing Yeast for Imaging
See Figure 2 for overview.
-
1.
Four days before imaging, streak yeast cells onto a YPD plate.
-
2.
On the day before imaging, in the morning: start a culture by inoculating one colony of yeast into 1 mL of SC medium (see protocol above). Grow at 30C while shaking/rotating.
-
3.In the evening, dilute cultures into prewarmed medium (30°C) to a density so that the cells have an optical density (OD600) of ~0.2–0.4 in the morning. If you are unsure how fast your strain grows, make a dilution series. The following dilutions can be used as starting points for strains exhibiting wildtype growth rates (90 min doubling time in glucose, 180 min doubling time in raffinose, 150 min doubling time in galactose):
-
a.In glucose, add 1 μL of culture in 5 mL of fresh medium.
-
b.In raffinose or galactose, add 2 μL of culture in 2 mL of fresh medium.
-
a.
CRITICAL: It is important that, during growth, yeast cells stay in mid-log phase (OD600 of 0.3–0.8). At higher OD, the carbon source becomes exhausted and cells will switch to a growth mechanism based on aerobic respiration. This is referred to as the diauxic shift, and during this process yeast cells become autofluorescent at various wavelengths, lower expression of the fluorescent proteins, and display uneven fluorescence distributions.
Optional: If fluorescence imaging requires labeling with a dye (such as HALO dyes; e.g., JF646 or JF552 (Grimm et al., 2015)) this dye can be added to the culture during the last part of the cell growth. In this case, add the dye after checking the culture OD. To acquire single-molecule resolution, generally you need to label sparsely, so only a small fraction of the molecules is fluorescently labeled. Too dense labeling results in too many labeled molecules in a small volume, which complicates resolving and tracking individual molecules. The time and dye concentration needed for sufficient labeling depends on the desired labeling ratio, which depends on the abundance of the protein to label. For example, 10 min labeling with 0.05 nM JF646 has been used for labeling abundant proteins such as histones and 2–3 h labeling with 5 nM JF646 has been used for imaging the low abundance transcription factor Gal4. Note that labeling with HALO dyes is only possible in yeast after deletion of the PDR5 gene and addition of the HALO tag to the gene of interest.
Figure 2.

Schematic of Steps for Culturing Yeast for Imaging
Preparing Sample Using Agar Pad
For details on this step, see Figure 3 and Methods Video S1.
-
4.Measure the OD600 of the yeast cultures using a spectrophotometer.
-
a.If the OD600 is below 0.2, grow cells for a few more hours.
-
b.If the OD600 is between 0.2 and 0.8, cells are ready for imaging, proceed to steps below.
-
c.If the OD600 is above 0.8, dilute culture to OD600 0.1–0.2 in prewarmed SC medium and grow them for two cell-cycles (around 180 min for strains with wildtype growth rates in glucose).
-
a.
-
5.
When the cells are ready for imaging, spin 1 mL of yeast imaging culture (see previous step) at 3,824 × g for 1 min.
-
6.
Remove supernatant.
-
7.
Resuspend cells in 4 μL of prewarmed SC medium with appropriate carbon source by shortly vortexing (see protocol above).
-
8.
Take one cleaned coverslip (stored in 70%, see above) and rinse it with 70% ethanol.
-
9.
Dry the coverslip over a flame. Do not put the coverslip in the flame, as it is likely to break and flaming may lead to cloudy coverslips. Just hold the coverslip close to the flame such that it dries quickly but the ethanol is not set on fire. Alternatively, you can air dry the coverslips at the bench or in small oven (set to max 50°C).
-
10.
Place coverslip in the Attofluor™ Cell Chamber.
-
11.
If cleaned coverslips are not necessary, you can skip steps 5–6 and use a glass-bottom dish.
-
12.
Pipet the entire volume (usually 4–10 μL) onto the coverslip.
-
13.
Cut agar slab. This is easily done by stabbing a circle out of the agar imaging pad (see protocol above) using a 14 mL growth tube.
-
14.
Put agar slab on top of the yeast cells. Press down gently with your thumb. Be careful not to break the coverslip.
-
15.
Optionally, an additional coverslip can be placed on top of the agar slab to prevent drying of the sample during the experiment. This is not necessary if the microscope used has an imaging chamber in which the humidity is controlled but it can be especially useful for cells that are imaged for several hours or more.
Optional: In case of labeling with a dye (such as the HALO dyes), this dye should be washed away before imaging. To do this, add a wash step after step 3: resuspend the cells in 1 mL prewarmed medium, spin cells for 1 min at 3,824 × g and remove supernatant. Then continue with steps 4–8 of the protocol above.
Figure 3.

Sample Preparation Using Agar Imaging Pad
(A) schematic and (B) photograph of sample prepared using an agar imaging pad.
Preparing Imaging Sample Using Concanavalin A
-
16.Measure the OD600 of the yeast cultures using a spectrophotometer.
-
a.If the OD600 is below 0.2, grow cells for a few more hours.
-
b.If the OD600 is between 0.2 and 0.8, cells are ready for imaging, proceed to steps below.
-
c.If the OD600 is above 0.8, dilute culture to OD 0.1–0.2 in prewarmed SC medium and grow them for two cell-cycles (around 180 min for strains with wildtype growth rates in glucose).
-
a.
-
17.
When the cells are ready for imaging, pipet 300 μL of yeast culture on a concanavalin A-coated coverslip (placed in a Attofluor™ Cell Chamber) or glass-bottom dish (see protocol above).
-
18.
Incubate 20 min at 30°C.
-
19.
Remove excess medium.
-
20.
Wash cells by adding 300 μL of prewarmed medium onto the cells and removing the fluid.
-
21.
Repeat wash step.
-
22.
Add 3 mL of prewarmed medium onto the cells. They are now ready for imaging.
Optional: In case of labeling with a dye (such as the HALO dyes), this dye should be washed away before imaging. To do this, add a wash step after step 1: resuspend the cells in 1 mL prewarmed medium, spin cells for 1 min at 3,824 × g and remove supernatant. Resuspend cells in 300 μL of medium and continue with steps 2–7 of the protocol above.
Image Acquisition
In this section we describe in detail the microscope settings for experiments aimed at imaging of transcription using the PP7 or MS2 systems (Bertrand et al., 1998; Chao et al., 2008). Both these systems are based on genetic integration of stem loop sequences in the gene of interest, which, when transcribed, are immediately bound by fluorescently tagged coat proteins to allow visualization of transcription of a single gene inside living cells in real time. Although each individual experiment will require some optimization of these parameters, we outline settings that can be used as a starting point for two types of experiments: either to visualize and localize individual RNAs within the yeast cell or to follow transcription of an individual gene over an extended amount of time. We also discuss how to adjust these setting for imaging single proteins or single genomic DNA loci in the notes.
-
23.
Turn on the microscope, camera, and fluorescence light source (lasers or LED).
-
24.
Turn on the stage incubator and set at correct temperature. Let the temperature of the incubator stabilize (stabilization time depends on the incubator, for the stage-top incubator from OKO labs described above it is around 30 min).
-
25.
Turn on the stage controller.
-
26.
Open the microscopy software.
-
27.
Focus on the cells using transmitted light.
-
28.
Switch to fluorescence excitation.
-
29.
Move the stage to find a field of view with cells that are suitable for imaging. In general, depending on the size of the field of view of the camera, aim for 50–100 cells in an area of 100 × 100 μm. Avoid cells that have high levels of autofluorescence. Yeast cells show more autofluorescence during diauxic shift, so it is important to keep them in mid-log phase during growth and imaging.
-
30.Set acquisition conditions in imaging software:
-
a.Excitation power; for tracking of individual RNAs, maximum excitation power of the Lumencor source described above is used. For imaging gene transcription over time, use 0.1%–1% of the maximum power. See notes below.
-
b.Exposure time; for tracking of individual RNAs, use 30–50 ms. For imaging gene transcription, use 150 ms. See notes below.
-
c.Z-stacks; for tracking individual RNAs, imaging of a single z-plane is required. For following gene transcription over time, multiple z-positions should be imaged as the transcription site may move in the z-dimension over time. The number of z-slices needed to cover the entire yeast nucleus depends on the numerical aperture of the microscope and the size of the yeast cells (ranging from 4 to 10 μm, depending on growth conditions and background strain). In general, we recommend 9–15 z-slices with 0.5 μm increments. For a 1.3 NA objective, this results in a slight undersampling of the imaging volume, but allows for proper localization of the transcription site within the cell. Decreasing the z-increment will lead to increased effects of photobleaching and phototoxicity which can be detrimental for the cells, especially when the cells are imaged for extended amounts of time.
-
d.Number of frames; depending on the stability of the fluorophore under the given excitation power and exposure time. For tracking individual RNAs, imaging around 100 frames gives sufficient insight in the RNA movement over time, while for imaging gene transcription, we aim for 240 frames.
-
e.Interval between frames; for fast processes such as tracking of individual RNAs, continuous imaging with no dead time between the frames is needed. For slower processes such as imaging of gene transcription over time, an interval of 10–30 s is used. See notes.
-
f.Autofocus: if available on the microscope, use the autofocus setting to prevent z-drift during the acquisition. This is most relevant for long acquisitions (more than 10 min).
-
a.
-
31.
Turn on the fluorescence excitation and focus on the center of the cells.
-
32.
Start acquisition.
Note: In general, choosing the optimal excitation power and exposure time requires a balance between good signal-to-noise and minimal photobleaching/phototoxicity. To a first approximation, each dye can emit a fixed number of photons before it photobleaches and turns dark (Lee et al., 2013). Increasing excitation power or exposure time will lead to a larger number of photons per acquisition, thus increasing the signal-to-noise ratio, but limiting the time until photobleaching. In addition, (high amounts of) fluorescent light can cause light-induced damage to the cell (phototoxicity). Be aware of signs of phototoxicity: sudden uneven fluorescence signal, sudden termination of the fluorescence signal, termination of transcription, and termination of cell division. In addition, balancing excitation power and exposure time depends on the mobility of the fluorophore within the cell. In general, for relatively static spots, such as transcription sites or single genomic loci, use longer exposure times of 150–200 ms, which allows the use of lower excitation powers. However, for a mobile spots, such as diffusing single RNAs, long exposure times result in blurring caused by motion of the fluorophore during the image acquisition, so we recommend imaging using 10–50 ms exposure times. Single proteins can be visualized using laser excitation and inclined illumination (HILO) with an TIRF angle of around 60°–65° and laser powers around 10 μW to 10 mW. Diffusing proteins can be observed at 10–30 ms exposure, but it is also possible to only vizualize DNA-bound proteins with 500–1,000 ms exposure (for example for DNA dwell time measurements). Exposure times above 1,000 ms are not recommended, because cell movement results in blurring of the spot.
Note: The interval between consecutive frames depends on the rate at which the biological process under study occurs. Because the total number of frames that a fluorophore can be imaged is limited by photobleaching, increasing the frame interval results in being able to follow the fluorophore for longer periods of time. For example, for fast processes such as tracking of diffusing proteins or individual RNA molecules, a high acquisition rate is needed. In this case, the frame interval can be set equal to the exposure time (i.e., no dead time between consecutive frames). Typically, an exposure time of 30–50 ms is needed to collect enough light to resolve the fluorophore of interest. In that case, the frame interval for such fast processes will thus be 30–50 ms. For slower processes such as DNA-protein dwell time measurements an interval of 200 ms to 5 s is suitable, and for gene transcription, an interval of 10–30 s is recommended. For even slower processes, such as following the yeast cell cycle, a time interval of around 5 min can be used.
Note: If the sample has been prepared on a concanavalin A-coated dish (rather than an agar slab), it is possible to add drugs or compounds during imaging. Examples of this could be adding galactose to drive expression of a protein under control of the GAL1-10 promoter, or by adding compounds for rapid depletion or degradation, such as addition of rapamycin for the anchor-away system (Haruki et al., 2008) or auxin for the AID system (Nishimura et al., 2009). For addition of such compounds, place the sample on the microscope stage without covering the imaging dish with the lid. At the moment of drug addition, gently open the microscope chamber and slowly add the compound using a micropipette. Preferably, add roughly equal volume as the volume already in the dish, to ensure good mixing and to prevent gradients or high local concentration. For example, add 1.5 mL of a 2× concentrated drug diluted in media to 1.5 mL of media. It may be necessary to adjust the focus of the microscope after drug addition. Alternatively, some stage-top incubator systems allow for drug addition via an injection lid.
Image Analysis
In order to obtain quantitative information from the acquired images, further analysis of these images is necessary. In general, the exact analysis pipeline will depend on the biological question and the application used. Basic principles behind image analysis are described extensively in (Waters, 2009). Image processing and quantification can be performed using the software package Fiji, which provides intuitive implementations of most common image processing tasks, as well as the possibility to develop macros for automation and scripting of the tasks necessary for a variety of imaging applications (Schindelin et al., 2012). For analyzing transcription data obtained with the PP7 or MS2 systems, an analysis pipeline will consist of several steps:
-
33.
Making a maximum intensity projection. This can be done using Fiji.
-
34.
Segmentation of cells and nuclei. This can be performed using CellProfiler (Lamprecht et al., 2007), which provides an intuitive, GUI-based pipeline for image segmentation.
-
35.
Detection and tracking individual RNAs or the transcription site over time. For tracking of individual RNAs, existing tracking and quantifaction packages such as the TrackMate package (Tinevez et al., 2017), which is available through Fiji or alternative approaches such as described and compared in (Chenouard et al., 2014) are sufficient. Tracking of single proteins and dwell time measurements can be done with Matlab software such as MatTrack (Mazza et al., 2013). Following and tracking the transcription site (as a measure of real-time gene transcription) over time is more challenging, as it requires appropriate gap-filling during periods of time when the gene is off. Typically, it is necessary to develop custom-written software for this purpose.
Expected Outcomes
In Figure 4, examples are given of possible fluorescence images obtained with this protocol. RNA of the GAL10 gene is labeled using the PP7 technology (Bertrand et al., 1998; Chao et al., 2008). Typically, a field of view with 50–100 cells is imaged (Figure 4A). Such cells are either imaged with short exposure times and high excitation power, to visualize individual RNAs moving within the cell (Figure 4B, Methods Video S2) or using longer exposure times, lower exposure times and longer intervals between the frames to measure GAL10 transcription by tracking the transcription site over time (Figure 4C, Methods Video S3). Other applications of the described technology include imaging the nuclear levels of a fluorescently tagged protein (Figure 5A), the tracking of single HALO-tagged proteins (Figure 5B) or localization of an individual DNA locus using the ParS DNA labeling system (Saad et al., 2014) (Figure 4D).
Figure 4.

Examples of Outcomes of Live-Cell Imaging Experiments
All examples are cells expressing PP7 coat protein fused with GFP Envy.
(A) Typical field of view of cells during an experiment. Cells shown are expressing PP7 coat protein fused to GFP Envy for visualizing transcription from 14 × PP7 loops at 5′GAL10, grown in galactose-rich media, imaged in widefield fluorescence excitation mode.
(B) Zoom in to several cells from the sample shown in (A), imaged at high power using short (50 ms) exposure time, showing individual RNAs in the cytoplasm.
(C) Zoom in to several cells from the sample shown in (A) imaged at lower power (500× less than in (B)) using longer (150 ms) exposure time, showing RNAs at the transcription sites.
Figure 5.

Other Applications of the Described Technology
(A and B) (A) Cells expressing Sth1-GFP showing nuclear localization of the protein, using widefield fluorescence excitation mode. Imaging settings: widefield illumination, exposure time 150 ms, excitation with 10% power using ND 1.0, 100× magnification, maximum intensity projection of 9 z-slices with a difference of 0.5 μm between consecutive slices. (B) Cells expressing Gal4-HALO. Bright spots represent individual Gal4 protein molecules. Imaging settings: HILO excitation mode, TIRF angle 62.33°, exposure time 1,000 ms, 12 μW laser power, 160× magnification, single z-plane.
(C) Cells imaged using the ParS system showing localization of the GAL10 DNA locus. Imaging settings: widefield illumination, exposure time 150 ms, excitation with 100% power using ND 2.0, 100× magnification, 9 z-slices with a difference of 0.5 μm between consecutive slices,
Limitations
Fluorophore Choice
A main limitation for fluorescence imaging is the photostability of the fluorophores that are used. A “good” fluorophore has a high brightness and a high photostability (Lee et al., 2013). For imaging inside living cells, there are additional requirements. First of all, the maturation time of the fluorophore must be sufficiently faster than the biological process under study. In addition, the fluorophore must not interfere with the biological process under study, and its cellular aggregation must be low. For imaging inside living yeast cells, fluorophorescent proteins with excitation around 400–500 nm are optimal. Lower excitation wavelength (toward blue) likely leads to increased phototoxicity, as radiation energy scales with the inverse of wavelength. In general, fluorescently encoded proteins with longer wavelengths (red/far red) show slightly lower fluorescence signals and photostability, leading to faster photobleaching (Botman et al., 2019). There are synthetic fluorescent dyes in the red/far-red regime with better photostability, but not all are suitable for live-cell imaging. However, we have good experience with labeling single proteins in living cells with the HALO dyes JF552 and JF646 (see below) (Grimm et al., 2015). In addition, yeast cells show increased autofluorescence when excited with higher wavelengths, which can significantly reduce the observed signal-to-noise ratio.
Although many fluorophorescent proteins are available, we recommend the use of yEGFP (Cormack et al., 1997) or GFP Envy (Slubowski et al., 2015), as it works well inside living yeast cells and has sufficient brightness and photostability. In addition, antibodies for GFP are commercially available, which allows the user to perform Western Blots or ChIP in addition to imaging experiments. If a second fluorophorescent protein is required (in the case of dual-color imaging), our recommendation is the use of ymScarlet-I (Botman et al., 2019). For in vivo FRET in yeast, a common fluorophore choice is CFP in combination with YFP (Skruzny et al., 2019).
In addition to conventional fluorophorescent proteins such as GFP and the others mentioned above, which are introduced into the cells by genetic fusion to a protein of interest, an alternative approach is the use of the HALO labeling system (Los et al., 2008). Here, a 297-amino acid HALO tag is fused to the protein of interest. This tag covalently binds to a HALO-dye and will emit a fluorescence signal which can be monitored in the same manner as the conventional fluorophores. The main advantages of using the HALO system are its flexibility and brightness; the excitation and emission wavelength can be varied by varying the dye used, and synthetic dyes are generally brighter and more photostable than fluorescent proteins. In addition, some dyes only fluoresce when they are bound to the HALO tag, limiting background fluorescence from unbound synthetic dyes. The main disadvantage of the HALO labeling system is the fact that it can be challenging to robustly introduce the dye into living yeast cells. The cell wall is difficult to penetrate, and the dye is actively exported from the cell. To prevent the latter, it is critical to delete the PDR5 exporter protein gene from the genome when using the HALO system (Ball et al., 2016), but even in cells lacking PDR5, we have not been able to achieve complete labeling. Although a large spectrum of dyes is available (Grimm et al., 2015), we have obtained best results with the JF552 and JF646 dyes for imaging in living yeast cells.
Troubleshooting
Problem
Too many cells in field of view; The field of view selected for imaging in the example in Figure 6A is too crowded, when compared to a correct sample shown in Figure 6D. The cells cannot be properly segmented in downstream analysis and the cells may enter diauxic shift.
Figure 6.

Examples of Possible Problems with Yeast Cultures during Imaging
All examples are cells expressing PP7 coat protein fused with GFP Envy.
(A) Cells from a field of view containing too many cells, leading to enhanced autofluorescence and making cells more difficult to segment in downstream analysis, due to potential overlapping of cells.
(B) Cells grown until stationary phase (culture grown for 12–18 h), lowering expression level of fluorescent proteins and more uneven staining in the nucleus.
(C) Indicated cell shows very high fluorescence levels, due to membrane staining which occurs in dead cells. Such cell should be omitted for quantification and analysis.
(D) Cells grown without any problems, for comparison to figures (A)−(C).
Potential Solution
Select a different field of view in this sample. If there are no fields with fewer cells, reduce the OD of the imaging culture or use less than 1 mL (in step 4 of Preparing sample using agar pad) or less than 300 μL (in step 2 of Preparing imaging sample using concanavalin A) of culture for sample preparation.
Problem
Uneven protein staining and lower protein expression levels; The cells imaged in Figure 6B exhibit a more uneven protein staining of the nucleus and lower protein expression levels when compared to a correct sample shown in Figure 6D. These lower, less even fluorescence levels may mask the fluorescence signal from the biological process under study. The cells in this example were grown until very high OD (saturated culture grown 12–18 h). In addition to the effects shown in this image, such cells may also show changed gene expression, which may influence dynamic measurements.
Potential Solution
The cells in the example in Figure 6B exhibit uneven staining in the nucleus. These cells were imaged while they were grown until very high OD (saturated culture grown 12–18 h) into diauxic shift, rather than kept in mid-log phase. Keeping them in mid-log phase will prevent this background fluorescence.
Problem
Individual cell exhibiting very high fluorescence: The cell imaged in Figure 6C and marked by the red circle, exhibits high fluorescence level throughout the entire cell (in both nucleus and cytoplasm). Such cells are typically dead cells. Their high fluorescence can interfere with image analysis of neighboring cells.
Potential Solution
Remove such cells and possibly neighboring cells from downstream analysis or image a different field of view.
Problem
Mobile cells during imaging on concanavalin A -coated dishes: The cells imaged in Methods Video S4 move during imaging. This is problematic for downstream analysis and cell segmentation.
During acquisition, some cells are not properly attached and move, making cell segmentation and downstream analysis more difficult.
Potential Solution
Use a lower cell density for the initial sample (by reducing the OD of the imaging culture). Alternatively, if it is not needed to change medium or add compounds during the experiment, samples can be prepared using agar imaging pads rather than concanavalin A-coated glass-bottom dishes.
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Tineke Lenstra (t.lenstra@nki.nl).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
This study did not generate datasets.
Acknowledgments
This work was supported by the Netherlands Organization for Scientific Research (NWO, 016.Veni.192.071 and gravitation program CancerGenomiCs.nl), Oncode Institute, which is partly financed by the Dutch Cancer Society, and the European Research Council (ERC Starting Grant 755695 BURSTREG).
Author Contributions
I.B., H.P.P., J.V.W.M., W.P., and T.L.L. conceived and optimized the protocols. I.B., H.P.P., and W.P. performed the experiments and analyzed the data. I.B. and T.L.L. wrote the manuscript. I.B., H.P.P., J.V.W.M., W.P., and T.L.L. revised and edited the manuscript.
Declaration of Interests
The authors declare no competing interests.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.xpro.2020.100142.
Contributor Information
Ineke Brouwer, Email: in.brouwer@nki.nl.
Tineke Laura Lenstra, Email: t.lenstra@nki.nl.
References
- Ball D.A., Mehta G.D., Salomon-Kent R., Mazza D., Morisaki T., Mueller F., McNally J.G., Karpova T.S. Single molecule tracking of Ace1p in Saccharomyces cerevisiae defines a characteristic residence time for non-specific interactions of transcription factors with chromatin. Nucleic Acids Res. 2016;44:1–12. doi: 10.1093/nar/gkw744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertrand E., Chartrand P., Schaefer M., Shenoy S.M., Singer R.H., Long R.M. Localization of ASH1 mRNA particles in living yeast. Mol. Cell. 1998;2:437–445. doi: 10.1016/s1097-2765(00)80143-4. [DOI] [PubMed] [Google Scholar]
- Botman D., de Groot D.H., Schmidt P., Goedhart J., Teusink B. In vivo characterisation of fluorescent proteins in budding yeast. Sci. Rep. 2019;9:1–14. doi: 10.1038/s41598-019-38913-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao J.A., Patskovsky Y., Almo S.C., Singer R.H. Structural basis for the coevolution of a viral RNA-protein complex. Nat. Struct. Mol. Biol. 2008;15:103–105. doi: 10.1038/nsmb1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chenouard N., Smal I., de Chaumont F., Maška M., Sbalzarini I.F., Gong Y., Cardinale J., Carthel C., Coraluppi S., Winter M. Objective comparison of particle tracking methods. Nat. Methods. 2014;11:281–289. doi: 10.1038/nmeth.2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cormack B.P., Bertram G., Egerton M., Gow N.A., Falkow S., Brown A.J. Yeast-enhanced green fluorescent protein (yEGFP): A reporter of gene expression in Candida albicans. Microbiology. 1997;143:303–311. doi: 10.1099/00221287-143-2-303. [DOI] [PubMed] [Google Scholar]
- Donovan B.T., Casillas L.N., Bury M.J., Mehlmann J.F., Singhaus R., Jr., Charnley A.K., Hughes T.V., DeMartino M.P., Wang G.Z., Romano J.J. Live-cell imaging reveals the interplay between transcription factors, nucleosomes, and bursting. EMBO J. 2019:1–18. doi: 10.15252/embj.2018100809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm J.B., English B.P., Chen J., Slaughter J.P., Zhang Z., Revyakin A., Patel R., Macklin J.J., Normanno D., Singer R.H., Lionnet T., Lavis L.D. A general method to improve fluorophores for live-cell and single-molecule microscopy. Nat. Methods. 2015;12:244–250. doi: 10.1038/nmeth.3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haruki H., Nishikawa J., Laemmli U.K. The anchor-away technique: rapid, conditional establishment of yeast mutant phenotypes. Mol. Cell. 2008;31:925–932. doi: 10.1016/j.molcel.2008.07.020. [DOI] [PubMed] [Google Scholar]
- Lamprecht M.R., Sabatini D.M., Carpenter A.E. CellProfilerTM: Free, versatile software for automated biological image analysis. BioTechniques. 2007;42:71–75. doi: 10.2144/000112257. [DOI] [PubMed] [Google Scholar]
- Lee S., Lim W.A., Thorn K.S. Improved blue, green, and red fluorescent protein tagging vectors for S. cerevisiae. PLoS One. 2013;8:e67902. doi: 10.1371/journal.pone.0067902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenstra T.L., Coulon A., Chow C.C., Larson D.R. Single-molecule imaging reveals a switch between spurious and functional ncRNA transcription. Mol. Cell. 2015;60:597–610. doi: 10.1016/j.molcel.2015.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Los G.V., Encell L.P., McDougall M.G., Hartzell D.D., Karassina N., Zimprich C., Wood M.G., Learish R., Ohana R.F., Urh M. HaloTag: A novel protein labeling technology for cell imaging and protein analysis. ACS Chem. Biol. 2008;3:373–382. doi: 10.1021/cb800025k. [DOI] [PubMed] [Google Scholar]
- Mazza D., Ganguly S., McNally J.G. Monitoring dynamic binding of chromatin proteins in vivo by single-molecule tracking. In: Shav-Tal Y., editor. Imaging Gene Expression: Methods and Protocols. Humana Press; 2013. pp. 117–137. [DOI] [PubMed] [Google Scholar]
- Nishimura K., Fukagawa T., Takisawa H., Kakimoto T., Kanemaki M. An auxin-based degron system for the rapid depletion of proteins in nonplant cells. Nat. Methods. 2009;6:917–922. doi: 10.1038/nmeth.1401. [DOI] [PubMed] [Google Scholar]
- Saad H., Gallardo F., Dalvai M., Tanguy-le-Gac N., Lane D., Bystricky K. DNA dynamics during early double-strand break processing revealed by non-intrusive imaging of living cells. PLoS Genet. 2014;10 doi: 10.1371/journal.pgen.1004187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B. Fiji: An open-source platform for biological-image analysis. Nat. Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skruzny M., Pohl E., Abella M. FRET microscopy in yeast. Biosensors. 2019;9:1–17. doi: 10.3390/bios9040122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slubowski C.J., Funk A.D., Roesner J.M., Paulissen S.M., Huang L.S. Plasmids for C-terminal tagging in Saccharomyces cerevisiae that contain improved GFP proteins, Envy and Ivy. Yeast. 2015;32:379–387. doi: 10.1002/yea.3065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tinevez J.Y., Perry N., Schindelin J., Hoopes G.M., Reynolds G.D., Laplantine E., Bednarek S.Y., Shorte S.L., Eliceiri K.W. TrackMate: An open and extensible platform for single-particle tracking. Methods. 2017;115:80–90. doi: 10.1016/j.ymeth.2016.09.016. [DOI] [PubMed] [Google Scholar]
- Tokunaga M., Imamoto N., Sakata-Sogawa K. Highly inclined thin illumination enables clear single-molecule imaging in cells. Nat. Methods. 2008;5:159–161. doi: 10.1038/nmeth1171. [DOI] [PubMed] [Google Scholar]
- Waters J.C. Accuracy and precision in quantitative fluorescence microscopy. J. Cell Biol. 2009;185:1135–1148. doi: 10.1083/jcb.200903097. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
During acquisition, some cells are not properly attached and move, making cell segmentation and downstream analysis more difficult.
Data Availability Statement
This study did not generate datasets.