

Summary
Kidney organoids represent a physiologically advanced model for studying the mechanisms of kidney development and disease. Here, we describe a simple two-step protocol for the differentiation of human pluripotent stem cells into kidney organoids. Our approach involves suspension culture that allows for rapid and cost-effective bulk production of organoids, which is well suited for large-scale assays such as drug screening. The organoids correspond to fetal human kidney tissue and may be of limited use for modeling adult kidney function.
For complete details on the use and execution of this protocol, please refer to Przepiorski et al. (2018).
Graphical Abstract
Highlights
-
•
Generation of kidney organoids from hPSCs in two simple steps
-
•
The method is cost-efficient and amendable for large-scale production
-
•
Organoid differentiation is optimal between day 12 and day 20
-
•
Organoid nephrons correspond to human fetal nephrons
Kidney organoids represent a physiologically advanced model for studying the mechanisms of kidney development and disease. Here, we describe a simple two-step protocol for the differentiation of human pluripotent stem cells into kidney organoids. Our approach involves suspension culture that allows for rapid and cost-effective bulk production of organoids, which is well suited for large-scale assays such as drug screening. The organoids correspond to fetal human kidney tissue and may be of limited use for modeling adult kidney function.
Before You Begin
Experiments using human pluripotent stem cells (hPSCs) must conform to relevant governmental and institutional regulations relating to human ethics, biosafety, and genetic modification, for which approvals have to be obtained. Standard cell culture procedures including sterile handling in Class II biosafety cabinets and incubation in humidified incubators at 37˚C and 5% CO2 are required for maintenance of hPSC lines. hPSC lines should be validated for pluripotency and normal karyotype prior to use.
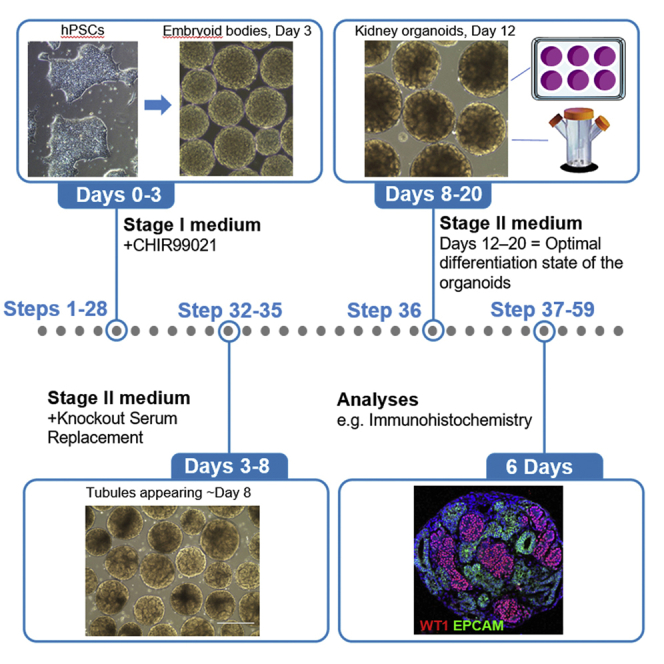
The principle of in vitro kidney organoid differentiation is based on mimicking fetal kidney development, for which Wnt signaling is one of the key factors. Our protocol is a two-stage approach, whereby the small molecule Wnt-agonist CHIR99021 is used in the first stage medium to promote mesoderm formation and initiate nephrogenesis in form of embryoid bodies. In the second stage, the supplementation of our “Stage II” medium with KnockOut Serum Replacement is sufficient to further differentiate the embryoid bodies into kidney tubule (nephron)-containing organoids. The formation of kidney organoids with our protocol is achieved without the need for additional growth factors, such as FGF9 and Activin A that are commonly used in other kidney organoid protocols. Thus, our protocol differs from other methods in that it is inexpensive, allowing kidney organoids to be grown in suspension culture in large scale. Despite the simpler approach, our organoids resemble those generated with other methods (Taguchi et al., 2014; Takasato et al., 2015; Morizane et al., 2015; Taguchi and Nishinakamura, 2017; Kumar et al., 2019) in that they develop the major renal cell types, i.e., podocytes, proximal and distal tubule cells, presumptive collecting duct cells, endothelial cells, and interstitial cells. For a more detailed comparison of protocols, please refer to Przepiorski et al. (2018).
Coating of Culture Dishes for hPSCs
Timing: 70 min
For feeder-free maintenance of hPSCs, culture dishes need to be coated with soluble extracellular matrix preparations such as growth factor-reduced Geltrex.
-
1.
Resuspend a frozen aliquot (e.g., 100 or 200 μL) of Geltrex in 1 mL DMEM then dilute to a final concentration of 1% in DMEM. Mix well by pipetting (avoiding bubbles), then add a volume of Geltrex/DMEM mix sufficient to cover the entire growth surface of the culture dish (e.g., ∼5 mL of 1% Geltrex for one 100 mm dish). Use the equivalent of 50 μL undiluted Geltrex per 100 mm dish.
-
2.
Leave to coat at room temperature (15°C–25°C) for 1 h or at 37˚C for 30 min. Aspirate Geltrex/DMEM immediately before use.
Note: Coated dishes can be stored at 4˚C for up to 4 days. For this, leave the Geltrex/DMEM mixture in the dish and seal with parafilm to prevent evaporation.
Alternatives: Growth factor-reduced Matrigel is commonly used for hPSC culturing instead of Geltrex. This protocol has successfully been applied to hPSCs grown on Matrigel.
CRITICAL: The combination of Geltrex (or Matrigel) coating and Dispase digestion of hPSC colonies is optimized in this protocol for lifting of whole hPSC colonies (step 7), which is required for efficient embryoid body/organoid formation. Alternative combinations of matrices and dissociation reagents may be less compatible and require adjustments.
Passaging of hPSCs
hPSCs grow in colonies and need to be passaged once they reach 70%–80% confluency (every 3 – 5 days). We use Gentle Cell Dissociation Reagent to perform non-enzymatic small aggregate-passaging. This method is recommended for routine passaging as it preserves the integrity of cell surface proteins, thus promoting cell survival. Aggregate-passaging also places less unwanted selective pressure on cell populations than single-cell passaging. This is important as selective pressure could lead to genetic aberrations in the culture.
-
3.
Pre-warm mTeSR1 medium to room temperature (15°C–25°C).
-
4.
Prepare mTeSR1 medium plus ROCK inhibitor Y27632 (final concentration of 5 μM). Use 8 mL of medium per 100 mm culture dish.
-
5.
Aspirate Geltrex/DMEM from pre-coated dish(es) and replace with mTeSR1 plus ROCK inhibitor.
-
6.
Wash the cells with 10 mL 1× DPBS.
-
7.
Aspirate DPBS and add 2 mL of Gentle Cell Dissociation Reagent (making sure the entire growth surface is covered), then incubate at room temperature for 6–8 min.
Note: Incubation time can vary between hPSC lines. Optimal dissociation is reached when the cells start to round up within the colonies (Figure 1A).
Figure 1.

hPSC Passaging
(A) For routine passaging, hPSC colonies are treated with Gentle Cell Dissociation Reagent for 6–8 min at room temperature (15°C–25°C; RT) until the cells start to round up within the colonies.
(B) Extended exposure to the reagent (shown here is 15 min at RT) leads to detachment of the colonies (red arrows) and increased cell death.
(C) Optimal size of aggregates (50–200 μm) when performing “aggregate-passaging” with Gentle Cell Dissociation Reagent.
(D−F) Colony densities on the day after splitting, showing the outcome of optimal (D), too sparse (E) and too dense (F) splitting.
(G) Optimal Accutase digestion (5 min at 37˚C) is achieved when cells start to round up within the colonies.
(H) Overdigestion with Accutase (shown here is 10 min at 37˚C) leads to detachment of the colonies (red arrows) and increased cell death.
Scale bars, 400 μm.
-
8.
Aspirate Gentle Cell Dissociation Reagent. Scrape cell aggregates off the dish using a cell lifter then wash the aggregates down with 4 – 6 mL of mTeSR1.
-
9.
Using a 10 mL serological pipette, gently pipette the aggregates up and down (up to 5×) to break into evenly sized aggregates of 50 – 200 μm (Figure 1C).
-
10.
Aspirate the aggregate suspension and split into prepared cultures dish(es) in a ratio of 1:4 – 1:8 (Figure 1D), e.g., if total aggregate suspension is 6 mL then transfer 1 mL (for a 1:6 ratio) to the new culture dish. Add dropwise, distributing aggregates evenly throughout the dish. Avoid too sparse or too dense splitting (Figures 1E and 1F).
-
11.
Transfer the dish(es) into the 37°C incubator and distribute the cell aggregates by side-to-side and figure-8 movements of the dish, then leave undisturbed for 16–24 h.
-
12.
Change mTeSR1 daily (without ROCK inhibitor) and visually check appearance of the cultures. We use an EVOS™ XL Core inverted Imaging System to monitor hPSC growth.
Optional: Accutase can be used instead of non-enzymatic passaging. Accutase treatment of hPSC colonies leads to dissociation of the colonies into single cells, which is required for cell counting or flow cytometry.
-
13.
Thaw an aliquot of Accutase and dilute 1:3 in 1× DPBS. Proceed through steps 3–6 as described above
-
14.
Aspirate DPBS and add 2 mL of diluted Accutase per 100 mm culture dish. Ensure that the entire growth surface is covered then incubate at 37°C for 3–5 min.
Note: Incubation time can vary between hPSC lines but must not exceed 6 min. Optimal dissociation is reached when the cells start to round up within the colonies (Figure 1G).
-
15.
Scrape cells off the dish using a cell lifter, wash the cells down with ∼5 mL of 1× DPBS and transfer into a 15 mL centrifugation tube.
-
16.
Centrifuge for 5 min at 103 × g at room temperature (15°C–25°C). We use a Heraeus Megafuge 8 with a TX-150 Swinging Bucket Rotor.
-
17.
Carefully aspirate the supernatant.
-
18.
Gently resuspend the cell pellet in 4 – 8 mL of mTeSR1.
-
19.
Split cells into prepared culture dish(es) (steps 4 and 5) in a ratio of 1:4 – 1:8. Avoid too dense or too sparse splitting (Figures 1D−1F).
-
20.
Transfer the dish(es) into the 37°C incubator and distribute the cells by side-to-side and figure-8 movements of the dish, then leave undisturbed for 16–24 h.
-
21.
Change mTeSR1 daily (without ROCK inhibitor) and visually check appearance of the cultures.
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies and Lectins | ||
| CALB1 rabbit (use 1:1,000). Labels distal tubules and collecting ducts | Sigma | Cat#C2724 |
| CDH1 (E-Cadherin) mouse IgG2a (use 1:300). Labels distal tubules and collecting ducts | BD Biosciences | Cat#610181 |
| CUBN rabbit IgG (use 1:500). Labels proximal tubules | Abcam | Cat#ab244274 |
| EPCAM rabbit (use 1:100). Labels all tubular epithelia | Sigma | Cat#HPA026761 |
| HNF1B rabbit (use 1:500). Labels nuclei of all tubular epithelia | Sigma | Cat#HPA002083 |
| Lotus Tetragonolobus Lectin (LTL), Fluorescein (use 1:300). Labels proximal tubules | Vectorlabs | Cat#FL-1321 |
| LRP2 (Megalin) mouse IgG1 (use 1:500). Labels proximal tubules | Novus Biologicals | Cat#NB110-96417 |
| NPHS1 sheep IgG (use 1:200). Labels podocytes | R&D Systems | Cat#AF4269 |
| SLC12A1 rabbit (use 1:1,000). Labels thick ascending limb | Sigma | Cat#HPA018107 |
| WT1 mouse IgG1 (use 1:25). Labels podocyte nuclei | Santa Cruz Biotechnology | Cat#sc-7385 |
| Hoechst 33258 (use 1:2,000/10 μM). Nuclear counterstain | Abcam | Cat#ab228550 |
| Anti-rabbit-Dylight 488 (use 1:600) | Abcam | Cat#ab96899 |
| Anti-mouse-Dylight 594 (use 1:600) | Abcam | Cat#ab96881 |
| Anti-sheep-Alexa Fluor 594 (use 1:600) | Thermo Fisher | Cat#A11016 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 1-Thioglycerol | Sigma | Cat#M6145 |
| 2-Mercaptoethanol | Thermo Fisher | Cat#21985023 |
| Accutase | STEMCELL Technologies | Cat#7920 |
| Agar | Sigma | Cat#A1296 |
| AlbumiNZ™ protease reduced, immunoassay (EIA) grade, ≥97% (BSA used for immunohistochemistry) | MP Biomedicals | Cat#219989880 |
| Chemically Defined Lipid Concentrate | Thermo Fisher | Cat#11905031 |
| CHIR99021 | STEMCELL Technologies | Cat#72054 |
| Dispase | STEMCELL Technologies | Cat#7923 |
| DMEM, low glucose, pyruvate | Thermo Fisher | Cat#11885084 |
| DPBS 1×, no calcium, no magnesium | Thermo Fisher | Cat#14190250 |
| Ethanol absolute | Sigma | Cat#1.07017 |
| Geltrex™ LDEV-Free Reduced Growth Factor Basement Membrane Matrix | Thermo Fisher | Cat#A1413202 |
| Gentle Cell Dissociation Reagent | STEMCELL Technologies | Cat#7174 |
| GlutaMAX Supplement | Thermo Fisher | Cat#35050061 |
| Ham's F12 Nutrient Mix | Thermo Fisher | Cat#11765054 |
| HEPES | Thermo Fisher | Cat#15630080 |
| Horse serum | Sigma | Cat#H1270 |
| HyAgarose™ LE Agarose | HydraGene | Cat#R9012LE |
| Hydrochloric acid | Sigma | Cat#320331 |
| IMDM | Thermo Fisher | Cat#12440053 |
| Insulin-Transferrin-Selenium-Ethanolamine (ITS -X) | Thermo Fisher | Cat#51500056 |
| K-252a (BDNF inhibitor) | Sigma | Cat#K1639 |
| KnockOut™ Serum Replacement - Multi-Species | Thermo Fisher | Cat#A3181502 |
| L-Ascorbic acid 2-phosphate sesquimagnesium salt hydrate | Sigma | Cat#A8960 |
| Leica Surgipath Paraplast (Paraffin) | bio-strategy | Cat#LEIS39601006 |
| Matrigel® Growth Factor Reduced Basement Membrane Matrix, LDEV-free | BD Biosciences | Cat#354230 |
| MEM Non-Essential Amino Acids Solution | Thermo Fisher | Cat#11140050 |
| mTeSR1 | STEMCELL Technologies | Cat#5850 |
| Paraformaldehyde 8% | Emgrid Australia | Cat#157-8 |
| Penicillin-Streptomycin | Thermo Fisher | Cat#15140122 |
| PFHM-II Protein-Free Hybridoma Medium | Thermo Fisher | Cat#12040077 |
| Plasmocin | InvivoGen | Cat#ant-mpt |
| Poly(vinyl alcohol) | Sigma | Cat#P8136 |
| Probumin® Bovine Serum Albumin | Sigma | Cat#821006 |
| ProLong™ Diamond Antifade Mountant | Thermo Fisher | Cat#P36961 |
| ROCK inhibitor Y-27632 | STEMCELL Technologies | Cat#72304 |
| Sodium chloride | Sigma | Cat#71376 |
| Trisodium citrate dihydrate | Sigma | Cat#W302600 |
| StemFlex™ Medium | Thermo Fisher | Cat#A3349401 |
| Sucrose | Sigma | Cat#S0389 |
| TeSR™-E8™ | STEMCELL Technologies | Cat#05990 |
| Tissue-Plus™ OCT Compound | Fisher Scientific | Cat#23-730-571 |
| Triton X-100 | Sigma | Cat#X100 |
| TRIzol™ Reagent | Thermo Fisher | Cat#15596026 |
| Tween 20 | Sigma | Cat#P1379 |
| UltraPure™ Tris Buffer | Thermo Fisher | Cat#15504020 |
| Xylene | Sigma | Cat#534056 |
| Other | ||
| Cell lifter | Corning | Cat#3008 |
| Corning tissue-culture treated culture dishes, D × H 100 mm × 20 mm | Merck Millipore | Cat#430167 |
| Corning disposable spinner flasks | Sigma | Cat#CLS3152 |
| Fisherbrand™ Multi Function 3D rotator | Fisher Scientific | Cat#15514080 |
| Fisherbrand™ Histosette™ II Tissue Processing/Embedding Cassettes | Fisher Scientific | Cat#15-182-708A |
| ImmEdge® Hydrophobic Barrier PAP Pen | Vector Labs | Cat#H-4000 |
| LockMailer™ - Microscope Mailer and Staining Jar | Ted Pella | Cat#21096 |
| Menzel™ Microscope Coverslips | Fisher Scientific | Cat#11778691 |
| Nunc® CryoTubes® 1.8 mL, Internal thread | Sigma | Cat#V7634 |
| pluriStrainer® 500 μm | Pluri Select | Cat#43-50500-03 |
| Stericup-GP Sterile Vacuum Filtration System (500 mL) | Merck Millipore | Cat#S2GPU05RE |
| Stericup-GP Sterile Vacuum Filtration System (200 mL) | Merck Millipore | Cat#SCGPU02RE |
| Superfrost Plus Microscope Slides | Thermo Fisher | Cat#4951PLUS4 |
| Tissue-Tek® Cryomold | VWR | Cat#4565 |
| Ultra-Low Attachment 24-well plates | Merck Millipore | Cat#CLS3473 |
| Ultra-Low Attachment 6-well plates | Merck Millipore | Cat#CLS3471 |
| Oligonucleotides | ||
| Primers for NPHS1 (marker for podocytes), fwd: AGTGTGGCTAAGGGATTACCC; rev: TCACCGTGAATGTTCTGTTCC | Przepiorski et al., 2018 | n/a |
| Primers for LRP2 (marker for proximal tubule), fwd: AAATTGAGCACAGCACCTTTGA; rev: TCTGCTTTCCTGACTCGAATAATG | Przepiorski et al., 2018 | n/a |
| Primers for SLC34A1 (marker for proximal tubule), fwd: CCATCATCGTCAGCATGGTCT; rev: GACAGCCAGTTAAAGCAGTCA | This paper | n/a |
| Primers for SLC12A1 (marker for thick ascending limb), fwd: AGTGCCCAGTAATACCAATCGC; rev: GCCTAAAGCTGATTCTGAGTCTT | Przepiorski et al., 2018 | n/a |
| Primers for UMOD (marker for thick ascending limb), fwd: CGGCGGCTACTACGTCTAC; rev: TGCCATCTGCCATTATTCGATTT | Przepiorski et al., 2018 | n/a |
| Primers for SLC12A3 (marker for distal tubule), fwd: CCTGGGTGGAGACCTTCATTC; rev: GAGCCCCAATTTACCTCTGGC | This paper | n/a |
| Primers for CDH1 (marker for distal tubule and collecting duct), fwd: ATTTTTCCCTCGACACCCGAT; rev: TCCCAGGCGTAGACCAAGA | Przepiorski et al., 2018 | n/a |
| Primers for CALB1 (marker for distal tubule and collecting duct), fwd: TCCAGGGAATCAAAATGTGTGG; rev: GCACAGATCCTTCAGTAAAGCA | Przepiorski et al., 2018 | n/a |
| Primers for GATA3 (marker for distal tubule and collecting duct), fwd: GCCCCTCATTAAGCCCAAG; rev: TTGTGGTGGTCTGACAGTTCG | Przepiorski et al., 2018 | n/a |
| Primers for HPRT1 (reference housekeeping gene), fwd: CATTATGCTGAGGATTTGGAAAGG; rev: CTTGAGCACACAGAGGGCTACA | Przepiorski et al., 2018 | n/a |
| Experimental Models: Cell Lines | ||
| MAFB-P2A-eGFP H9 (ESC, female, labels podocytes) | Tran et al., 2019 | n/a |
| HPSI0214i-wibj_2 (iPSC, female) | HipSci | n/a |
| CRL1502 (clone C32) (iPSC, female) | n/a | RBK/GUDMAP Resources |
| LRP2:mTagBFP2 (iPSC, male, labels proximal tubules) | Howden et al., 2018; Vanslambrouck et al., 2019 | RBK/GUDMAP Resources |
| BJ RiPS (iPSC, male) | Warren et al., 2010; Przepiorski et al., 2018 | n/a |
| MANZ-2-2 (iPSC, female) |
Przepiorski et al., 2018; Oh et al., 2020. |
n/a |
| MANZ-4-37 (iPSC, male) |
Przepiorski et al., 2018; Oh et al., 2020. |
n/a |
| HNF1B-/-, 3 clones (iPSC, female, isogenic to MANZ-2-2) | Przepiorski et al., 2018 | n/a |
| CTNS-mutant lines, 3 clones (iPSC, male, patient-derived) | Hollywood et al., 2020 | n/a |
| CTNS knockout lines, 3 clones (iPSC, female, isogenic to CRL1502 (clone C32)) | Hollywood et al., 2020 | n/a |
Materials and Equipment
hPSC Culture and Organoid Generation
The aliquot sizes suggested here correspond to the volumes required for preparing 250 mL of mBPEL medium or 500 mL of Stage II medium.
mTeSR1 hPSC Culture Medium
Thaw 5× mTeSR1 Supplement then combine with 400 mL mTeSR1 Basal Medium. Add 50 μL Plasmocin (dilute 1:10,000 for a final concentration of 2.5 μg/mL) and 5 mL Penicillin-Streptomycin 100× (for final concentration of 1×). Store at 4°C for up to 2 weeks.
Alternatives: Other hPSC culture media such as StemFlex or TeSR-E8 media can be used.
Probumin Bovine Serum Albumin (BSA)
Prepare 10% w/v in IMDM medium, aliquot into 6.25 mL and store at −20°C for up to 1 year. The lyophilized stock powder is stable for 2 years at 4°C.
Alternatives: Other low endotoxin and low IgG BSA preparations that are labeled as suitable for cell culture can be used.
L-Ascorbic Acid 2-Phosphate Sesquimagnesium Salt Hydrate (AA2P)
Prepare 5 mg/mL in ddH2O, aliquot into 2.5 mL and store at −20°C for up to 1 year.
Penicillin-Streptomycin (P/S)
Prepare 2.5 or 5 mL aliquots of the 100× stock and store at −20°C for up to 1 year.
Poly(Vinyl Alcohol) (PVA)
Prepare 10% w/v in 1× DPBS. Leave to dissolve in 95°C water bath (∼8 h). Store at 4˚C for up to 6 months.
Knockout Serum Replacement (KOSR)
Prepare 50 mL aliquots of the 100× stock and store at −20°C for up to 18 months. Avoid additional freeze-thaw cycles.
Dispase
Prepare 2 or 4 mL aliquots and store at −20°C until expiry date indicated on the label. Thawed aliquots are stable for up to 2 weeks at 4°C. Do not re-freeze.
Accutase
Prepare 2 or 4 mL aliquots and store at −20°C until expiry date indicated on the label. Thawed aliquots are stable for up to 2 months at 4°C.
CHIR99021
Prepare 10 mM stock in DMSO, aliquot (e.g., in 50 μL) and store at −20°C for up to 6 months.
ROCK Inhibitor Y-27632
Prepare 10 mM stock in ddH2O, aliquot (e.g., in 50 μL) and store at −20°C for up to 1 year.
Geltrex
Thaw Geltrex at 4°C for 16–20 h. Prepare ice-cold (−20°C) pipette tips and cryotubes. Place thawed Geltrex on ice, mix carefully and aliquot (e.g., into 100 or 200 μL aliquots), then freeze immediately. Store at −20°C for up to 18 months.
Note: We recommend preparing Geltrex aliquots in cryotubes with internal thread and wide-diameter opening (e.g., Nunc CryoTubes™) to facilitate sterile pipetting.
Modified Bovine Serum Albumin (BSA) Polyvinylalcohol Essential Lipids (mBPEL) Medium
BPEL medium was originally developed for embryoid body-based differentiation of hPSCs (Ng et al., 2008). Our modified (m)BPEL medium is a variation of the original in that it contains 0.1× ITS-X (adapted from Orlova et al., 2014).
| Reagent | Stock Concentration | Final Concentration | Amount |
|---|---|---|---|
| IMDM | 1× | 0.41× | 101.25 mL |
| Ham's F-12 Nutrient Mix | 1× | 0.45× | 113.46 mL |
| PFHM-II Protein-Free Hybridoma Medium | 1× | 0.05× | 12.5 mL |
| Probumin BSA | 10% | 0.25% | 6.25 mL |
| ITS-X | 100× | 0.1× | 250 μL |
| 1-Thioglycerol | ≥97% | 0.0039% | 9.75 μL |
| AA2P | 5 mg/mL | 50 μg/mL | 2.5 mL |
| P/S | 100× | 1× | 2.5 mL |
| GlutaMAX | 100× | 1× | 2.5 mL |
| PVA | 10% | 0.25% | 6.25 mL |
| Lipid concentrate | 100× | 1× | 2.5 mL |
| Plasmocin | 25 mg/mL | 2.5 μg/mL | 25 μL |
| Total | n/a | n/a | 250 mL |
Alternatives: Albumin Polyvinylalcohol Essential Lipids (APEL) medium, the animal-product free formulation (Ng et al., 2008) of this medium, can be used instead of BPEL.
Complete mBPEL Medium
Freshly prepare complete mBPEL medium on day 0 and day 2 of organoid generation (starting from steps 2 and 13, respectively). For one assay, add 8 μM CHIR99021 (14.4 μL), 3.3 μM ROCK inhibitor Y-27632 (6 μL) and 100 μM 2-Mercaptoethanol (32.7 μL) to 18 mL of mBPEL to make complete mBPEL medium for day 0. Add 8 μM CHIR99021 (9.6 μL) to 12 mL of mBPEL to make complete mBPEL medium for day 2.
Stage II Medium
Note: We changed from high-glucose DMEM (as used in the original protocol) to low-glucose DMEM. This modification was made as the low-glucose DMEM formulation (containing 5.5 mM glucose) more closely resembles physiological fasting glucose levels of 5.5 mM, whereas high-glucose DMEM (containing 25 mM glucose) exposes the organoids to hyperglycemia.
| Reagent | Stock Concentration | Final Concentration | Amount |
|---|---|---|---|
| DMEM (low glucose) | 1× | 0.79× | 392.5 mL |
| KOSR | 100% | 15% | 75 mL |
| Non-Essential Amino Acids | 100× | 1× | 5 mL |
| P/S | 100× | 1× | 5 mL |
| HEPES | 1 M | 10 mM | 5 mL |
| GlutaMAX | 100× | 1× | 5 mL |
| PVA | 10% | 0.25% | 12.5 mL |
| Plasmocin | 25 mg/mL | 2.5 μg/mL | 50 μL |
| Total | n/a | n/a | 500 mL |
Histology
Agarose/Agar Embedding Medium
Dissolve 1% low-melting agarose, 1% agar and 5% sucrose in ddH2O by heating in a microwave. Prepare 10 – 20 mL aliquots and store at 4°C for up to 1 year.
Citrate Buffer for Antigen Retrieval
Dissolve 2.94 g trisodium citrate in 1 L ddH2O (for a 10 mM solution), adjust pH to 6.0 with 1 N HCl, then add 0.5 mL Tween 20 and mix well. Store at room temperature (15°C–25°C) for 3 months or at 4°C for up to 1 year.
10× Tris-Buffered Saline (TBS)
For 1 L of 10× TBS, dissolve 24 g of Tris base and 88 g of NaCl in 900 mL ddH2O. Adjust pH to 7.6 with 12 N HCl. Adjust final volume with ddH2O. 10× TBS is stable for 2 years at room temperature (15°C–25°C). Freshly dilute to 1× TBS with ddH2O and use within 1 week.
Blocking Solution for Immunohistochemistry
Prepare 2% horse serum and 10% immunoassay grade BSA (w/v) in 1× TBS plus 0.5% Triton X-100. Store at 4°C and use within 1 week.
Paraformaldehyde
Freshly prepare 4% paraformaldehyde (v/v) in ddH2O.
Step-By-Step Method Details
Passaging hPSCs (Day −2)
This split is performed to achieve 40%–50% confluency at day 0 of the organoid generation protocol. In the original publication of this protocol, we determined the average cell number in a 40%–50% confluent 100 mm culture of MANZ-2–z2 iPSCs to be 6.6E+06 ± 2.3E+05 (Przepiorski et al., 2018).
-
1.
Passage hPSCs into one or more 100 mm Geltrex-coated culture dishes as described above. Splitting the cultures at 1:4–1:6 usually leads to the desired confluency after 2 (maximum 3) days.
Starting the Organoid Assay (Day 0)
This step describes how hPSC colonies are prepared for differentiation. Our protocol starts with a 3-day treatment with 8 μM CHIR99021 to induce mesoderm formation in form of spherical embryoid bodies in suspension culture (Figure 2A). The volumes given are for one assay that is initiated from one 100 mm culture dish.
-
2.
Check for 40%–50% confluency of the culture (Figure 2B). The colonies should be mostly discrete.
Note: If the optimal confluency is missed, we recommend splitting the culture and starting organoid differentiation with the subsequent passage.
-
3.
Pre-warm mBPEL medium to room temperature (15°C–25°C) and thaw an aliquot of Dispase.
-
4.
Prepare 18 mL of complete mBPEL medium in a 50 mL centrifuge tube (add 14.4 μL CHIR99021, 6 μL ROCK inhibitor Y-27632 and 32.7 μL 2-Mercaptoethanol to 18 mL mBPEL). Mix well by inverting the tube.
-
5.
Aliquot 2 mL of complete mBPEL medium into each well of an ultra-low attachment 6-well plate.
-
6.
Wash hPSCs twice with 10 mL 1× DPBS.
-
7.
Aspirate DPBS and add 2 mL of Dispase dropwise (ensuring that all colonies are covered) then incubate at 37°C for 6 min. Upon this treatment, the edges of the hPSC colonies should start to curl up but not detach from the dish (Figure 2C).
Note: Depending the type of culture medium used, this timing may need to be adjusted.
-
8.
Aspirate Dispase, then wash 3× with 10 mL 1× DPBS each.
-
9.
After the last wash, aspirate DPBS and scrape the colonies off the culture dish using a cell lifter.
-
10.
Use a 10 mL serological pipette to wash down the colonies with the remaining 6 mL of complete mBPEL. Pipette up and down (3–5×) to break up large colonies into evenly sized fragments of ∼100 μm (Figure 2E), then distribute 1 mL to each well of the 6-well plate. Troubleshooting 2 (Figure 4B)
-
11.
Place the 6-well plate into a 37°C incubator, move side-to-side for even distribution of the colonies.
-
12.
Leave the plate undisturbed for 48 h.
Figure 2.

Differentiation of hPSCs into Kidney Organoids
(A) Schematic of the protocol.
(B) Bright-field images of ~50% confluent hPSC colonies on day 0.
(C) High magnification of desired appearance of hPSC colonies after Dispase-treatment (6 min at 37˚C) showing the edge of the colony starting to curl up (white arrowhead).
(D) Upon overdigestion with Dispase (shown here is 12 min at 37˚C) the colonies are peeling off the growth surface (red arrowheads) and detach (red arrows), leading to increased cell death and low efficiency in organoid formation.
(E) Fragments of hPSC colonies in suspension after Dispase digest.
(F and F′) Day 2 embryoid bodies.
(G) Tilted 6-well plate for day 2 half-medium change.
(H) Orbital shaker used from day 2 onward.
(I and I′) Day 3 embryoid bodies.
(J) A spinning bioreactor can be used for embryoid body / organoid culture from day 3 onward.
(K) Tubule formation is visible on the surface of day 8 organoids (arrows in K′).
(L) Day 12 kidney organoids.
Scale bars, 1 mm (B, F, and I); 400 μm (C−E, F′, I′, K, and L).
Figure 4.

Troubleshooting
(A) Low efficiency of tubule formation.
(B) Absence of tubule formation in small organoids.
(C) Occasional fusing of embryoid bodies at day 3 (asterisks); high magnification of fused embryoid bodies is shown in (C′).
(D) Large embryoid bodies due to clumping.
(E) Elimination of large embryoid bodies through a 500 μm strainer.
(F) Severe clumping of day 3 embryoid bodies.
(G) Areas of non-renal outgrowth (i.e., the uniformly colored areas lacking the outlines of tubules, arrows) in day 20 organoids.
Scale bars, 1 mm (C, D, and F); 400 μm (A, B, C′, and G).
Half-Medium Change (Day 2)
On day 2, a half-medium change is performed to supply fresh mBPEL and CHIR99021.
-
13.
Pre-warm mBPEL to room temperature (15°C–25°C), then prepare 12 mL of complete mBPEL (with 9.6 μL CHIR99021 added).
-
14.
Check the 6-well plate for embryoid body formation. At this stage, the embryoid bodies should be obvious, appearing as spheres of 50 – 150 μm in diameter among floating single cells (Figures 2F and 2F′). We use an EVOS™ XL Core inverted Imaging System to monitor embryoid body (and organoid) appearance.
-
15.
Place 6-well plate into the tissue-culture hood and tilt to achieve a ∼45° angle (e.g., place on the edge of a tube rack, Figure 2G). Let embryoid bodies sediment on the bottom (3–5 min).
-
16.
Carefully aspirate approximately half of the medium from each 6-well.
-
17.
Return 6-well plate into flat position and add 2 mL of complete mBPEL to each well.
-
18.
Place 6-well plate on an orbital shaker in the incubator (Figure 2H).
Transfer to Stage II Medium (Day 3)
On day 3, mBPEL is replaced by Stage II medium. Stage II medium contains 15% KnockOut Serum Replacement in DMEM. This is sufficient to drive renal tubule formation, which becomes apparent by day 7 to day 9 of the protocol.
-
19.
Pre-warm DMEM and Stage II media to room temperature (15°C–25°C).
-
20.
Check appearance of day 3 embryoid bodies. The embryoid bodies should be spherical, light-golden in color with smooth edges (Figures 2I and 2I′). Troubleshooting 4
-
21.
Aspirate embryoid bodies from all six wells using a 10 or 25 mL serological pipette and combine in a 50 mL centrifuge tube.
-
22.
Rinse the six wells with 1 mL DMEM per well to collect remaining embryoid bodies then add these to the 50 mL tube.
-
23.
Leave the embryoid bodies to sediment at the bottom of the tube (3–5 min) at room temperature.
-
24.
Carefully aspirate the supernatant then wash with 15 mL of DMEM. Repeat step 23.
-
25.
Aliquot 2 mL of Stage II medium into each well of the 6-well plate.
-
26.
Carefully aspirate the supernatant from the sedimented embryoid bodies.
-
27.
Take up the embryoid bodies in 6 mL of Stage II medium and redistribute into the 6-well plate, 1 mL per well.
-
28.
Return plate to the orbital shaker.
Alternatives: Instead of continuing to culture the embryoid bodies in the 6-well plate format, they can be transferred into a spinning bioreactor, e.g., the Corning spinner flask (Figure 2J) from day 3 onward.
-
27a.
Take up embryoid bodies in Stage II medium and transfer into a spinner flask, then fill up to 45 mL with Stage II medium.
-
28a.
Place on magnetic stirrer (at 90 rpm) in the incubator (Figure 2J).
Optional: Size-Filtration of Embryoid Bodies / Organoids
Occasional fusion may lead to abnormally large embryoid bodies and organoids. (Troubleshooting 3). We have observed that organoids exceeding a diameter of 700 μm display signs of core apoptosis and necrosis, perhaps due to insufficient levels of oxygen and/or nutrients (Przepiorski et al., 2018). To eliminate large specimens and achieve a uniformly sized organoid culture, size-filtration can be performed from day 3 onward.
-
29.
Place a 500 μm cell strainer onto a 50 mL centrifugation tube (Figure 4E).
-
30.
Take up embryoid bodies / organoids with a 10 or 25 mL serological pipette and slowly drop onto the cell strainer then rinse with 5 mL Stage II medium.
-
31.
Leave embryoid bodies / organoids in the flow-through to sediment, then redistribute into 6-well plate or transfer into spinner flask.
Maintaining Embryoid Body Cultures (Day 3 to Day 8)
From day 3 onward, Stage II medium needs to be changed every 2–3 days.
-
32.
Pre-warm Stage II medium to room temperature (15°C–25°C).
-
33.
Remove spinner flask or 6-well plate from incubator, leave embryoid bodies to sediment (in tilted position for 6-well plates) at room temperature.
-
34.
Carefully aspirate most of the medium from each 6-well and roughly half of the medium from a spinner flask.
-
35.
Replace with 2 - 3 mL fresh Stage II medium per 6-well, or adjust to 45 mL per spinner flask.
Note: For spinner flasks, it is sufficient to perform medium changes every 3 days. For 6-well plates, the amount of Stage II medium per well can be adjusted to the frequency of medium changes, i.e., add 3 mL when changing every 3 days (e.g., over the weekend), add 2 mL when changing every other day.
Maintaining Kidney Organoid Cultures (Day 8 to Day 20+)
Between day 7 and day 9, the outlines of the kidney tubules should become visible on the surface of the embryoid bodies (here forth referred to as kidney organoids; Figure 2K) when observed by bright-field microscopy. (Troubleshooting 6).
-
36.
Keep changing Stage II medium every 2–3 days as described in steps 32–35.
The organoids will reach their optimal state of differentiation by day 12 (Figure 2L), when gene expression for specific markers of the renal cell types (i.e., podocytes, proximal tubule cells, distal tubule and collecting duct cells) can be detected by quantitative (q)PCR, and marker proteins are detectable by immunohistochemistry (Figure 3; Przepiorski et al., 2018). Validated oligonucleotide primers and antibodies recommended for determining optimal organoid maturity are listed in the Key Resources Table. The organoids maintain expression of these gene and protein markers through to approximately day 20. We recommend performing assays, such as drug treatments, during this period (Digby et al., 2020).
Pause Point: For gene expression analysis, wash organoids once in 1× DPBS and then transfer into TRIzol reagent. Store at −80°C until RNA isolation, cDNA synthesis, and qPCR analysis. For detailed protocols and expected results, please refer to our previous publications (Przepiorski et al., 2018; Digby et al., 2020). For immunohistochemistry, fix whole organoids in 1 mL 4% Paraformaldehyde for 20 min at room temperature (15°C–25°C), then wash once with 1× DPBS and store at 4°C until further processing (for up to 2 weeks).
Figure 3.

Expected Outcomes
(A) Yield of a single assay. Shown are day 12 organoids distributed into 24 wells.
(B) Single 24-well of the same assay.
(C) High efficiency of tubule formation, shown are day 12 organoids.
(D) Confocal images of immunohistological stainings on sections of day 12 organoids (derived from the MANZ-2-2 iPSC line) showing WT1+ podocytes, EPCAM+ or HNF1B+ tubular epithelia, LRP2+ or CUBN+ proximal tubules, and CHD1+ distal tubules and connecting/collecting duct epithelia. Scale bars, 0.5 cm (B); 1 mm (C); 100 μm (D).
Paraffin Embedding and Microtome Sectioning
For histological analyses, the organoids need to be embedded into paraffin and sectioned on a microtome. This work can be performed on a regular laboratory work bench. We recommend wearing personal protective equipment including safety glasses, lab coat, and gloves.
Note: A minimum of 20 organoids is advisable for paraffin processing.
-
37.
Transfer organoids into a plastic cryomold (using a 1 mL tip-cut pipette or transfer pipette to avoid damaging the organoids) then carefully take off all of the DPBS with a 200 μL pipette.
-
38.
Melt agarose/agar embedding medium in the microwave on low power (∼20 s), then slowly fill up the mold with the warm medium, avoiding bubbles.
-
39.
Leave embedding medium to solidify at room temperature (15°C–25°C) for 10–15 min, then carefully remove the agarose/agar block from the cryomold and transfer into ∼5 mL 70% ethanol in a 15 mL centrifuge tube (or 50 mL tube if using larger cryomolds). Leave at room temperature for 30 min, then transfer to 4°C and leave for 16–20 h.
-
40.
Melt paraffin in a 65°C oven (takes several hours depending the size of container). Perform a dehydration series using ∼5 mL (∼10× the volume of the size of the block): 90% ethanol, 95% ethanol, two changes of 100% ethanol, 50% ethanol : 50% xylene, 100% xylene. Leave for 1 h per step on an orbital shaker or rotator at room temperature. Finish with 50% xylene:50% molten paraffin (add liquid paraffin to xylene), incubate in the 65°C oven for 16–20 h.
-
41.
Replace with 100% paraffin, then leave to infiltrate for ≥24 h with at least three more paraffin changes (∼10× the volume of the size of the block).
-
42.
Use an embedding station to transfer the agarose/agar block into a paraffin-filled mold and attach the block to a tissue embedding cassette. Leave paraffin to solidify for at least 30 min.
-
43.
Heat a water bath to 47°C. Remove paraffin block from the mold.
-
44.
Trim the block, then cut 6 μm-thin sections on a microtome. Place paraffin ribbons to float (and flatten) on the water bath surface.
-
45.
Collect sections on Superfrost Plus microscope slides and leave to dry in a slide rack at room temperature for 16–20 h.
Alternatives: Embedding into Optimal Cutting Temperature (OCT) compound followed by cryosectioning is an option for histology. We prefer paraffin-sectioning over cryosectioning for better morphological preservation of the tissue. We do not observe reduced antigenicity of proteins in paraffin-embedded organoids.
Immunohistochemistry of Kidney Organoids
We routinely perform fluorescent immunostainings on paraffin sections of the organoids to analyze expression patterns of kidney markers. During the following steps, avoid complete drying of the sections and do not touch them. This work can be performed on a regular laboratory work bench. We recommend wearing personal protective equipment including safety glasses, lab coat, and gloves.
-
46.
De-paraffinize sections in two changes of xylene, for a minimum of 10 min each (or overnight, 16–20 h), then rinse 3× with 1× TBS plus 0.1% Triton X-100 (TBST). This can be done by either immersing the slides into a slide-container (e.g., Coplin staining jar), or by gently pipetting TBST onto slides that are placed horizontally on a rack. Either way, ensure that the sections are covered with TBST for the entire incubation time.
Optional: Antigen retrieval. Pre-heat water bath to 95°C. Fill a slide jar with Citrate buffer and place into the heating water bath. Once it has reached 95°C, transfer slides into the jar and incubate for 30 min. Following the incubation leave the slide jar to cool on the bench for another 30 min. After this incubation, take out the slides and rinse them 3× with 1× TBST. We perform this type of antigen retrieval by default. However, one should be aware that some antibodies may not be compatible with this procedure.
-
47.
Prepare the blocking solution.
-
48.
Tilt the slides and tap off the TBST onto a tissue.
-
49.
While sections are still slightly wet, draw circles around them using a PAP pen to create a hydrophobic barrier. This minimizes the use of antibody solutions and allows multiple antibody stainings to be performed on serial sections on the same slide.
-
50.
Add a small droplet (∼50 μL) of blocking solution onto the sections (within the hydrophobic circle) then incubate for ≥ 1 h at room temperature (15°C–25°C) in a humid chamber.
-
51.
Prepare primary antibody dilutions (see Key Resources Table and Przepiorski et al., 2018; Digby et al., 2020) in blocking solution.
-
52.
Tap off blocking solution and add antibody dilutions directly onto the sections (depending the size of the hydrophobic circle, 20–50 μL per section is usually enough). Incubate for 16–20 h at 4°C in a humid chamber.
-
53.
Wash 3× 15 min with 1× TBST.
Note: If using primary antibodies or lectins that are conjugated to fluorescent dyes, perform these washes protected from light.
-
54.
Prepare fluorescently labeled antibodies in 1× TBST and drop onto the sections. Incubate for 2 h at room temperature in a humid chamber protected from light.
-
55.
Wash 3× 15 min with 1× TBST, protected from light.
-
56.
Dilute Hoechst 33258 1:2,000 for a final concentration of 10 μM in 1× TBST. Drop onto the sections and incubate for 5 min, then briefly rinse with 1× TBST.
-
57.
Mount slides using Prolong Diamond mounting medium then place a coverslip onto the sections, avoiding air bubbles.
-
58.
Leave slides to dry for approximately 30 min, then seal with nail polish. Store at 4°C protected from light. Fluorescent signals are stable for at least 2 weeks.
-
59.
Image immunolabeled organoid sections by fluorescent microscopy (Figure 3D). We use a Zeiss LSM710 confocal microscope.
Expected Outcomes
This protocol produces kidney organoids within less than 2 weeks. An assay starting from a 100 mm culture dish of hPSCs typically yields 500–1,000 organoids (Figures 3A and 3B, showing a highly efficient assay of >1,000 day 12 organoids distributed into ten wells of a 24-well plate for compound testing). Organoid efficiency, i.e., the number of organoids that contain nephron-like structures within one assay, is typically >90% of the total number of organoids (Figure 3C; Troubleshooting 1). Our protocol has successfully been applied to >30 human induced pluripotent stem cell and embryonic stem cell lines, including isogenic lines. Commercially available and published hPSC lines are listed in the Key Resources Table. Organoid production with this protocol is very robust, i.e., no adjustments in CHIR99021 concentration, media composition, or treatment periods are required to achieve the above-mentioned high efficiency of organoid formation.
Immunostainings on paraffin sections confirm the presence of the main kidney tissues in the organoids, i.e., nephrons and their sub-segmentation into podocyte clusters (resembling primitive glomeruli), proximal tubules, distal tubules and connecting/collecting duct epithelia. Figure 3D shows representative images of organoid sections at day 12 with WT1-labeled podocytes, EPCAM, and HNF1B-labeled tubules, CUBN- and LRP2-labeled proximal tubules and CDH1-labeled distal tubules and connective/collecting duct epithelia. For more examples of immunohistologically labeled organoids, please refer to our previous publications (Przepiorski et al., 2018; Digby et al., 2020).
Limitations
Comparative analyses with fetal human kidney tissue revealed that the maturity of our kidney organoids corresponds to the “late capillary loop” stage of first trimester human kidneys (Przepiorski et al. 2018). As such, markers of nephron segments that form at later stages of fetal kidney development are not detectable. The fetal-like state of the organoid kidney tissues may limit studies that aim to address structural aspects and functions of fully differentiated human kidneys, as well as adult-onset disease modeling. Nonetheless, our kidney organoids (and organoids with comparable maturity generated with other protocols) have been shown to recapitulate congenital kidney diseases (Forbes et al., 2018; Tanigawa et al., 2018; Combes et al., 2019; Przepiorski et al., 2018; Freedman et al., 2015) and acute kidney injury (Morizane et al., 2015; Hale et al., 2018; Digby et al., 2020). As an alternative to the fetal-like kidney tissues derived from hPSCs, organoids can be generated from adult human kidney biopsies or urine-derived epithelial cells (Schutgens et al., 2019; Jun et al., 2018). These so-called tubuloids provide a rapidly achievable and stable system that could be more suitable for personalized disease modeling of e.g., kidney malignancies. However, in contrast to hPSC-derived organoids that are comprised of multiple renal cell types and reflect the structural complexity of the kidney, tubuloids only contain epithelial cells that grow in a tubulocystic configuration, thus have their own limitations.
Extended culture (>20 days) of kidney organoids generated with our protocol may lead to expansion of non-renal cell types, including neuronal cells and myofibroblasts (Figure 4G; Troubleshooting 7), leaving the organoids less representative of healthy human kidney tissue. This limits the optimal time frame for experiments on the organoids to ∼day 10 to day 20.
Troubleshooting
Problem 1
Low-efficiency organoid formation (Figure 4A).
Potential Solution
Using bright-field microscopy, specimens without tubules are clearly distinguishable from tubule-containing organoids by day 8 or day 9 of the protocol. Low efficiency (<50%) of tubule formation is most likely due to sub-optimally maintained hPSC cultures. It is most critical that cultures are passaged regularly and before reaching 80% confluency, as high-density cultures can lead to spontaneous differentiation and low-yield organoid assays. Do not start organoid differentiation if an hPSC culture has become over-confluent. Instead, passage the cells twice, then resume with organoid differentiation. Furthermore, ensure that ROCK inhibitor is added when passaging, the split ratio is not too high (we do not recommend higher than 1:8) and the cultures have tested negative for mycoplasma.
Problem 2
Unevenly-sized embryoid bodies / organoids (Figure 4B).
Potential Solution
Avoid excessive trituration of Dispase-treated colonies (step 10), as cell aggregates smaller than ∼50 μm are less likely to form tubular organoids. On the other hand, aggregates >200 μm will likely develop into large embryoid bodies / organoids that may form apoptotic or necrotic cores (Przepiorski et al., 2018).
Problem 3
Clumping of embryoid bodies (Figures 4C−4F).
Potential Solution
Occasional merging of two or more embryoid bodies can occur during the first 48 h of the protocol (Figure 4C). This can result in large specimens (Figure 4D) that can be removed by size-filtration (steps 29–31; Figure 4E). To prevent severe clumping of embryoid bodies, ensure that the cultures are not left to sediment for more than 5 min, e.g., during half-medium change at day 2 (step 15), transfer into Stage II medium at day 3 (step 23) or medium changes at later stages of the protocol. Severe clumping will also occur if the orbital shaker or spinner flask is accidentally left switched off (Figure 4F).
Problem 4
Sub-ideal appearance of embryoid bodies.
Potential Solution
Based on our experience, embryoid bodies that appear bean-shaped, dark-colored or exhibit rough edges by day 5 to day 7 of the protocol are unlikely to develop into tubular kidney organoids. We recommend discontinuing such assays and instead starting over, using healthy, non-differentiating hPSC cultures. Non-spherical shaped embryoid bodies can be a result of differentiating hPSCs. See also Problem 1.
Problem 5
High density of embryoid bodies/organoids.
Potential Solution
The optimal number of embryoid bodies or organoids per 6-well is ≤150. If too dense, distribute into several 6-well plates or use a spinner flask, which can hold up to 6,250 organoids (Przepiorski et al., 2018). For short-term culture up to 72 h, e.g., drug treatments after day 12, ∼100 organoids can be cultured per 24-well (as shown in Figures 3A and 3B).
Problem 6
Delayed tubule formation, or inconsistent maturation of organoids generated from different (isogenic) lines.
Potential Solution
This could be due to intrinsic differences between hPSC lines. If observed consistently, refer to Phipson et al. (2019) and check organoid maturation state by qPCR or RNA sequencing.
Problem 7
Outgrowth of non-renal cells (Figure 4G).
Potential Solution
We observed that some hPSC lines trend toward forming large areas of non-renal cells, including cells of neuronal and myofibroblast appearance, when cultured >20 days. For example, the CRL1502 (clone C32) iPSC line shows a tendency toward neuronal differentiation, which is also noticeable by single-cell RNA sequencing (Wu et al., 2018). These organoids are usually large and can be removed by size-filtration (steps 29–31). Alternatively, organoid cultures could be treated with K-252a, an inhibitor of neuronal differentiation, as described in Wu et al. (2018). We also observed that more stringent hPSC culture conditions (e.g., using TeSR-E8 medium instead of mTeSR1) helped with reducing non-renal outgrowths on organoids derived from some hPSC lines.
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Veronika Sander (v.sander@auckland.ac.nz).
Materials Availability
Not applicable.
Data and Code Availability
Not applicable.
Acknowledgments
We thank G. Chang and T. Perreau for critical reading of the manuscript and L. Conrad for helpful input on the troubleshooting. This work was supported by the Health Research Council of New Zealand (17/425), Auckland Medical Research Foundation (1116018), Cystinosis Research Foundation USA, Cystinosis Research Ireland Foundation (MRCG2014-8), the National Institute of Diabetes and Digestive and Kidney Diseases (R01-DK-069403), the United States Army Medical Research and Development Command (W81XWH-17-1-0610), and Valrae Collins Philanthropic support for A.P.
Author Contributions
A.P. established the protocol, A.P. and V.S. optimized the protocol, A.E.C. validated the benefits of using low-glucose DMEM in this protocol, T.M.H., A.J.D., and N.A.H. supervised the study, V.S. wrote the manuscript and A.J.D., N.A.H., and V.S. acquired funding.
Declaration of Interests
The authors declare no competing interests.
Contributor Information
Veronika Sander, Email: v.sander@auckland.ac.nz.
Alan J. Davidson, Email: a.davidson@auckland.ac.nz.
References
- Combes A.N., Zappia L., Er P.X., Oshlack A., Little M.H. Single-cell analysis reveals congruence between kidney organoids and human fetal kidney. Genome Med. 2019;11:3. doi: 10.1186/s13073-019-0615-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Digby J.L.M., Vanichapol T., Przepiorski A., Davidson A.J., Sander V. Evaluation of cisplatin-induced injury in human kidney organoids. Am. J. Physiol. Renal Physiol. 2020;318:F971–F978. doi: 10.1152/ajprenal.00597.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forbes T.A., Howden S.E., Lawlor K., Phipson B., Maksimovic J., Hale L., Wilson S., Quinlan C., Ho G., Holman K. Patient-iPSC-derived kidney organoids show functional validation of a ciliopathic renal phenotype and reveal underlying pathogenetic mechanisms. Am. J. Hum. Genet. 2018;102:816–831. doi: 10.1016/j.ajhg.2018.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freedman B.S., Brooks C.R., Lam A.Q., Fu H., Morizane R., Agrawal V., Saad A.F., Li M.K., Hughes M.R., Werff R.V. Modelling kidney disease with CRISPR-mutant kidney organoids derived from human pluripotent epiblast spheroids. Nat. Commun. 2015;6:8715. doi: 10.1038/ncomms9715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hale L.J., Howden S.E., Phipson B., Lonsdale A., Er P.X., Ghobrial I., Hosawi S., Wilson S., Lawlor K.T., Khan S. 3D organoid-derived human glomeruli for personalised podocyte disease modelling and drug screening. Nat. Commun. 2018;9:5167. doi: 10.1038/s41467-018-07594-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollywood J.A., Przepiorski A., D'Souza R.F., Sreebhavan S., Wolvetang E.J., Harrison P.T., Davidson A.J., Holm T.M. Use of human induced pluripotent stem cells and kidney organoids to develop a cysteamine/mtor inhibition combination therapy for cystinosis. J. Am. Soc. Nephrol. 2020;31:962–982. doi: 10.1681/ASN.2019070712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howden S.E., Thomson J.A., Little M.H. Simultaneous reprogramming and gene editing of human fibroblasts. Nat. Protoc. 2018;13:875–898. doi: 10.1038/nprot.2018.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jun D., Kim S.Y., Na J.C., Lee H.H., Kim J., Yoon Y.E., Hong S.J., Han W.K. Tubular organotypic culture model of human kidney. PLoS One. 2018;13:e0206447. doi: 10.1371/journal.pone.0206447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S.V., Er P.X., Lawlor K.T., Motazedian A., Scurr M., Ghobrial I., Combes A.N., Zappia L., Oshlack A., Stanley E.G., Little M.H. Kidney micro-organoids in suspension culture as a scalable source of human pluripotent stem cell-derived kidney cells. Development. 2019;146:dev172361. doi: 10.1242/dev.172361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morizane R., Lam A.Q., Freedman B.S., Kishi S., Valerius M.T., Bonventre J.V. Nephron organoids derived from human pluripotent stem cells model kidney development and injury. Nat. Biotechnol. 2015;33:1193–1200. doi: 10.1038/nbt.3392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng E.S., Davis R., Stanley E.G., Elefanty A.G. A protocol describing the use of a recombinant protein-based, animal product-free medium (APEL) for human embryonic stem cell differentiation as spin embryoid bodies. Nat. Protoc. 2008;3:768–776. doi: 10.1038/nprot.2008.42. [DOI] [PubMed] [Google Scholar]
- Oh J.K., Przepiorski A., Chang H.-H., Dodd R.C., Sander V., Sorrenson B., Shih J.-H., Hollywood J.A., De Zoysa J., Shepherd P.R. Derivation of induced pluripotent stem cell lines from New Zealand donors. J. R. Soc. New Zealand. 2020 doi: 10.1080/03036758.2020.1830808. [DOI] [Google Scholar]
- Orlova V.V., van den Hil F.E., Petrus-Reurer S., Drabsch Y., Ten Dijke P., Mummery C.L. Generation, expansion and functional analysis of endothelial cells and pericytes derived from human pluripotent stem cells. Nat. Protoc. 2014;9:1514–1531. doi: 10.1038/nprot.2014.102. [DOI] [PubMed] [Google Scholar]
- Phipson B., Er P.X., Combes A.N., Forbes T.A., Howden S.E., Zappia L., Yen H.-J., Lawlor K.T., Hale L.J., Sun J. Evaluation of variability in human kidney organoids. Nat. Methods. 2019;16:79–87. doi: 10.1038/s41592-018-0253-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Przepiorski A., Sander V., Tran T., Hollywood J.A., Sorrenson B., Shih J.-H., Wolvetang E.J., McMahon A.P., Holm T.M., Davidson A.J. A simple bioreactor-based method to generate kidney organoids from pluripotent stem cells. Stem Cell Reports. 2018;11:470–484. doi: 10.1016/j.stemcr.2018.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schutgens F., Rookmaaker M.B., Margaritis T., Rios A., Ammerlaan C., Jansen J., Gijzen L., Vormann M., Vonk A., Viveen M. Tubuloids derived from human adult kidney and urine for personalized disease modeling. Nat. Biotechnol. 2019;37:303–313. doi: 10.1038/s41587-019-0048-8. [DOI] [PubMed] [Google Scholar]
- Taguchi A., Kaku Y., Ohmori T., Sharmin S., Ogawa M., Sasaki H., Nishinakamura R. Redefining the in vivo origin of metanephric nephron progenitors enables generation of complex kidney structures from pluripotent stem cells. Cell Stem Cell. 2014;14:53–67. doi: 10.1016/j.stem.2013.11.010. [DOI] [PubMed] [Google Scholar]
- Taguchi A., Nishinakamura R. Higher-order kidney organogenesis from pluripotent stem cells. Cell Stem Cell. 2017;21:730–746.e6. doi: 10.1016/j.stem.2017.10.011. [DOI] [PubMed] [Google Scholar]
- Takasato M., Er P.X., Chiu H.S., Maier B., Baillie G.J., Ferguson C., Parton R.G., Wolvetang E.J., Roost M.S., de Lopes S.M., Little M.H. Kidney organoids from human iPS cells contain multiple lineages and model human nephrogenesis. Nature. 2015;526 doi: 10.1038/nature15695. nature15695. [DOI] [PubMed] [Google Scholar]
- Tanigawa S., Islam M., Sharmin S., Naganuma H., Yoshimura Y., Haque F., Era T., Nakazato H., Nakanishi K., Sakuma T. Organoids from nephrotic disease-derived iPSCs identify impaired NEPHRIN localization and slit diaphragm formation in kidney podocytes. Stem Cell Reports. 2018;11:727–740. doi: 10.1016/j.stemcr.2018.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran T., Lindström N.O., Ransick A., Brandine G.D.S., Guo Q., Kim A.D., Der B., Peti-Peterdi J., Smith A.D., Thornton M. In vivo developmental trajectories of human podocyte inform in vitro differentiation of pluripotent stem cell-derived podocytes. Dev. Cell. 2019;50:102–116.e6. doi: 10.1016/j.devcel.2019.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanslambrouck J.M., Wilson S.B., Tan K.S., Soo J.Y.-C., Scurr M., Spijker H.S., Starks L.T., Neilson A., Cui X., Jain S. A toolbox to characterize human induced pluripotent stem cell–derived kidney cell types and organoids. J. Am. Soc. Nephrol. 2019;30:1811–1823. doi: 10.1681/ASN.2019030303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren L., Manos P.D., Ahfeldt T., Loh Y.-H., Li H., Lau F., Ebina W., Mandal P.K., Smith Z.D., Meissner A. Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell Stem Cell. 2010;7:618–630. doi: 10.1016/j.stem.2010.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H., Uchimura K., Donnelly E.L., Kirita Y., Morris S.A., Humphreys B.D. Comparative analysis and refinement of human PSC-derived kidney organoid differentiation with single-cell transcriptomics. Cell Stem Cell. 2018;23:869–881.e8. doi: 10.1016/j.stem.2018.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.