

Summary
Aquaporins (AQPs) are membrane channel proteins that facilitate the movement of water down osmotic gradients across biological membranes. This protocol allows measurements of AQP-mediated water transport across the plasma membrane of live mammalian cells. Calcein is a fluorescent dye that is quenched in a concentration-dependent manner. Therefore, on short timescales, its concentration-dependent fluorescence can be used as a probe of cell volume, and therefore a probe of water transport into or out of cells.
For complete details on the use and execution of this protocol, please refer to Kitchen et al. (2020) and Kitchen and Conner (2015). For the underlying methodology development, please refer to Fenton et al. (2010) and Solenov et al. (2004).
Graphical Abstract
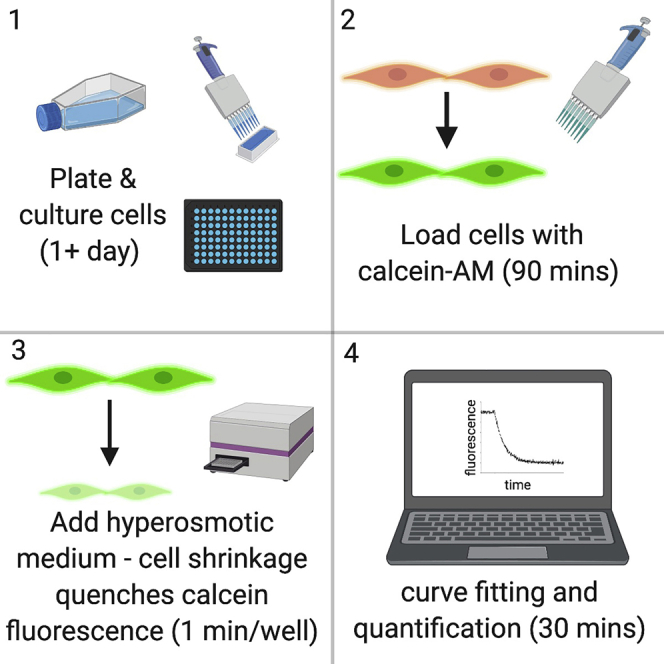
Highlights
-
•
Calcein fluorescence quenching can be used to monitor cell volume
-
•
This can be used to quantify plasma membrane water permeability
-
•
The protocol is suitable for all adherent cell types
Aquaporins (AQPs) are membrane channel proteins that facilitate the movement of water down osmotic gradients across biological membranes. This protocol allows measurements of AQP-mediated water transport across the plasma membrane of live mammalian cells. Calcein is a fluorescent dye that is quenched in a concentration-dependent manner. Therefore, on short timescales, its concentration-dependent fluorescence can be used as a probe of cell volume, and therefore a probe of water transport into or out of cells.
Before You Begin
Plate Adherent Cells
Timing: 1+ days
This protocol is intended to be applicable to any adherent cell type. We have successfully used it in primary astrocytes, Madin-Darby canine kidney (MDCK) cells, human embryonic kidney 293 (HEK293) cells and breast cancer cell lines (MDA-MB-231 and MCF7). In principle any adherent cell type can be used, although if using a different cell type to those we have used, the reader should pay special attention to the notes about cell adherence and calcein sequestration. The generic culture protocols below may need to be adapted to the cell type of interest.
-
1.
Collect cells of interest using standard techniques appropriate to that cell type (e.g., trypsin, Accutase, Versene), and count.
-
2.
Plate cells in a tissue-culture-treated, sterile, clear-bottomed, black-walled 96-well plate, aiming for a cell density of approximately 90% at the time of the experiment. If the cell type of interest requires it, the plate should be pre-coated with a substrate to facilitate adherence (e.g., poly-lysine, gelatin, laminin, etc). We recommend at least 24 h between plating and permeability measurements, both to allow the cells time to adhere, and to allow the re-expression of membrane proteins (including aquaporins) that may have been degraded by the cell collection enzyme(s).
Note: For overexpression experiments, we strongly recommend using a stably transfected subclone, not transiently transfected cells. We have successfully used this approach to measure subtle differences in water permeability between multiple mutants of human AQP4 expressed in MDCK cells (Kitchen and Conner, 2015), and differences in water and glycerol permeability in breast cancer cell lines stably transfected with an anti-AQP3 small hairpin RNA (Arif et al., 2018). Following transient transfection, there will be a heterogeneous population of cells, some with very high expression of the transfected construct, and some with no expression at all. This plate reader-based method will average over all cells – to avoid spurious results it is therefore important that the cell population is relatively homogeneous in terms of cell-to-cell variability in water permeability. Similar considerations apply to experiments utilizing RNA interference for knockdowns.
CRITICAL: Care should be taken to ensure even seeding of cells within wells and equal numbers of cells between wells. Regular movement of the plate in a “cross” motion (i.e., back-and-forth and left-to-right) can help to ensure even distribution of cells in individual wells. We recommend use of a multichannel pipette for seeding, to minimize the settling of suspended cells during seeding. Where this is not available, the tube or vessel containing the cells should be inverted regularly to ensure a homogeneous suspension.
Prepare Stock Solutions
-
3.
Prepare a 5 mM calcein acetoxymethyl ester (calcein-AM) stock from lyophilized powder. Centrifuge the tube before opening to ensure that all powder is at the bottom. Add 201.0 μL dimethylsulfoxide (DMSO) per 1 mg of calcein-AM (for practicality, this could be rounded down to 200 μL). We typically use 50 μg microtubes (ThermoFisher C3100MP), and we dissolve the powder in 10.0 μL DMSO. Each 50 μg tube provides just enough calcein-AM to load a full 96-well plate, so there is no need for aliquoting. If the calcein-AM is in a transparent tube, wrap it in aluminum foil to protect from light. Store at −20°C until use. The calcein-AM DMSO stock can be refrozen and reused 1–2 times after thawing. If a large batch is prepared, aliquot to avoid repeated freeze-thaw cycles. Alternatively, some suppliers offer a pre-dissolved DMSO stock, although this is typically more expensive.
-
4.
Prepare a 175 mM stock of probenecid. Dissolve 50 mg of probenecid powder in 1 mL of 200 mM NaOH. Dissolution may take several minutes and is helped by vortex mixing. Store at −20°C in 100 μL aliquots. Probenecid is an organic anion transporter (OAT) and ATP-binding cassette (ABC) transporter inhibitor, which will prevent calcein from leaving the cell via these transporters.
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, Peptides, and Recombinant Proteins | ||
| Probenecid | Sigma-Aldrich | P8761 |
| Calcein-AM | ThermoFisher | C3100MP |
| D-mannitol | Sigma-Aldrich | M4125 |
| DMEM HEPES formulation | Sigma-Aldrich | D1152 |
| Other | ||
| Clear-bottomed, black-walled, flat, sterile, cell culture treated microplates | Corning | 3904 |
| Fluorescence plate reader | BMG Labtech | FLUOstar Omega |
| Freezing point depression osmometer | Löser | Micro-Digital i-Osmometer Basic 7i |
| Coulter counter | Beckman Coulter | Z1 |
| 0.2 μm syringe filter | Any supplier | N/A |
| Experimental Models: Cell Lines | ||
| Madin-Darby canine kidney (MDCK) cells | ATCC | CRL-2936 |
| HEK293 cells | ATCC | CRL-1573 |
| Primary astrocytes (rat) | ThermoFisher | N7745100 |
| Primary astrocytes (human) | ScienCell | 1800 |
| Breast cancer cell lines (MDA-MB-231) | ATCC | HTB-26 |
| Breast cancer cell lines (MCF7) | ATCC | HTB-22 |
Materials and Equipment
Alternatives: In the Key Resources Table, we have listed the instrument makes and models used in our lab. However, these specific models are not crucial for success of the protocol. Any Coulter counter and freezing point depression osmometer will suffice, as well as any fluorescence plate reader with a green filter (usually ex:485/em:525), internal temperature control, and an injector. This protocol is, in principle, applicable for any adherent cell type (primary cells or cell lines). In the Key Resources Table, we have listed some of the cell types we have used successfully – this is not intended as an exhaustive list and we encourage the reader to experiment with whatever cell type(s) they are interested in.
Step-By-Step Method Details
Load Cells with Calcein
In this step, cells are loaded with calcein-AM, which they will metabolize to calcein. Calcein fluorescence is quenched by protein in a concentration-dependent manner and can therefore be used as a probe of protein concentration. On short timescales during which the number of intracellular protein molecules is constant, calcein fluorescence can therefore be used as a probe of cell volume. At higher concentrations (in the mM range), calcein is known to self-quench. At the concentrations used here (μM loading concentrations leading to nM intracellular free calcein concentrations (Solenov et al., 2004)), self-quenching does not occur, and the observed fluorescence changes are caused by protein-dependent quenching.
-
1.
Prepare calcein-AM loading medium by taking pre-warmed full cell culture medium and adding probenecid to a final concentration of 1 mM (from a 175 mM stock in 200 mM NaOH), and calcein-AM to a final concentration of 5 μM (from a 5 mM DMSO stock). The non-fluorescent, membrane-permeable calcein-AM will be metabolized to fluorescent non-membrane-permeable calcein by intracellular esterases.
-
2.
Remove all culture medium from cells and add 100 μL of calcein-AM loading medium per well. For speed and reproducibility, we recommend use of a multichannel pipette where possible. Return cells to the incubator at 37°C for 90 min. During this 90 min incubation, switch on the plate reader and set the internal temperature to 37°C. How soon into the 90 min this needs to be done will depend on the specific plate-reader model, and how quickly it heats up. The experimenter should check this beforehand.
Note: In some cell types, (e.g., some neuronal cells), calcein can be sequestered in intracellular organelles (Crowe et al., 1995; Kovacs et al., 2011). This may lead to artifacts in the measurements described here. This can be checked by observing calcein-loaded cells under a fluorescence microscope; in order for this protocol to be used, the calcein fluorescence should be uniformly distributed through the cytoplasm.
Wash Cells and Prepare Hypertonic Medium
In this step, the calcein-AM loading medium is removed, the cells are washed to remove residual calcein-AM, and the hypertonic medium used to induce cell shrinkage is prepared.
-
3.
Prepare washing medium by taking pre-warmed HEPES-buffered full cell culture medium and adding probenecid to a final concentration of 1 mM. HEPES-buffered variants of most cell culture medium formulations are available from a variety of suppliers. For example, for cells cultured in DMEM, we would use HEPES-buffered DMEM (e.g., Sigma-Aldrich D1152). A full 96-well plate will require 275 × 96 6 = 26,400 μL (26.4 mL). This can be rounded up to 30 mL for simplicity and to allow for pipetting losses. Wash cells twice by removing all medium and replacing with 100 μL of washing medium. On the third wash, replace with 75 μL of washing medium. Return cells to the incubator for at least 10 min to equilibrate with the new medium. An alternative to using HEPES-buffered medium in this step is to use standard bicarbonate-buffered medium and a plate reader with environmental gas control set to 5% CO2. Bicarbonate buffer requires CO2 control, whereas HEPES buffer does not.
-
4.
Prepare hypertonic D-mannitol medium by dissolving D-mannitol powder directly in washing medium (full HEPES-buffered culture medium + probenecid) to give a final concentration of 400 mM. Sterilize using a 0.2 μm filter. We recommend preparing at least 10 mL more than is required, to prevent the formation of bubbles in the injector tubing toward the end of the experiment. For example, 1 full 96-well plate will require 75 × 96 = 7,200 μL (7.2 mL), therefore 17.2 mL should be prepared. This could be rounded up to 20 mL for simplicity. The D-mannitol concentration (and the osmotic gradient used in the experiment) can be checked using a freezing point depression osmometer. The D-mannitol medium should have an osmolality 400 mOsm higher than the mannitol-free washing medium, which will give an osmotic gradient of 200 mOsm when mixed 1:1 with mannitol-free medium in the next step of the protocol.
-
5.
Prime the plate-reader injector system with the hypertonic D-mannitol medium. To maintain the temperature of this solution, it can be stored in a heating block or water bath set to 37°C or placed inside the plate-reader housing (depending on the model).
Alternatives: The experimenter should consider whether D-mannitol is an appropriate non-permeant solute for their cell type and experiment. There is evidence that some membrane channels, such as mammalian AQP9 (Tsukaguchi et al., 1998) are mannitol permeable. If the cells of interest may express such a channel, alternative solutes should be considered, e.g., sucrose.
Note: Other similar protocols recommend the use of phenol red-free cell culture medium for experiments with calcein-AM. We find that the absorbance and fluorescence of phenol red in standard cell culture medium at the wavelengths in question (ex: 485 nm, em: 525 nm) is negligible. We therefore use standard culture medium with phenol red. If the experimenter wishes to convince themselves of this, they can use a phenol red-free washing medium and compare the fluorescence intensity between wells of calcein-AM-loaded cells in standard medium and phenol red-free medium. In our hands, using bottom optics, they are indistinguishable.
Note: The incubation times for steps with probenecid are 90 min for the calcein loading, and 10 min for the post-loading equilibration.
Measure Water Permeability
In this step, calcein fluorescence is measured continuously following injection of hypertonic medium. Cell shrinkage will lead to calcein fluorescence quenching.
-
6.
Remove the cells from the incubator, remove the lid from the cell culture plate, and insert the plate into the pre-warmed plate reader. Set the plate reader to read each well for 5 s, followed by injection of 75 μL hypertonic D-mannitol medium at a speed of 300 μL/s (see Troubleshooting section for further discussion of injection speed), and a further 50 s of continuous reading, with a read interval of 50 ms using bottom optics. If the cells of interest have especially high or especially low water permeability, the length of the second read step can be increased or decreased from 50 s.
Note: The plate can be left to run at this point. A full 96-well plate will take 96 x 55 s (88 min).
Expected Outcomes
The outcome of the experiment will be a set of cell shrinkage curves, and either quantified relative changes in plasma membrane water permeability, or absolute water permeability, under the experimental conditions. We have previously used this technique to study changes in AQP4-dependent astrocyte water permeability under hypoxia (Kitchen et al., 2020), changes in breast cancer cell water permeability following knockdown of AQP3 (Arif et al., 2018), and changes in AQP4 water permeability following mutagenesis of selectivity filter amino acid residues and stable transfection of mutant constructs in MDCK cells (Kitchen and Conner, 2015). Representative data from astrocytes cultured under hypoxia, and MDCK cells with overexpressed AQP4 are shown in Figure 1, demonstrating that hypoxic astrocytes have elevated membrane water permeability, and that overexpression of AQP4 increases the membrane water permeability of MDCK cells.
Figure 1.

Calcein Fluorescence Quenching in Live Mammalian Cells
(A) Calcein fluorescence quenching in primary astrocytes cultured under normoxia or hypoxia for 6 h shows a hypoxia-induced increase in membrane water permeability. Figure reprinted with permission from Kitchen et al., 2020.
(B) Overexpression of AQP4 increases plasma membrane water permeability of MDCK cells by approximately sevenfold. Figure reprinted with permission from Kitchen and Conner, 2015.
Quantification and Statistical Analysis
In this step, shrinkage rate constants will be calculated. These can be used to calculate relative changes in membrane water permeability (Kitchen et al., 2020), or to estimate the membrane water permeability (Pf).
To measure relative changes in membrane permeability:
-
1.
Subtract background (average fluorescence from wells of calcein-free cells) from all fluorescence readings.
-
2.
For each well of the plate, normalize the background-subtracted fluorescence readings to the average of the 5 s pre-injection fluorescence. At this point, the experimenter should check that the pre-injection fluorescence is stable and does not show any time-dependent trend.
-
3.
Fit curves of the form y = 1 – Ce-kt to the normalized post-injection data, with the time of the injection set to t=0. The rate constant, k, is proportional to plasma membrane water permeability. We typically report these data normalized to the permeability of control cells, with the exact nature of the control depending on the scientific question (e.g., for inhibitor studies, this might be a DMSO vehicle control).
Note: If the experimenter wishes to calculate the osmotic permeability of the membrane (Pf), a calibration curve is required to convert relative fluorescence data to relative volume data (Fenton et al., 2010). We note that the Pf calculation requires an estimate of the surface area/volume ratio of the cell type of interest, which can be difficult to measure due to membrane ruffling and folding. The geometrical surface area (i.e., the expected surface area based on a sphere with the same volume as the cell) is an underestimate and can be incorrect by at least an order of magnitude. For example, in Xenopus oocytes, the ratio between the actual membrane surface area and the geometrical surface area (i.e., A = π1/3(6V)2/3), has been estimated as 9 (Zampighi et al., 1995). However, estimates for this ratio do not exist in the literature for the vast majority of mammalian cell types. We therefore caution that any Pf measured in this way should be considered a crude estimate, unless a robust measurement of the membrane surface area can be made.
In the following steps, a Coulter counter is used to measure cell volume, and a calcein fluorescence standard curve is constructed to convert relative fluorescence values to relative cell volumes.
-
4.
Prepare 2 96-well plates of cells (aiming for 90% confluence at the time of the experiment, as above), and load one with calcein-AM following the instructions above, including the washing step.
-
5.
Prepare cell culture medium containing concentrations of D-mannitol from 0 to 500 mM, in steps of 25 mM. This can be done by making a 500 mM stock and mixing with mannitol-free culture medium in the appropriate ratios. The volume needed depends on the minimum volume required in the electrolyte chamber of the Coulter counter.
-
6.
Where available, the osmolality of these standards should be checked using a freezing point depression osmometer.
-
7.
Collect the cells from the first (calcein-AM-free) plate by whatever method is appropriate for the cell type of interest (e.g., trypsin, Accutase, Versene) and fully dissociate the cells. Complete dissociation can be checked by phase contrast microscopy.
-
8.
Centrifuge cells at the standard speed used for the cell type of interest, resuspend in 6 mL of standard culture medium and thoroughly resuspend to achieve a single cell suspension. Make 500 μL aliquots of cells in 11 tubes labeled 0,25,50…250.
-
9.
Prepare the Coulter counter. Add the 0 mM-labeled D-mannitol medium to the electrolyte feed chamber of the Coulter counter. Add 500 μL of 0 mM-labeled D-mannitol medium to the tube labeled “0.” Mix by inverting, incubate for 60 s, then run these cells through the Coulter counter and record the mean cell volume. Upper and lower size limits can be set for the mean cell size calculation, to avoid counting dead cell debris or cell clumps.
-
10.
Add 25 mM D-mannitol medium to the electrolyte feed chamber of the Coulter counter. Add 500 μL of 50 mM D-mannitol medium to the tube labeled “25” (note the different concentrations – the medium added to the electrolyte chamber has half the D-mannitol concentration of that added to the cells), mix by inverting, incubate for 60 s, and run these cells through the Coulter counter, again recording the mean cell volume.
-
11.
Repeat step 10 for all D-mannitol concentrations.
-
12.
To avoid bacterial or fungal growth on remnants of culture medium, the Coulter counter should be thoroughly flushed and cleaned after these measurements.
-
13.
Take the second (calcein-AM-loaded) plate from the incubator. To the first three wells, add 75 μL of 0 mM D-mannitol medium. Mix by very gentle pipetting. After 60 s, measure calcein-AM fluorescence from these three wells in the plate reader.
-
14.
To the second three wells, add 75 μL of 50 mM D-mannitol medium, and repeat the step above (step 13).
-
15.
Repeat step 14 for all D-mannitol concentrations.
-
16.
The experimenter should now have two datasets: cell volume as a function of extracellular osmolality, and calcein-AM fluorescence as a function of extracellular osmolality. As before, subtract the background from the fluorescence data. Normalize the volume and fluorescence data to 0 mM D-mannitol, i.e., calculate V/V0 and F/F0. Plot V/V0 as a function of F/F0 (i.e., V on the y axis and F on the x axis). Fit a function of the form y = A + Be-C(x-D), where A-D are the fit parameters (Fenton et al., 2010). For full details of the physical interpretation of these parameters, the interested reader can refer to equation 3 in Fenton et al. This will give an equation to convert relative fluorescence to relative volume. We include representative datasets from MDCK cells for the reader to practice this procedure (Supplemental File Sets; using these datasets, we get, to 3 significant figures, A = 0.646, B = 0.0118, C = −11.3, D = 0.701, R2 = 0.997). The experimenter can now convert experimental fluorescence data to volume data, fit a function of the form y = 1 – Ce-kt to the volume time series and use the equation Pf = kCV0/ΔΠAVw where k and C are the rate constant and amplitude fit parameters, V0 is the cell volume at 0 mM D-mannitol, taken from the Coulter counter data, ΔΠ is the osmotic gradient, A is the cell surface area (see discussion above), and Vw is the partial molar volume of water (1.8 × 10−5 m3/mol).
Limitations
The cell types that can be used in this protocol are limited to those that adhere well to culture vessels and can withstand injection without being dislodged, and those in which calcein is homogeneously distributed through the cytoplasm and not sequestered in organelles.
Accurate quantification of the water permeability of the plasma membrane (Pf) requires accurate quantification of the cell surface area; in the absence of an accurate area measurement, the calculated Pf values should be considered rough estimates (order of magnitude), and should not be compared between cell types with potentially different surface area to volume ratios. In addition, different cell types are likely to have different background (i.e., aquaporin-independent) membrane water permeabilities due to differences in membrane lipid and sterol composition, so we urge caution in the direct comparison of results between different cell types.
Troubleshooting
Problem 1
Discontinuous changes in fluorescence intensity following injection (step 6 of the Step-By-Step Method Details).
Potential Solution
We find that this is usually due to an injection speed that is too fast. This leads to either (a) dislodging of cells from the plate surface, or (b) generation of bubbles that can scatter both excitation and emission light.
In some cases, (a) can be solved by optimizing cell density. In particular, we find that the commonly used human embryonic kidney (HEK)-293 cell line is especially challenging due to its poor adhesion to plastic, but a reduced cell density (50%–70% at the time of the experiment) can help to prevent cell loss upon injection. Coating of the culture vessel with extracellular matrix (ECM) proteins can also help to improve cell adhesion. Only when cell culture optimization does not solve the problem would we recommend reducing the injection speed, as a slower injection speed will lead to slower mixing, and the protocol assumes a near-instantaneous change in extracellular osmolality.
(b) can be greatly improved by switching from top optics to bottom optics, where this is available. Cleaning the injector tubing with 70% isopropanol, following by thorough flushing with sterile distilled water can help to reduce bubbles if dirty tubing is the cause. The experimenter should also check that the injector inlet tubing is placed as close to the bottom of the feed solution as possible to prevent intake of air into the tubing. Again, reducing injection speed should be a last resort.
Problem 2
Large variation in fluorescence intensity between wells (step 6 of the Step-By-Step Method Details).
Potential Solution
This is usually due to differences in cell density across the plate and/or inhomogeneous distribution of cells within individual wells. Cells should be seeded at equal density across the plate. Cells should be thoroughly resuspended before plating to ensure that cells grown in monolayers are broken down to a single cell suspension. If an entire 96-well (or 384-well) plate is to be seeded, a multichannel pipette should be used where available, to minimize cells settling out of suspension. Where a multichannel pipette is not available, the tube containing the cells should be inverted several times at regular intervals during seeding to maintain a homogenous cell density.
Many modern plate readers have a “well scan” mode, which allows qualitative and quantitative analysis of within-well and between-well heterogeneity. This can be used along with visual inspection by phase contrast or epifluorescence microscopy to ensure even seeding of cells. Wells with inhomogeneous cell populations can be excluded from subsequent analysis. The well scan can also be repeated after the experiment to check for dislodging of cells during medium injection.
Problem 3
Time-dependent trends in fluorescence intensity before injection (step 6 of the Step-By-Step Method Details).
Potential Solution
This may be due to calcein leakage from cells. Ensure that probenecid is added to culture media.
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Philip Kitchen (p.kitchen1@aston.ac.uk).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
This study did not generate/analyze datasets/code.
Acknowledgments
We acknowledge the UK Biotechnology & Biosciences Research Council (to R.M.B., A.C.C., and P.K. through BB/P025927/1); the Swedish Research Council (to S.T.-H. through 2013-05945); the Crafoord Foundation (to S.T.-H. through 20140811 and 20180916); the Magnus Bergvall Foundation (to S.T.-H. through 2015-01534); M.A.-A. is supported by a studentship co-funded by Aston University and the UK Engineering and Physical Sciences Research Council (EP/R512889/1) to R.M.B.; P.K. is the recipient of an Aston University 50th Anniversary Prize Fellowship: M.M.S. is supported through funding from Biogen to Prof. Tomas Kirchhausen. The graphical abstract was created with Biorender.com.
Author Contributions
R.M.B., A.C.C., P.K., and M.M.S. conceived the study and devised the experimental strategy with M.A.-A., P.K. and M.A.-A. performed the experiments, and with M.M.S., A.C.C. and R.M.B., analyzed the data. All authors wrote the manuscript and approved the final version.
Declaration of Interests
The authors declare no competing interests.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.xpro.2020.100157.
Contributor Information
Philip Kitchen, Email: p.kitchen1@aston.ac.uk.
Roslyn M. Bill, Email: r.m.bill@aston.ac.uk.
Supplemental Information
References
- Fenton R.A., Moeller H.B., Nielsen S., De Groot B.L., Rutzler M. A plate reader-based method for cell water permeability measurement. Am. J. Physiol. Renal Physiol. 2010;298:F224–F230. doi: 10.1152/ajprenal.00463.2009. [DOI] [PubMed] [Google Scholar]
- Kitchen P., Conner A.C. Control of the aquaporin-4 channel water permeability by structural dynamics of aromatic/arginine selectivity filter residues. Biochemistry. 2015;54:6753–6755. doi: 10.1021/acs.biochem.5b01053. [DOI] [PubMed] [Google Scholar]
- Kitchen P., Salman M.M., Halsey A.M., Clarke-Bland C., Macdonald J.A., Ishida H., Vogel H.J., Almutiri S., Logan A., Kreida S. Targeting aquaporin-4 subcellular localization to treat central nervous system edema. Cell. 2020;181:784–799.e19. doi: 10.1016/j.cell.2020.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukaguchi H., Shayakul C., Berger U.V., Mackenzie B., Devidas S., Guggino W.B., Van Hoek A.N., Hediger M.A. Molecular characterization of a broad selectivity neutral solute channel. J. Biol. Chem. 1998;273:24737–24743. doi: 10.1074/jbc.273.38.24737. [DOI] [PubMed] [Google Scholar]
- Zampighi G.A., Kreman M., Boorer K.J., Loo D.D., Bezanilla F., Chandy G., Hall J.E., Wright E.M. A method for determining the unitary functional capacity of cloned channels and transporters expressed in Xenopus laevis oocytes. J. Membr. Biol. 1995;148:65–78. doi: 10.1007/BF00234157. [DOI] [PubMed] [Google Scholar]
- Kovacs R., Raue C., Gabriel S., Heinemann U. Functional test of multidrug transporter activity in hippocampal-neocortical brain slices from epileptic patients. J. Neurosci. Methods. 2011;200:164–172. doi: 10.1016/j.jneumeth.2011.06.032. [DOI] [PubMed] [Google Scholar]
- Solenov E., Watanabe H., Manley G.T., Verkman A.S. Sevenfold-reduced osmotic water permeability in primary astrocyte cultures from AQP-4-deficient mice, measured by a fluorescence quenching method. Am. J. Physiol. Cell Physiol. 2004;286:C426–C432. doi: 10.1152/ajpcell.00298.2003. [DOI] [PubMed] [Google Scholar]
- Arif M., Kitchen P., Conner M.T., Hill E.J., Nagel D., Bill R.M., Dunmore S.J., Armesilla A.L., Gross S., Carmichael A.R. Downregulation of aquaporin 3 inhibits cellular proliferation, migration and invasion in the MDA-MB-231 breast cancer cell line. Oncol. Lett. 2018;16:713–720. doi: 10.3892/ol.2018.8759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowe W.E., Altamirano J., Huerto L., Alvarez-Leefmans F.J. Volume changes in single N1E-115 neuroblastoma cells measured with a fluorescent probe. Neuroscience. 1995;69:283–296. doi: 10.1016/0306-4522(95)00219-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate/analyze datasets/code.