

Summary
Since its discovery, several ligands of the ZZ domain have been identified; however, molecular and structural information underlying binding of these ligands remains limited. Here, we describe a protocol for biochemical and structural analysis of the ZZ domain of human E3 ubiquitin ligase HERC2 (HERC2ZZ) and its interaction with its ligands: the N-terminal tails of histone H3 and SUMO1. This methodology could be applied for characterization of binding activities of other histone readers.
For complete details on the use and execution of this protocol, please refer to Liu et al. (2020).
Graphical Abstract
Highlights
-
•
Optimized protocol to purify HERC2ZZ from bacteria and mammalian cells
-
•
Strategy to characterize binding of HERC2ZZ to histone tails in vitro and in cells
-
•
Biochemical analysis and structure determination of histone reader
Since its discovery, several ligands of the ZZ domain have been identified; however, molecular and structural information underlying binding of these ligands remains limited. Here, we describe a protocol for biochemical and structural analysis of the ZZ domain of human E3 ubiquitin ligase HERC2 (HERC2ZZ) and its interaction with its ligands: the N-terminal tails of histone H3 and SUMO1. This methodology could be applied for characterization of binding activities of other histone readers.
Before You Begin
This protocol integrates complementary biochemical, molecular biology and structural biology approaches, enabling a rapid and straightforward evaluation of HERC2ZZ. Histone peptide pull-down assays, NMR titration experiments, and tryptophan fluorescence measurements are commonly used to characterize interactions between proteins and their ligands (Klein et al., 2016; Musselman and Kutateladze, 2016). Co-IP assays are performed to confirm the binding in cells (Mi et al., 2018). This protocol could be applicable for studying other histone readers.
In this protocol, we first detail the procedure for expression and purification of His8-SUMO tagged HERC2ZZ, GST-tagged HERC2ZZ, HERC2ZZ linked to H31–6 (residues 1–6 of histone H3) and HERC2ZZ linked to SUMO11–6 (residues 1–6 of SUMO1). We then describe the methodology for characterization of the interactions between HERC2ZZ and the H3 and SUMO1 peptides using peptide pull-down, tryptophan fluorescence, and NMR titration experiments. Lastly, we report the procedure allowing for crystallization of HERC2ZZ with the substrates. To begin, we need to generate the expression plasmids by performing DNA cloning, mutagenesis and transformation, and prepare Tobacco Etch Virus (TEV) Protease and Ubiquitin-like-specific protease 1 (Ulp1).
Prepare the Plasmids that Encode HERC2ZZ, H31–6-HERC2ZZ, SUMO11–6-HERC2ZZ, and Full-Length SUMO1
Timing: 4–5 days
-
1.
Obtain the sequence of human HERC2 (UniProt ID: O95714) from the UniProt database, https://www.UniProt.org (UniProt, 2007) and determine boundaries of the ZZ domain using the secondary structure elements prediction server JPred4 (https://www.compbio.dundee.ac.uk/jpred/) and by aligning the HERC2 sequence with the sequences of several ZZ domain-containing proteins using the Clustal Omega multiple sequence alignment tool on the EMBL-EBI server (https://www.ebi.ac.uk). Specifically, we aligned sequences of the ZZ domains of histone acetyltransferase p300 (UniProt ID: Q09472), Sequestosome-1 (UniProt ID: Q13501), E3 ubiquitin ligase MIB1 (UniProt ID: Q86YT6) and the ZZ-type zinc finger-containing protein 3 (UniProt ID: Q8IYH5). Based on this analysis, the region encompassing residues 2,702–2,755 of HERC2 was chosen for HERC2ZZ expression.
-
2.
Obtain the pCIOX vector (Addgene). The pCIOX vector is modified from a pET SUMO backbone which encodes the N-terminal His8x-SUMO tag and contains 5,643 bp of DNA.
-
3.Design the forward and reverse primers for HERC2ZZ and amplify the following sequence (ATTCATCCTGGGGTTACGTGTGATGGATGTCAGATGTTTCCTATCAATGGATCCAGATTCAAATGCAGAAACTGTGATGACTTTGATTTTTGTGAAACGTGTTTCAAGACCAAAAAACACAATACCAGGCATACATTTGGCAGAATAAATGAACCAGGTCAG) by PCR. The sequences of the primers are summarized in Key Resources Table.
-
a.Prepare the mix below:
Reagent Amount Platinum™ II Hot-Start PCR Master Mix (2X) (Thermo Fisher Scientific) 25 μL 10 μM forward primer 1 μL 10 μM reverse primer 1 μL template (20–50 ng/μL) 1 μL ddH2O up to 50 μL -
b.Use the following parameters to perform the PCR in a thermocycler.
PCR Cycling Conditions
Steps Temperature Time Cycles Initial Denaturation 95°C 3 min 1 Denaturation 95°C 30 s 30 cycles Annealing 56°C 30 s Extension 68°C 1 min Final Extension 68°C 10 min 1 Hold 10°C ∞
-
a.
-
4.
Digest the PCR product and the pCIOX vector (Addgene) with Nde I (NEB) and EcoR I (NEB) restriction endonucleases. Incubate the reaction mixture at 37°C for 1 h.
| Reagent | Amount |
|---|---|
| PCR product or pCIOX vector | 1–2 μg |
| CutSmart buffer (NEB) | 5 μL |
| EcoR I | 1 μL |
| Nde I | 1 μL |
| ddH2O | up to 50 μL |
-
5.
Run a 1% agarose gel to verify the digestion and purify both the digested PCR product and vector using the MinElute Gel Extraction Kit (Qiagen) according to the manufacturer’s protocol.
-
6.
Ligate the purified PCR product into the pCIOX vector. Mix the following reagents and products. Incubate the reaction mixture at 16°C for 1 h.
| Reagent | Amount |
|---|---|
| T4 DNA Ligase Buffer (10X) (NEB) | 1 μL |
| digested vector DNA | 20 ng |
| digested PCR product | 60 ng |
| T4 DNA Ligase | 1 μL |
| ddH2O | up to 10 μL |
Note: The digested PCR product is used at a 3:1 molar ratio over digested vector.
Pause Point: the ligation product can be stored at 4°C for 1–2 days.
-
7.Transform the ligation product into DH5α competent cells (Thermo Fisher Scientific).
-
a.Thaw the competent cells on ice and pipette 50 μL cells to a pre-chilled 0.6 mL Eppendorf tube.
-
b.Add 10 μL ligation product into competent cells.
-
c.Keep the cells on ice for 30 min.
-
d.Heat shock at 42°C for 45 s.
-
e.Put the competent cells back on ice for 2 min.
-
f.Add 100 μL LB media to the tube.
-
g.Incubate the tube at 37°C in a Forma Scientific Model 4580 Orbital Shaker (Forma Scientific) with shaking at 220 rpm for 60 min.
-
h.Equilibrate agar plate containing 100 μg/mL kanamycin to 23°C–25°C.
-
i.Spread the cells and media on the plate.
-
j.Incubate the plate at 37°C for ∼12–18 h
-
a.
CRITICAL: Competent cells are very sensitive to heat. Avoid touching the bottom of the tube with competent cells. Do not mix cells with the ligation product by pipetting up and down.
-
8.
Pick a colony with a sterile tip from the agar plate and inoculate into 5 mL LB media. Incubate at 37°C with shaking at 220 rpm 14–18 h.
-
9.
Extract the plasmids from the cells by a QIAprep Spin Miniprep Kit (QIAGEN) using the manufacturer’s protocol.
-
10.
Verify the construct by DNA sequencing.
-
11.Generate the H31–6 linked and SUMO11–6 linked HERC2ZZ constructs (H31–6-HERC2ZZ and SUMO11–6-HERC2ZZ) through insertional mutagenesis using QuikChange Lightning Site-Directed Mutagenesis Kit (Agilent Technologies).
-
a.Prepare the mixture below:
Reagent Amount 10X QuikChange Lightning Buffer 2.5 μL 10 μM forward primer 0.6 μL 10 μM reverse primer 0.6 μL dNTP Mix 0.5 μL QuikSolution 0.7 μL DNA template (20–50 ng/μL) 1 μL QuikChange Lightning Enzyme 0.5 ddH2O up to 25 μL -
b.Use the following parameters to perform the PCR in a T100™ Thermal Cycler (Bio-Rad).
PCR Cycling Conditions
Steps Temperature Time Cycles Initial Denaturation 95°C 2 min 1 Denaturation 95°C 20 s 20 cycles Annealing 60°C 10 s Extension 68°C 4 min Final Extension 68°C 5 min 1 Hold 10°C ∞
-
a.
-
12.
Add 1 μL of Dpn I (Agilent Technologies) restriction enzyme to the reaction product and incubate at 37°C for 10 min to digest the parental template.
-
13.
Transform 10 μL of the reaction product into DH5α competent cells (Thermo Fisher Scientific) using the protocol described above.
-
14.
Pick a colony from the agar plate and inoculate to 5 mL LB media. Incubate at 37°C with shaking at 220 rpm 14–18 h.
-
15.
Extract the DNA plasmid from the cells using a QIAprep Spin Miniprep Kit (QIAGEN) and the manufacturer’s protocol.
-
16.
Verify the constructs by DNA sequencing.
-
17.
Design the forward and reverse primers for full-length SUMO1 and subclone the sequence into a pDEST-15 vector using Gateway cloning (Thermo Fisher Scientific) and the manufacture’s protocol to be able to express the protein with the N-terminal GST tag and the TEV cleavage site.
-
18.
Design the forward and reverse primers to introduce the FLAG tag at the C terminus of histone H3 and subclone the sequence into a pcDNA vector.
-
19.
Design the forward and reverse primers for the C-terminal half of HERC2 (aa 2,600–4,834) and subclone the sequence into a pcDNA vector.
Prepare the Antibiotics and IPTG Stocks
-
20.Make the following antibiotics and IPTG (Goldbio) stock solutions to use in cell culture and induce expression.
-
a.Dissolve 4.76 g IPTG (Goldbio) in 16 mL Milli-Q water. Bring the final volume to 20 mL. Filter the solution with 0.22 μm syringe filter. Store at −20°C.
-
b.Dissolve 5 g Ampicillin (Goldbio) in 45 mL Milli-Q water. Bring the final volume to 50 mL. Filter the solution with 0.22 μm syringe filter. Store at −20°C.
-
c.Dissolve 2.5 g Kanamycin (Goldbio) in 45 mL Milli-Q water. Bring the final volume to 50 mL. Filter the solution with 0.22 μm syringe filter. Store at −20°C.
-
d.Dissolve 1.7 g Chloramphenicol (Goldbio) in 45 mL 100% EtOH (Decon Labs, Inc). Bring the final volume to 50 mL. Store at −20°C.
-
a.
| Antibiotics and IPTG Stocks | Stock Concentration | Solvent | Sterilization |
|---|---|---|---|
| IPTG | 1.0 M | Milli-Q water | Filter sterilize |
| Ampicillin | 100 mg/mL | Milli-Q water | Filter sterilize |
| Kanamycin | 50 mg/mL | Milli-Q water | Filter sterilize |
| Chloramphenicol | 34 mg/mL | Ethanol | N/A |
Prepare LB Agar Plates
-
21.
Weigh out 7.4 g LB Agar, Miller (Thermo Fisher Scientific) and dissolve in 180 mL Milli-Q water.
-
22.
Heat and mix until LB Agar is completely dissolved. Cool down to ∼25°C.
-
23.
Add Milli-Q water up to 200 mL and sterilize by autoclaving at 121°C for 15 min.
-
24.
Let agar cool down to 50°C–55°C (you should be able to hold the flask without burning yourself). Add 200 μL antibiotic stocks to make different types of plates according to need.
-
25.
Pour ∼10–15 mL LB Agar into each plate.
-
26.
Let the plates cool down until solid.
-
27.
Seal plate edges with parafilm or foil and store the plates at 4°C.
Purification of Tobacco Etch Virus (TEV) Protease and Ubiquitin-like-Specific Protease 1 (Ulp1)
-
28.
Transform pRK793 plasmid (Addgene # 8827) (Kapust et al., 2001) that encodes the TEV protease into Escherichia coli BL21-CodonPlus (DE3) RIL chemically competent cells (Agilent Technologies) and grow colonies on LB agar plates containing 100 μg/mL ampicillin and 34 μg/mL chloramphenicol.
-
29.
Transform pFGET19_Ulp1 plasmid (Addgene # 64697) (Guerrero et al., 2015) that encodes the Ulp1 into Escherichia coli BL21-CodonPlus (DE3) RIL chemically competent cells (Agilent Technologies) and grow colonies on LB agar plates containing 50 μg/mL kanamycin and 34 μg/mL chloramphenicol.
-
30.
Inoculate a single colony into 100 mL Luria-Bertani (LB) media supplemented with corresponding antibiotics in a 250 mL PYREX® Round Media Storage Bottle (The Lab Depot). Grow the cells in a Forma Scientific Model 4580 Orbital Shaker (Forma Scientific) at 37°C with shaking at 200 rpm, 14–18 h.
-
31.
Dilute 100 mL of cell culture into 2 L of LB media in a 6 L Narrow-Mouth Erlenmeyer Flask (Thermo Fisher Scientific) and continue growing cells in a MaxQ™ 5000 Model Shaker (Thermo Fisher Scientific) at 37°C with shaking at 200 rpm until the optical density of a 1 mL sample reaches 1.0 at an absorbance of 600.
-
32.
Reduce the temperature of the shaker to 30°C and slow down the shaker speed to 180 rpm. Protein expression is induced by adding 2 mL of 1.0 M IPTG and incubation at 30°C with shaking for 4 h.
-
33.
Harvest cells by centrifugation at 5,000 rpm (4,424 × g) using a F10-6 x 500y Fixed-Angle Rotor (Thermo Fisher Scientific) in Sorvall RC-5B Refrigerated Superspeed Centrifuge (Sorvall) for 10 min and discard supernatant.
-
34.
Resuspend cell pellet with 150 mL lysis buffer A. Lyse the cells on ice by sonication for 10 min at 60% power with intervals of 5 s on and 10 s off.
-
35.
Spin down the cell lysate at 15,000 rpm (26,964 × g) in a SS-34 Fixed-Angle Rotor (Thermo Fisher Scientific) for 30 min to remove cell debris and insoluble fractions.
-
36.
Incubate the supernatant with 1 mL Ni-NTA resin (QIAGEN) in a 50 mL Falcon tube at 25°C for 1 h with rocking on a Unico Test Tube Rocker.
-
37.
Wash the resin with 60 mL lysis buffer A containing 30 mM imidazole.
-
38.
Elute the protein off the resin with 10 mL Ni-NTA elution buffer and then dialyze the elution to 1 L storage buffer using a Regenerated Cellulose Dialysis Tubing (Thermo Fisher Scientific).
-
39.
Concentrate the protein to 10–15 mg/mL using a 10 kDa MWCO centrifugal filter (Millipore) at 3,000 × g on an A-4-44 rotor (Eppendorf) in Eppendorf 5804 R Centrifuge (Eppendorf) and store the protein at −80°C.
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-HERC2 antibody | Abcam | Cat. # Ab85832 |
| Anti-Flag antibody | Sigma-Aldrich | Cat. # F3165 |
| Anti-GST antibody | Santa Cruz | Cat. # sc-459 |
| Bacterial and Virus Strains | ||
| Escherichia coli BL21-CodonPlus (De3) RIL | Agilent Technologies | N/A |
| DH5α Competent Cells | Thermo Fisher Scientific | Cat. # 18265017 |
| Critical Commercial Assays | ||
| X-tremeGENE™ HP DNA Transfection Reagent | Roche | Cat: # 6366546001 |
| Platinum™ II Hot-Start PCR Master Mix (2×) | Thermo Fisher Scientific | 14000013 |
| QuikChange Lightning Site-Directed Mutagenesis Kit | Agilent Technologies | Cat. # 210518 |
| MinElute Gel Extration Kit | QIAGEN | Cat. # 28604 |
| QIAprep Spin Miniprep Kit | QIAGEN | Cat # 27106 |
| JCSG-plus™ | Molecular Dimensions | MD1-37 |
| PACT premierTM | Molecular Dimensions | MD1-29 |
| Other | ||
| Cell filter | Corning | Cat: # 3008 |
| Regenerated Cellulose Dialysis Tubing | Thermo Fisher Scientific | Cat. # 21-152-14 |
| 1,000 μL Universal Pipetter Tips: wide bore | Axygen | Cat: # 14-222-703 |
| HiPrep™ 16/60 Sephacryl® S-100 HR column | GE Healthcare | Cat. # 17-1165-01 |
| Econo-Pac® Chromatography Columns | Bio-Rad | Cat. # 7321010 |
| Amicon Ultra 15 mL 3K NMWL centrifugal filter unit | Millipore | Cat. # UFC900308 |
| Amicon Ultra 15 mL 10K NMWL centrifugal filter unit | Millipore | Cat. # UFC901024 |
| Millex-GV Syringe Filter Unit, 0.22 μm | Millipore | Cat. # SLGU033RS |
| Fluoromax-3 spectrofluorometer | HORIBA Scientific | N/A |
| Sorvall RC-5B Refrigerated Superspeed Centrifuge | Sorvall | N/A |
| Unico Test Tube Rocker | TEquipment | L-TTR200 |
| Microplate Titer Plate Shaker | LAB-LINE INSTRUMENTS, Inc | N/A |
| Eppendorf 5804 R Centrifuge | Eppendorf | 5804 R |
| F10-6 × 500y Fixed-Angle Rotor | Thermo Fisher Scientific | Cat. # 78510TS |
| SS-34 Fixed-Angle Rotor | Thermo Fisher Scientific | Cat. # 28020TS |
| Rotor A-4-44 | Eppendorf | Cat. # 05-400-115 |
| 6 L Reusable Glass Narrow-Mouth Erlenmeyer Flasks | Thermo Fisher Scientific | Cat. # FB5006000 |
| MaxQ™ 5000 Floor-Model Shaker | Thermo Fisher Scientific | Cat. # 11-675-208 |
| Forma Scientific Model 4580 Orbital Shaker Incubator | Forma Scientific | Cat. # 25423 |
| Quartz Fluorometer Cell | Starna Cells, Inc | Cat. # 3-5.45-Q-5 |
| Varian INOVA 600 MHz NMR spectrometer | Agilent Technologies | N/A |
| T100™ Thermal Cycler | Bio-Rad | N/A |
| Shigemi 5 mm Symmetrical NMR microtube | SHIGEMI, INC | BMS-005B |
| Falcon 50 mL Conical Centrifuge Tubes | Thermo Fisher Scientific | Cat. # 14-432-22 |
| 250 mL PYREX® Round Media Storage Bottle | The Lab Depot | Cat. # 1395-250 |
| INTELLI-PLATE® 96 Well | Art Robbins Instruments | Cat. # 102-0001-00 |
| Branson SFX250 sonicator | Branson | SFX250 |
| Tube Rotators | Lab Quake | 415110 |
| Magnetic stirrers | Corning | PC-410 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Dithiothreitol | Gold Biotechnology | 27565-41-9 |
| β-mercaptoethanol | Sigma-Aldrich | Cat. # M6250 |
| Phenylmethanesulfonyl fluoride | Sigma-Aldrich | Cat. # P7626 |
| Lysozyme | Thermo Fisher Scientific | Cat. # 89833 |
| DNase I | Sigma-Aldrich | Cat. # 11284932001 |
| NP-40 | Sigma-Aldrich | Cat. # 127087-87-0 |
| Ampicillin | Goldbio | Cat. # A-301 |
| Kanamycin | Goldbio | Cat. # K-120 |
| Chloramphenicol | Goldbio | Cat. # C-105 |
| Imidazole | Alfa Aesar | Cat. # A10221 |
| 100% Ethanol 200 PROOF | Decon Labs, Inc | Cat. # V1016 |
| Microbiology Media: LB Broth, Miller | Thermo Fisher Scientific | Cat. # BP1426-2 |
| LB Agar, Miller | Thermo Fisher Scientific | Cat. # BP9724-500 |
| NdeI restriction endonucleases | New England Biolabs | Cat. # R0111L |
| EcoRI restriction endonucleases | New England Biolabs | Cat. # R0101L |
| Dpn I Enzyme | Agilent Technologies | Cat. # 210518-52 |
| T4 DNA ligase | New England Biolabs | Cat. # M0202L |
| Gateway™ BP Clonase™ II Enzyme mix | Thermo Fisher Scientific | Cat. # 11789100 |
| Gateway™ LR Clonase™ II Enzyme mix | Thermo Fisher Scientific | Cat. # 11791020 |
| Reduced glutathione | Sigma-Aldrich | G4251 |
| 15NH4Cl | Sigma-Aldrich | Cat. # 299251 |
| Multivitamin Tablet | Centrum | Walmart # 573963323 |
| ZnCl2 | Sigma-Aldrich | Cat. # 229997 |
| Deuterium oxide | Sigma-Aldrich | Cat. # D4501 |
| IPTG | Goldbio | Cat. # I2481 |
| Sodium dodecyl sulfate | Sigma-Aldrich | Cat. # L3771 |
| Quick Start™ Bradford 1× Dye Reagent | Bio-Rad | Cat. # 500-0205 |
| Ni-NTA beads | QIAGEN | Cat. # 30210 |
| Glutathione Sepharose 4B beads | GE Healthcare | Cat. # 17-0756-01 |
| Streptavidin magnetic beads | Thermo Fisher Scientific | Cat. # 88816 |
| ANTI-FLAG M2 Magnetic Beads | Sigma-Aldrich | Cat. # A2220 |
| H31–12, SUMO11–6 peptides | Synpeptide | N/A |
| H31–22, H41–24, H2A1–22, H2B1–22 peptides | Synpeptide | N/A |
| Biotinylated H31-22 peptides | Synpeptide | N/A |
| TEV and Ulp1 proteases | Home expressed | N/A |
| DMEM | Corning | Cat: # 10-013-CV |
| Opti-MEM | Gibco | Cat: # 31985062 |
| cOmplete™, Mini, EDTA-free Protease Inhibitor Cocktail | Roche | Cat: # 11836170001 |
| Triton X-100 | Calbiochem | Cat: # 9410-1L |
| Deposited Data | ||
| Crystal structure of SUMO11–6 linked HERC2ZZ | This study | PDB: 6WW3 |
| Crystal structure of H31–6 linked HERC2ZZ | This study | PDB: 6WW4 |
| Crystal structure of p300 ZZ domain in complex with histone H3 peptide | Zhang et al., 2018 | PDB: 6DS6 |
| Experimental Models: Cell Lines | ||
| Human: HEK293T | ATCC | Cat. # CRL-3216 |
| Oligonucleotides | ||
| Primer: HERC2_ZZ_W Forward: GAACCAGGTCAGTGGTAAGAATTCGAGCT |
This study - IDT | N/A |
| Primer: HERC2_ZZ_W Reverse: AGCTCGAATTCTTACCACTGACCTGGTTC |
This study - IDT | N/A |
| Primer: HERC2_2600-4834 Forward: ATTTGATCAATGGATGGATTGCATGATCTCAATGTGC |
This study - IDT | N/A |
| Primer: HERC2_2600-4834 Reverse: CCGCTCGAGTTAGTGTCCTGTTAAATAATCTTG |
This study - IDT | N/A |
| Primer: D2730A Forward: TGCAGAAACTGTGATGACTTTGCTTTTTGTGAAAC GTGTTTCAAG |
This study - IDT | N/A |
| Primer: D2730A Reverse: CTTGAAACACGTTTCACAAAAAGCAAAGTCATCACA GTTTCTGCA |
This study - IDT | N/A |
| Primer: D2709A Forward: GGGTTACGTGTGCGGGATGTCAGAT |
This study - IDT | N/A |
| Primer: D2709A Reverse: ATCTGACATCCCGCACACGTAACCC |
This study - IDT | N/A |
| Primer: H3_ZZ Forward: GCGCGTACCAAACAGACCATTCATCCTGGGGTTA CGTGTG |
This study - IDT | N/A |
| Primer: H3_ZZ Reverse: GGTCTGTTTGGTACGCGCACCACCAATCTGTTCTC TGTGA |
This study - IDT | N/A |
| Primer: SUMO1_ZZ Forward: AGCGATCAGGAAGCGAAAATTCATCCTGGGGTTA CGTGTG |
This study - IDT | N/A |
| Primer: SUMO1_ZZ Reverse: TTTCGCTTCCTGATCGCTACCACCAATCTGTTCTC TGTGA |
This study - IDT | N/A |
| Recombinant DNA | ||
| Plasmid: pRK793 | Addgene | Cat. # 8827 |
| Plasmid: pDEST15 | Thermo Fisher Scientific | Cat. # 1180214 |
| Plasmid: pCIOX | Andrea Mattevi lab, University of Pavia | Addgene, Cat. # 51300 |
| Plasmid: pcDNA3 | Xiaobing Shi lab Van Andel Institute |
Addgene (unavailable) |
| Plasmid: pGEX-6p-1 | Xiaobing Shi lab Van Andel Institute |
Addgene, Cat. # 27459701 |
| Software and Algorithms | ||
| HKL2000 | Otwinowski and Minor, 1997 | https://www.hkl-xray.com/download-instructions-hkl-2000 |
| Phenix | Adams et al., 2010 | http://www.phenix-online.org/ |
| Coot | Emsley et al., 2010 | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ |
| KaleidaGraph 4.5 | Synergy Software | https://www.synergy.com/ |
| Prism 8 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| OriginPro | OriginLab | https://www.originlab.com |
| NMRPipe | Delaglio et al., 1995 | https://www.ibbr.umd.edu/nmrpipe/ |
Materials and Equipment
Luria-Bertani (LB) Media
Dissolve the reagents below in 1 L Milli-Q water and autoclave the media at 121°C for 30 min. 1 L LB media contains 10 g tryptone, 5 g yeast extract, and 10 g NaCl.
-
•
25 g LB Broth, Miller Molecular Genetics Power
M9 Minimal Media
Dissolve the reagents below in 1 L Milli-Q water and autoclave the media at 121°C for 30 min:
| Reagent | Amount | Final Concentration |
|---|---|---|
| Na2HPO4•7H2O | 12.8 g | 47.7 mM |
| KH2PO4 | 3.0 g | 22 mM |
| NaCl | 0.5 g | 8.56 mM |
| 15NH4Cl | 1 g | 18.4 mM |
| 1 M MgSO4 | 2 mL | 2 mM |
| 1 M CaCl2 | 200 μL | 200 μM |
| 0.1M ZnCl2 | 0.5 mL | 50 μM |
Add the following reagents before applying to cell culture:
| Reagent | Amount |
|---|---|
| multivitamin solution (1 multivitamin tablet (Centrum) dissolved in 50 mL water, mixed, and filtered) | 5 mL |
| 20% (w/v) glucose (filtered) | 25 mL |
The M9 minimal media can be stored at 25°C for 1 week.
Lysis Buffer A
Lysis buffer A contains 50 mM Tris pH 7.5, 500 mM NaCl, 1 mM phenylmethanesulfonyl fluoride (PMSF) and 10 mM β-mercaptoethanol. To prepare lysis buffer A, add the reagents below to 800 mL Milli-Q water:
| Reagent | Amount | Final Concentration |
|---|---|---|
| Tris-base | 6.06 g | 50 mM |
| NaCl | 29.22 g | 500 mM |
Adjust the pH of the buffer to 7.5 at 25°C. Add Milli-Q water up to 1 L and filter the buffer using a 0.22 μm filter. The buffer can be stored at 4°C for 1 day.
Note: PMSF can produce chemical burn of skin and eyes, therefore precaution must be taken to prevent direct contact with the reagent. β-mercaptoethanol is toxic and volatile. Add these reagents to the buffer in the hood.
Lysis Buffer B
Lysis buffer B contains 20 mM Tris, pH 7.5, 200 mM NaCl, 5 mM dithiothreitol (DTT). To prepare lysis buffer B, add the reagents below to 800 mL Milli-Q water:
| Reagents | Amount | Final Concentration |
|---|---|---|
| Tris-base | 2.42 g | 20 mM |
| NaCl | 11.7 g | 200 mM |
| DTT | 0.77 g | 5 mM |
Adjust the pH of the buffer to 7.5 at 25°C. Add Milli-Q water up to 1 L and filter the buffer using a 0.22 μm filter. The buffer can be stored at 4°C for 14 days.
Note: Avoid contact and inhalation when handling DTT.
Lysis Buffer C
Lysis buffer C contains 50 mM Tris, pH 7.5, 150 mM NaCl, 0.05% NP-40. To prepare lysis buffer C, add the reagents below to 800 mL Milli-Q water:
| Reagents | Amount | Final Concentration |
|---|---|---|
| Tris-base | 6.06 g | 50 mM |
| NaCl | 8.77 g | 150 mM |
| 10% NP-40 | 5 mL | 0.05% |
Adjust pH of the buffer to 7.5 at 25°C. Add Milli-Q water up to 1 L and filter the buffer using a 0.22 μm filter. The buffer can be stored at 4°C for 14 days.
Note: Avoid contact, ingestion, and inhalation when handling NP-40.
Gel-Filtration Buffer
Gel-filtration buffer contains 20 mM Tris, pH 8.0, 200 mM NaCl, 5 mM DTT. To prepare gel-filtration buffer, add the reagents below to 800 mL Milli-Q water:
| Reagents | Amount | Final Concentration |
|---|---|---|
| Tris-base | 2.42 g | 20 mM |
| NaCl | 11.7 g | 200 mM |
| DTT | 0.77 g | 5 mM |
Adjust pH of the buffer to 8.0 at 25°C. Add Milli-Q water up to 1 L and filter the buffer using a 0.22 μm filter. The buffer can be stored at 4°C for 14 days.
Binding Buffer A
Binding buffer contains 20 mM Tris, pH 6.8, 100 mM NaCl, 2 mM DTT. To prepare binding buffer A, add the reagents below to 800 mL Milli-Q water:
| Reagents | Amount | Final Concentration |
|---|---|---|
| Tris-base | 2.42 g | 20 mM |
| NaCl | 5.85 g | 100 mM |
| DTT | 0.31 g | 2 mM |
Adjust pH of the buffer to 6.8 at 25°C. Add Milli-Q water up to 1 L and filter the buffer using a 0.22 μm filter. The buffer can be stored at 4°C for 14 days.
Binding Buffer B
Binding buffer B contains 50 mM Tris, pH 7.5, 150–300 mM NaCl, 0.05% NP-40. To prepare binding buffer B, add the reagents below to 800 mL Milli-Q water:
| Reagents | Amount | Final Concentration |
|---|---|---|
| Tris-base | 6.06 g | 50 mM |
| NaCl | 8.77 g | 150 mM |
| NP-40 | 5 mL | 0.05% |
Adjust pH of the buffer to 7.5 at 25°C. Add Milli-Q water up to 1 L and filter the buffer using a 0.22 μm filter. The buffer can be stored at 4°C for 14 days.
Ni-NTA Wash Buffer
Wash buffer contains 50 mM Tris pH 7.5, 500 mM NaCl, 200 mM imidazole. To prepare wash buffer, add the reagents below to 800 mL Milli-Q water:
| Reagents | Amount | Final Concentration |
|---|---|---|
| Tris-base | 6.06 g | 50 mM |
| NaCl | 29.22 g | 500 mM |
| Imidazole | 13.62 g | 200 mM |
Adjust pH of the buffer to 7.5 at 25°C. Add Milli-Q water up to 1 L and filter the buffer using a 0.22 μm filter. The buffer can be stored at 4°C for 14 days.
Note: Wash buffer could be stored at 25°C. Mix wash buffer with lysis buffer A to make desirable concentration imidazole solutions, which are used to remove proteins that non-specifically bind Ni-NTA resin. For example, mix 4 mL wash buffer with 36 mL lysis buffer A to make 40 mL of 20 mM imidazole solution.
Ni-NTA Elution Buffer
Elution buffer contains 50 mM Tris pH 7.5, 500 mM NaCl, 500 mM imidazole. To prepare elution buffer, add the reagents below to 800 mL Milli-Q water:
| Reagents | Amount | Final Concentration |
|---|---|---|
| Tris-base | 6.06 g | 50 mM |
| NaCl | 29.22 g | 500 mM |
| Imidazole | 34.04 g | 500 mM |
Adjust pH of the buffer to 7.5 at 25°C. Add Milli-Q water up to 1 L and filter the buffer using a 0.22 μm filter. The buffer can be stored at 4°C for 14 days.
Storage Buffer
The storage buffer for TEV and Ulp1 enzyme contains 20 mM Tris, pH 7.5, 500 mM NaCl, 5 mM β-mercaptoethanol and 5% glycerol. To prepare the buffer, add the reagents below to 800 mL Milli-Q water:
| Reagents | Amounts | Final Concentration |
|---|---|---|
| Tris-base | 2.42 g | 20 mM |
| NaCl | 29.22 g | 500 mM |
| Glycerol | 50 mL | 5% |
| β-mercaptoethanol | 350 μL | 5 mM |
Adjust pH of the buffer to 7.5 at 25°C. Add Milli-Q water up to 1 L and filter the buffer using a 0.22 μm filter. The buffer can be stored at 4°C for 1 day.
GST Elution Buffer
Elution buffer contains 100 mM Tris, pH 8.0. To prepare elution buffer, add the reagents below to 800 mL Milli-Q water:
-
•
12.1 g Tris-base
Add 10–15 mg reduced glutathione (Sigma) per 1 mL elution buffer. Adjust pH of the buffer to 8.0 at 25°C. Bring the volume to 1 L with Milli-Q water and filter the buffer using a 0.22 μm filter. The buffer can be stored at 4°C for 14 days.
Cell Lysis Buffer
Cell lysis buffer contains 50 mM Tris, pH 7.4, 250 mM NaCl, 0.5% Triton X100 and 10% glycerol. To prepare elution buffer, add the reagents below to 800 mL Milli-Q water:
| Reagents | Amounts | Final Concentration |
|---|---|---|
| Tris-base | 6.06 g | 50 mM |
| NaCl | 14.61 g | 250 mM |
| Triton X100 | 5 mL | 0.5% |
| Glycerol | 100 mL | 10% |
Adjust pH of the buffer to 7.4 at 25°C. Add Milli-Q water up to 1 L and filter the buffer using a 0.22 μm filter. Add freshly 1 mM DTT, 1 mM PMSF, and 1 tablet of protease inhibitors per 1 L buffer. The buffer can be stored at 4°C for 14 days.
Note: Avoid contact, ingestion, and inhalation when handling Triton X100.
Tips: Measure glycerol using 100 mL cylinder and rinse the cylinder with Milli-Q water to be sure no glycerol left in the cylinder. To ensure homogeneity, stir the buffer using a magnetic stirrer for 4–24 h.
5× SDS Sample Buffer
5 x SDS sample buffer contains 250 mM Tris, pH 6.8, 10% SDS, 30% Glycerol, 5% β-mercaptoethanol. To prepare this buffer, add the reagents below to 50 mL Milli-Q water. Adjust pH of the buffer to 6.8 at 25°C. Add Milli-Q water up to 100 mL.
| Reagents | Amount | Final Concentration |
|---|---|---|
| Tris-base | 30.3 g | 250 mM |
| SDS | 10 g | 10% |
| Glycerol | 30 mL | 30% |
| β-mercaptoethanol | 5 mL | 5% |
For 2 x SDS sample buffer, dilute the above 5 x SDS sample buffer with Milli-Q water.
Note: Avoid contact, ingestion and inhalation when handling SDS and β-mercaptoethanol. The buffer can be stored at 4°C for 1 day.
Step-By-Step Method Details
HERC2ZZ and Full-Length SUMO1 Expression and Purification
Prepare the protein samples for NMR, tryptophan fluorescence and crystallization experiments (Figure 1).
Note: The plasmids and corresponding antibiotics for selection are summarized as bellow:
| Plasmids | Antibiotic for Selection |
|---|---|
| pCIOX HERC2ZZ (Wild-type and mutants) | Kanamycin |
| pDEST-15 SUMO1 | Ampicillin |
| pRK793 | Ampicillin |
| pFGET19 Ulp1 | Kanamycin |
| pGEX-6p-1-HERC2ZZ (wild-type and mutants) | Ampicillin |
Note: DH5α chemically competent cells carry no resistance to antibiotics while BL21-CodonPlus (DE3) RIL chemically competent cells are chloramphenicol-resistant.
-
1.
Transform the expression plasmids that encode HERC2ZZ, H31–6-HERC2ZZ, SUMO11–6-HERC2ZZ and full-length SUMO1 into BL21 (DE3) RIL chemically competent cells (Agilent Technologies) following the protocol described above and plate onto LB agar plates supplemented with 34 μg/mL chloramphenicol and 100 μg/mL kanamycin (PCIOX HERC2ZZ) or ampicillin (pDEST-15 SUMO1).
-
2.
Inoculate single colony into 200 mL LB media supplemented with corresponding antibiotics. Culture the cells in a Forma Scientific Model 4580 Orbital Incubator Shaker (Forma Scientific) at 37°C and 200 rpm, 14–18 h.
-
3.
Dilute the overnight culture 1:100 in 2L LB media supplemented with 34 μg/mL chloramphenicol and 100 μg/mL kanamycin (pCIOX HERC2ZZ) or ampicillin (pDEST-15 SUMO1), and 50 μM ZnCl2 in a 6 L Narrow-Mouth Erlenmeyer Flask (Thermo Fisher Scientific). Culture the cells at 37°C in a MaxQ™ 5000 Floor-Model Shaker (Thermo Fisher Scientific) with an agitation speed of 200 rpm until the optical density of a 1 mL sample reaches 0.6–0.8 at an absorbance of 600.
-
4.
Reduce the shaker temperature to 16°C and the speed to 180 rpm, while cooling down the cell culture for 30 min to 23°C–25°C. Protein expression is induced by adding IPTG (at a final concentration of 0.2 mM), along with agitation and incubation at 16°C for 20 h.
-
5.
Harvest cells by centrifugation at 5,000 rpm (4,424 × g) using a F10-6 × 500y Fixed-Angle Rotor (Thermo Fisher Scientific) in Sorvall RC-5B Refrigerated Superspeed Centrifuge (Sorvall) for 10 min at ∼4°C and discard supernatant.
-
6.
For HERC2ZZ and H31–6 linked and SUMO11–6 linked HERC2ZZ purification, resuspend the cell pellets in 50 mL lysis buffer A per liter cell culture by pipetting up and down using 10 mL transfer pipette. For SUMO1 purification, resuspend the cell pellets in 50 mL lysis buffer B per liter cell culture.
-
7.
Lyse the resuspended cells in a beaker on ice by sonication for 10 min at 60% power with intervals of 5 s on and 10 s off.
Note: 0.1–1 mg/mL Lysozyme (Thermo Fisher Scientific) can be added to the buffer to increase the lysis efficiency. Additionally, 1 tablet of EDTA-free protease inhibitor cocktail (Roche) per 1 L buffer and 10 μg/mL DNAse I (Sigma-Aldrich) could be added to inhibit the protease activity and remove DNA that may be bound by the protein of interest.
-
8.
Spin down the cell lysate at 15,000 rpm (26,964 × g) using a SS-34 Fixed-Angle Rotor (Thermo Fisher Scientific) for 30 min to remove cell debris and insoluble fractions.
-
9.
For HERC2ZZ, H31–6 linked and SUMO11–6 linked HERC2ZZ purification, incubate the supernatant with 3 mL Ni-NTA resin (QIAGEN) in a 50 mL Falcon tube for 1 h rocking on a test tube rocker (Unico) at 4°C.
-
10.
Apply the suspension to an Econo-Pac® Chromatography Column (Bio-Rad) to collect the protein bound Ni-NTA resin.
-
11.
To remove impurities, wash the resin with 20 mL lysis buffer A, followed by 40 mL lysis buffer A that contains 20 mM and 30 mM imidazole (pH 7.5) respectively.
-
12.
Analyze a 15 μL sample of each purification step by SDS Polyacrylamide gel electrophoresis (PAGE). Before loading the gel, add 5 μL of 4X SDS loading buffer and denature the samples by heating to 90°C for 10 min before loading onto a 15-well 15% SDS PAGE gel (15% resolving, 5% stacking). The gel and 4X SDS loading buffer were prepared according to standard Bio-rad gel making protocol. http://www.bio-rad.com/webroot/web/pdf/lsr/literature/Bulletin_6201.pdf
-
13.
Resuspend the Ni-NTA resin in 10 mL lysis buffer A, add the prepared Ulp1 enzyme (14 mg/mL, 30 μL), and incubate the reaction mixture rocking on a Microplate Titer Plate Shaker (LAB-LINE INSTRUMENTS, Inc.) for 4–24 h at 4°C to cleave the His8-SUMO tag.
-
14.
Analyze a 15 μL sample of the Ulp1 cleavage reaction by polyacrylamide gel electrophoresis (PAGE) as described in step 12 (Figure 1B). Once the desirable extent of the cleavage reaction is achieved (∼60%–100%), collect the cleavage supernatant, which contains the cleaved protein. Concentrate the protein to 2 mL using a 3 kDa MWCO centrifugal filter (Millipore) at 3,000 × g in an Eppendorf 5804 R Centrifuge with an A-4-44 rotor.
-
15.
Inject the concentrated protein onto a HiPrep™ 16/60 Sephacryl® S-100 HR column (GE Healthcare) pre-equilibrated in gel-filtration buffer at 0.5 mL/min. HERC2ZZ elutes at ∼85 mL on this column.
-
16.
Pool protein-containing fractions and concentrate the protein to 5 mg/mL using a 3 kDa MWCO centrifugal filter (Millipore) at 3,000 × g in an Eppendorf 5804 R Centrifuge with an A-4-44 rotor (Eppendorf).
-
17.
For SUMO1 purification, incubate the supernatant with 3 mL Glutathione Sepharose 4B beads (GE Healthcare) for 3 h rocking on a test tube rocker (Unico) at 4°C.
-
18.
Apply the suspension to an Econo-Pac® Chromatography Column (Bio-Rad) to collect the protein bound GST resin.
-
19.
Wash the resin by adding 50 mL lysis buffer B through the column to remove impurities.
-
20.
Resuspend the GST resin in 10 mL lysis buffer B, add the prepared TEV enzyme (14 mg/mL, 30 μL), and incubate the reaction mixture gently rocking for 4–24 h at 4°C to cleave off the GST tag. Analyze a 15 μL sample of cleavage supernatant as in step 14 to assess MW difference corresponding to protein cleavage.
-
21.
Concentrate untagged SUMO1 to 2 mL using a 10 kDa MWCO centrifugal filter (Millipore) at 3,000 × g in an Eppendorf 5804 R Centrifuge with an A-4-44 rotor (Eppendorf) and inject the protein onto an HiPrep™ 16/60 Sephacryl® S-100 HR column (GE Healthcare) pre-equilibrated in gel-filtration buffer at 0.5 mL/min.
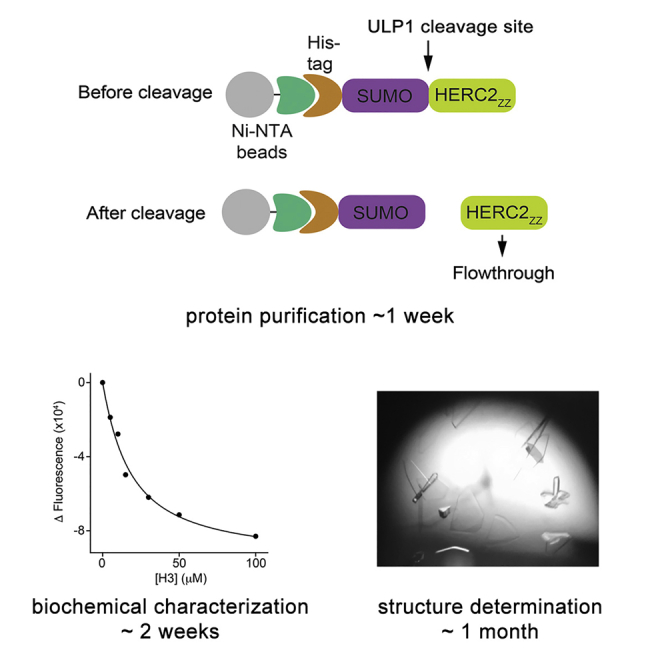
Figure 1.

Diagram of the Procedure for Purification of HERC2ZZ
(A) Preparation of cells expressing HERC2ZZ.
(B) Affinity purification and removal of the His8-SUMO tag from HERC2ZZ.
(C) Further purification by size exclusion chromatography.
Purification of GST-Tagged Protein
Prepare the protein samples for histone peptide pull-down
-
22.
Express HERC2ZZ following the steps 1–4 in the section of HERC2ZZ and full-length SUMO1 expression and purification.
-
23.
Resuspend the pellet from a 100 mL culture in 10 mL pre-chilled lysis buffer C.
-
24.
Add 100 μL of 50 mg/mL lysozyme solution to the cells, and keep the cells on ice for 30 min, invert occasionally.
-
25.
Sonicate the cells on ice using a Branson SFX250 sonicator at 18% power for 20 s with intervals of 1 s on and 1 s off. Repeat this step twice.
-
26.
Transfer the lysate into a 15 mL disposable tubes and spin down at 12,000 rpm (13,000 × g) at 4°C for 15 min. Transfer the supernatant (∼10 mL) of lysed bacteria to a new 15 mL tube.
-
27.
Prepare glutathione beads. For each 10 mL cell lysate, use 100 μL of glutathione agarose slurry for purification.
Note: Mix the glutathione agarose slurry before taking.
-
28.
Add 10 mL cold lysis buffer C, and wash the beads at 4 ˚C for 2 min.
-
29.
Centrifuge at 2,000 rpm (376 × g) at 4°C for 2 min.
-
30.
Remove the supernatant and wash the beads two more times with lysis buffer C.
-
31.
After the final wash, bring the final volume of beads to 200 μL with lysis buffer C (2X the beads volume).
-
32.
Add 200 μL of washed 50% slurry of glutathione beads into the cell lysates from step 24. Incubate the supernatant with beads at 4°C with gentle rotation on tube rotator (Lab Quake) at 20 rpm for 4 h.
-
33.
Centrifuge the tube at 2,000 rpm (376 × g) at 4°C for 2 min. Remove the supernatant.
-
34.
Add 10 mL of pre-chilled lysis buffer C, incubate at 4 ˚C with rotation for 5 min, and spin down the beads. Repeat this step twice.
-
35.
Add 10 mL pre-chilled GST elution buffer (without glutathione) to the beads. Centrifuge at 4,000 rpm (1,500 × g) for 1 min to remove the supernatant.
-
36.
Add 350 μL GST elution buffer (with glutathione) and incubate at 4°C with rotation for 4 h.
-
37.
Centrifuge at 4,000 rpm (1,500 × g) for 2 min and transfer 300 μL supernatant into a new 1.5 mL Eppendorf tube.
-
38.
Determine protein concentration by Bio-Rad Bradford assay.
Histone Peptide Pull-Down
Detect the interaction between HERC2ZZ and histones through in vitro pull-down method (Figure 2).
-
39.
Thaw GST-tagged proteins on ice and spin down at 13,000 rpm (15,871 × g) for 10 min.
-
40.
Dilute proteins for binding: each pull-down requires 1–2 μg of GST-tagged protein in 300 μL of binding buffer. Prepare two extra tubes of diluted GST-tagged protein: one for input and one for pull-down without histone peptide as a negative control.
-
41.
Add 1 μg of each biotinylated histone peptides (1 mg/mL) (Synpeptide) into each tube. Incubate at 4˚C with rotation for 4 h.
-
42.
Prepare Streptavidin Magnetic beads (Pierce). Use 5 μL Streptavidin beads for each binding assay. Wash the beads two more times with binding buffer with agitating for 5 min on a tube rotator (Lab Quake) at 20 rpm. After the final wash, resuspend the beads with 10 μL (2-fold of beads volume) of binding buffer.
-
43.
Add 10 μL of 50% beads slurry into each tube and incubate at 4°C with agitating on tube rotator (Lab Quake) at 20 rpm for 1 h.
-
44.
Wash the beads with 1 mL of binding buffer for three times at 4˚C with rotation for 5 min.
-
45.
Resuspend beads in 60 μL of 2X SDS sample buffer.
-
46.
To replace 10% input, take 30 μL of samples from the saved input tube, add 30 μL 2X SDS sample buffer and heat the samples in boiling water.
-
47.
Load 10 μL of each sample for Western blot analysis.
Figure 2.

A Step-By-Step Peptide Pull-Down Procedure to Identify Histone Substrates for HERC2ZZ
NMR Titration Experiments Using HERC2ZZ and the H31–12 or SUMO11–6 Peptides
Validate the interaction between HERC2ZZ and its ligands by NMR spectroscopy (Figure 3A)
-
48.
Concentrate the 15N-labeled WT HERC2ZZ and mutant proteins to 0.1 mM in binding buffer. Add 22 μL Deuterium oxide (Sigma-Aldrich) to 198 μL protein sample and transfer the sample to the Shigemi 5 mm Symmetrical NMR microtube (SHIGEMI Co., LTD).
-
49.
Record 1H,15N heteronuclear single quantum coherence (HSQC) spectra of the protein apo-state at 298K on a Varian INOVA 600 MHz NMR spectrometer using 48 increments, 32 scans, and 1.1 s relaxation delay. Sweep widths of 13.2 ppm and 28 ppm and offsets of 4.63 ppm and 119 ppm are used for the 1H and 15N dimension, respectively. The total acquisition time for each experiment is 65 min.
-
50.
Add increasing amounts of the H31–12 or SUMO11–6 peptide to the NMR sample and record 1H,15N HSQC spectra of the sample after each addition of the ligand until saturation is reached.
-
51.
Process the spectra using the NMRPipe software (Delaglio et al., 1995). Overlay of these spectra will reveal resonance perturbations induced in HERC2ZZ by the peptide binding.
Figure 3.

Characterization of the Interaction between HERC2ZZ and the Histone H3 Peptide
The workflow of biochemical assays: NMR titration (A) and tryptophan fluorescence (B) used to qualitatively and quantitatively characterize the binding of HERC2ZZ to the H3 peptide. The 1H,15N HSQC spectra were recorded at protein:peptide molar ratios of 1:0, 1:1, 1:2, and 1:5.
Measurements of Binding Affinities of HERC2ZZ to H31–12 and SUMO11–6 Peptides by Tryptophan Fluorescence
Quantitative analysis of the interaction of HERC2ZZ with the H31–12 and SUMO11–6 peptides (Figure 3B)
-
52.
Experiments are performed at 25°C on a Fluoromax-3 spectrofluorometer (HORIBA Scientific).
-
53.
Fill a quartz fluorometer cuvette (Starna Cells, INC) with 400 μL binding buffer A and record the spectrum of control. The sample is excited at 295 nm, and tryptophan emission spectra are recorded over a range of wavelengths between 330 nm and 360 nm with a 0.5 nm step size and a 1 s integration time and averaged over three scans.
-
54.
Add HERC2ZZ into the binding buffer to have the final protein concentration of 5 μM. Before adding the protein, remove equal volume of buffer from the cuvette to minimize change of the sample volume. Record spectra using the same parameters as above at 0 min, 5 min and 10 min to make sure that the emission curves are stable before adding ligands into the sample.
-
55.
Add increasing amounts of the H31–12 or SUMO11–6 peptide to the protein sample and record spectra after each addition of the peptide.
-
56.
Analyze the data using a KaleidaGraph 4.5 software (Synergy Software). Other programs such as Prism 8 (GraphPad) and OriginPro (OriginLab) could also be used to analyze the data. The binding affinity is determined using a nonlinear least-squares analysis and the equation:
where ΔI is the observed change of signal intensity, and ΔImax is the difference in signal intensity of the free and bound states of HERC2ZZ. [L] is the concentration of the ligands, [P] is the concentration of HERC2ZZ. The Kd value is averaged over three separate experiments, with error calculated as the standard deviation between three runs.
Co-immunoprecipitation
Detect the interaction between HERC2ZZ and histone H3 in cells (Figure 4)
-
57.
Culture HEK 293T (ATCC CRL-3216) cells in DMEM (Corning, 10-013-CV) medium at 37°C in a 5% CO2 incubator. Perform plasmid transfection when HEK 293T cells reach 50%–60% confluence.
-
58.
Perform transfection reactions in 500 μL Opti-MEM (Gibco). For example, use 2 μg of pcDNA-H3-3Flag, 4 μg of pcDNA-HERC (aa 2,702–2,755) and 18 μL X-tremeGENE™ HP DNA transfection reagent (Roche); for control, use 2 μg of pcDNA-flag vector, 4 μg of pcDNA-HERC (aa 2,702–2,755) plasmids and 18 μL X-tremeGENE™ HP DNA transfection reagent (Roche).
Note: For plasmids, run an agarose gel before transfection to make sure the plasmids are of good quality with high percentage of a supercoil form.
-
59.
Scrape the cells 48 h after transfection and resuspend cells in 1 mL cell lysis buffer. Keep the cell lysate on ice for 30 min, flipping tubes gently occasionally.
-
60.
Sonicate the cells on ice using a Branson SFX250 sonicator using 15% output and intervals of 1 s on and 1 s off for 30–40 s.
-
61.
Spin cells at 12,000 rpm (13,000 × g) for 10 min. Gently transfer supernatant into a new 1.5 mL reaction tube and determine protein concentration. Save 50 μL of lysate as 5% input.
-
62.
Use 30 μL of Flag M2 beads for each binding. Wash flag beads three times with 10X volume of cell lysis buffer containing PMSF. Rock the tubes on tube rotator (Lab Quake) at 4˚C for 5 min each time and spin down the beads at 3,000 rpm (845 × g) for 2 min.
-
63.
Resuspend the beads with same volume of cold cell lysis buffer after the last wash.
-
64.
Use wide bore pipette tips (Axygen) to add 30 μL of FLAG M2 beads into 950 μL of lysate and incubate the lysate with beads at 4˚C with rotation for 4 h. When mixing the beads with the cell lysate, make sure to mix well by gently flipping the bottle up and down until the beads are fully resuspended (∼20 times).
-
65.
Wash the beads for 4–5 times with 1 mL of cold cell lysis buffer.
-
66.
After final wash, add 200 μL of 2X SDS sample buffer into the pellet. For input, add 150 μL of 2X SDS sample buffer.
-
67.
Heat all samples at 95°C for 5 min. Load 10 μL sample for Western blot analysis.
Figure 4.

Cross-validation of the Interaction between HERC2ZZ and Histone H3 Tail Using Co-IP Experiments
Crystallization and Structure Determination of HERC2ZZ in Complex with H3 and SUMO1
Generate the crystals of H31–6-HERC2ZZ (aa 1–6 of histone H3, aa 2,702–2,755 of HERC2ZZ, no linker between) and SUMO11–6-HERC2ZZ (aa 1–6 of SUMO1, aa 2,702–2,755 of HERC2ZZ, no linker between), collect data sets on these crystals and determine the structures of the complexes (Figure 5).
-
68.
The crystallization screening is performed using commercially available JCSG plusTM and PACT premierTM kits (Molecular Dimensions).
-
69.
Concentrate the H31–6-HERC2ZZ and SUMO11–6-HERC2ZZ proteins to 3–5 mg/mL and spin down to remove any precipitates before setting trays. Crystals are grown by equilibrating a mixture containing 1 μL protein solution and 1 μL reservoir solution against 50 μL reservoir solution at 25°C using sitting drop vapor diffusion methods.
-
70.
The H3-HERC2ZZ crystals are obtained in 0.1 M ammonium acetate, 0.1 M Bis-Tris, pH 5.5 and 17% PEG 10,000 condition. The SUMO1-HERC2ZZ crystals were obtained in 0.2 M ammonium chloride, 0.1 M Tris, pH 8.0 and 20% PEG 6,000 condition. Plate-shaped crystals appear within 24 h and grow to their full size within 2–3 days.
-
71.
Loop the crystals using appropriate loops under the optical microscope and cryoprotect the crystals with 25% glycerol before freezing in liquid nitrogen.
-
72.
Collect the crystallization data sets using a high-frequency microfocus X-ray generator. The Advanced Light Source beamline 4.2.2 or a campus X-ray homesource: Rigaku Micromax 007 high-frequency microfocus X-ray generator were utilized for these experiments.
-
73.
Use HKL2000 program (Otwinowski and Minor, 1997) for indexing, integration, and scaling of crystallographic data.
-
74.
Determine the structures by molecular replacement using the Molrep program in CCP4 (Emsley et al., 2010) and a search model with ≥25% sequence similarity. P300ZZ (PDB code: 6DS6) was used to determine phase solutions for the HERC2ZZ structures (Zhang et al., 2018).
-
75.
Perform the model building by using the Coot program (Emsley et al., 2010) and refine the structures with the Phenix Refine program (Adams et al., 2010).
-
76.
Further validate the final model using Worldwide Protein Data Bank OneDep SYSTEM (https://validate-rcsb-1.wwpdb.org/), fix any issues that may arise, if possible.
-
77.
Deposit data in Worldwide Protein Data Bank OneDep SYSTEM (https://deposit-2.wwpdb.org/deposition/)
Note: Timing of crystallization and structure determination may vary based on the protein and data quality.
Figure 5.

Elucidation of the Molecular Mechanism by which HERC2ZZ Recognizes H3 Tail
The N terminus of H3 is shown as green sticks, and residues of HERC2ZZ involved in the interaction with H3 are shown in pink sticks. The crystallization process consists of a broad screening using sitting drop method, generating diffracting crystals, data collection, and structure determination.
Expected Outcomes
In-depth analysis of histone readers and their binding partners is vital to our understanding of complex mechanisms underlying chromatin regulation (Andrews et al., 2016; Musselman et al., 2012) While this methodology was developed to study the ZZ domain of HERC2, it can be broadly used to produce and characterize other ZZ domain-containing proteins or other histone readers in general. In our study, 2 L cell culture grown in LB media yields 1–2 mg of HERC2ZZ, H31–6-HERC2ZZ, and SUMO11–6-HERC2ZZ. We note that cell culture grown in 2 L M9 minimal media produces ∼0.5 mg of 15N-labeled HERC2ZZ.
Quantification and Statistical Analysis
The crystal structures of HERC2ZZ linked to H31–6 and SUMO11–6 were determined using materials and software listed in the Key Resources Table. Statistics generated from X-ray crystallography data processing, refinement, and structure validation is described in (Liu et al., 2020).
Tryptophan fluorescence assays were performed in three independent replicates. The Kd values were determined by a nonlinear least-squares analysis using KaleidaGraph 4.5 (Synergy Software) and represent means ± SD.
Limitations
While the purification steps described here and applications of biochemical methods, including peptide pull-down, NMR, tryptophan fluorescence, co-immunoprecipitation, and crystallization, are suitable for characterization of soluble and folded proteins, this protocol may not work well for unstable, unfolded, insoluble proteins. In addition, expression conditions, such as temperature, duration, IPTG concentration, the cell line often vary for individual proteins and need to be optimized to increase the yield. Tryptophan fluorescence assay requires the presence of at least one tryptophan residue in the protein. If the protein does not contain tryptophan, a tryptophan residue can be introduced through site-directed mutagenesis with QuikChange Lightning Site-Directed Mutagenesis Kit and thermal cycling conditions mentioned above (The primer sequences of HERC2_ZZ_W Forward and HERC2_ZZ_W Reverse are summarized in Key Resources Table). The site for mutagenesis has to be chosen carefully – tryptophan should be positioned near the binding site to detect changes in the fluorescence signal but not too close to the binding site to avoid impeding the interaction. Generally, adding the tryptophan residue at the N terminus or C terminus is a good starting point, and a single mutation should be adequate for such a small size protein (∼6 kDa). NMR titration experiments has its own limitation and can only be applied to a relatively small protein (MW <35 KDa). It also remains challenging to acquire a meaningful NMR spectrum with dispersed crosspeaks for the proteins containing more than one domain.
Troubleshooting
Problem 1
Protein expresses but remains in the cell pellet after lysis (step 8).
Potential Solution
The lysis buffer could be incompatible with the protein. Alter the buffer (for example change Tris to HEPES or PBS), pH, and/or salt concentration. Protein may precipitate if the pH of the buffer is close to the pI of the protein. Ideally, the difference between pI and pH should generally be larger than 1.0 pH unit. Increasing the salt concentration will inhibit hydrophobic interactions, which may cause aggregation and precipitation of the protein. Increase the lysis time, or apply another lysis method, such as high-pressure homogenization. The homogenization process provides a more complete cell disruption, increasing the yield, and producing less heat that may denature the protein of interest. If the protein misfolds during expression, slow down the rate of expression, lower concentration of IPTG to 0.1 mM and lower induction temperature to 12°C–16°C. Further potential solution includes optimization of the construct boundaries by extending or shortening constructs.
Problem 2
Cleavage reaction is incomplete (steps 14 and 20).
Potential Solution
The cleavage site could be occluded. Try to cleave the protein at 25°C or perform an off-resin cleavage. Elute the protein off the resin with 10 mL Ni-NTA elution buffer, and then dialyze the elution back to 1 L lysis buffer A to remove the imidazole. Add the cleavage enzyme (TEV) to the protein solution to remove the tag at 4°C or 25°C. for 20 h. After cleavage, the His tag or GST tag can be removed by incubating with corresponding resin for 30 min or more. The latter procedure increases the cleavage efficiency based on the experience in our lab.
Problem 3
The signal to noise ratio in NMR spectra is low (step 49).
Potential Solution
Increase the concentration of the 15N-labeled protein or increase the number of scans.
Problem 4
The tryptophan fluorescence curve is bouncy (steps 53–55).
Potential Solution
This could arise from the presence of bubbles in buffer. Bubbles can be easily formed when using chilled buffer. Make fresh buffer and sonicate at 25°C for 10 min or centrifuge the buffer at 13,000 rpm (15,871 × g) in an Eppendorf 5424 R Centrifuge with an FA-45-24-11 rotor for 20 min to remove bubbles.
Problem 5
Protein does not crystallize (steps 69 and 70).
Potential Solution
Screen as many conditions as possible, try different protein buffers, alter temperature (between 4 to 25°C) and crystallization method (hanging versus sitting drop, drop ratio, or crystal seeding). Add another purification step, such as ion exchange chromatography to increase purity of the protein.
Problem 6
Binding background shows in beads sample (steps 47 and 67).
Potential Solution
Use magnetic beads to reduce background signals. Try different fragments or different batches of protein.
Problem 7
The results of histone peptide binding assays with different modifications are not consistent between repeats (step 41–47).
Potential Solution
Make sure that the incubation time is the same for all the reactions.
Problem 8
The expression level of transiently transfected protein is not consistent (step 58).
Potential Solution
Run an agarose gel before transfection to make sure the plasmids are of good quality and the concentration is accurate. Count cells with a cytometer prior to plating cells and make the proper dilution needed to ensure seed the same number of cells are plated.
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Tatiana Kutateladze (tatiana.kutateladze@cuanschutz.edu).
Materials Availability
All reagents generated in this study will be made available on reasonable request.
Data and Code Availability
Coordinates and structure factors have been deposited in the Protein Data Bank under ID codes 6WW3 and 6WW4.
Acknowledgments
This work was supported by grants from NIH GM135671, GM125195, CA252707, and HL151334 to T.G.K., and CA204020 to X.S. X.S. is a Leukemia & Lymphoma Society Career Development Program Scholar. Some icons in the figures were created with BioRender.com.
Author Contributions
All authors contributed to writing the manuscript.
Declaration of Interests
The authors declare no competing interests.
Contributor Information
Xiaobing Shi, Email: xiaobing.shi@vai.org.
Tatiana G. Kutateladze, Email: tatiana.kutateladze@cuanschutz.edu.
References
- Adams P.D., Afonine P.V., Bunkoczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.W., Kapral G.J., Grosse-Kunstleve R.W. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews F.H., Strahl B.D., Kutateladze T.G. Insights into newly discovered marks and readers of epigenetic information. Nat. Chem. Biol. 2016;12:662–668. doi: 10.1038/nchembio.2149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaglio F., Grzesiek S., Vuister G.W., Zhu G., Pfeifer J., Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- Emsley P., Lohkamp B., Scott W.G., Cowtan K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrero F., Ciragan A., Iwai H. Tandem SUMO fusion vectors for improving soluble protein expression and purification. Protein Expr. Purif. 2015;116:42–49. doi: 10.1016/j.pep.2015.08.019. [DOI] [PubMed] [Google Scholar]
- Kapust R.B., Tozser J., Fox J.D., Anderson D.E., Cherry S., Copeland T.D., Waugh D.S. Tobacco etch virus protease: mechanism of autolysis and rational design of stable mutants with wild-type catalytic proficiency. Protein Eng. 2001;14:993–1000. doi: 10.1093/protein/14.12.993. [DOI] [PubMed] [Google Scholar]
- Klein B.J., Wang X., Cui G., Yuan C., Botuyan M.V., Lin K., Lu Y., Wang X., Zhao Y., Bruns C.J. PHF20 readers link methylation of histone H3K4 and p53 with H4K16 acetylation. Cell Rep. 2016;17:1158–1170. doi: 10.1016/j.celrep.2016.09.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J., Xue Z., Zhang Y., Vann K.R., Shi X., Kutateladze T.G. Structural insight into binding of the ZZ domain of HERC2 to histone H3 and SUMO1. Structure. 2020 doi: 10.1016/j.str.2020.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi W., Zhang Y., Lyu J., Wang X., Tong Q., Peng D., Xue Y., Tencer A.H., Wen H., Li W. The ZZ-type zinc finger of ZZZ3 modulates the ATAC complex-mediated histone acetylation and gene activation. Nat. Commun. 2018;9:3759. doi: 10.1038/s41467-018-06247-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musselman C.A., Kutateladze T.G. Preparation, biochemical analysis, and structure determination of methyllysine readers. Methods Enzymol. 2016;573:345–362. doi: 10.1016/bs.mie.2015.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musselman C.A., Lalonde M.E., Cote J., Kutateladze T.G. Perceiving the epigenetic landscape through histone readers. Nat. Struct. Mol. Biol. 2012;19:1218–1227. doi: 10.1038/nsmb.2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z., Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- UniProt C. The universal protein resource (UniProt) Nucleic Acids Res. 2007;35:D193–D197. doi: 10.1093/nar/gkl929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Xue Y., Shi J., Ahn J., Mi W., Ali M., Wang X., Klein B.J., Wen H., Li W. The ZZ domain of p300 mediates specificity of the adjacent HAT domain for histone H3. Nat. Struct. Mol. Biol. 2018;25:841–849. doi: 10.1038/s41594-018-0114-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Coordinates and structure factors have been deposited in the Protein Data Bank under ID codes 6WW3 and 6WW4.