

Summary
Alternative splicing greatly expands the coding capacity of the human genome, but how many alternative transcripts are translated as proteins or carry functional importance remains unknown and awaits experimental verification. Here, we describe a protocol that combines transcriptomics (RNA-seq) and proteomics (mass spectrometry [MS]) analyses to identify alternative isoforms in proteomes. This workflow is applicable to custom-generated RNA-seq and MS data from matching samples, as well as the reanalysis of existing transcriptomics and proteomics datasets in public repositories.
For complete details on the use and execution of this protocol, please refer to Lau et al. (2019).
Graphical Abstract
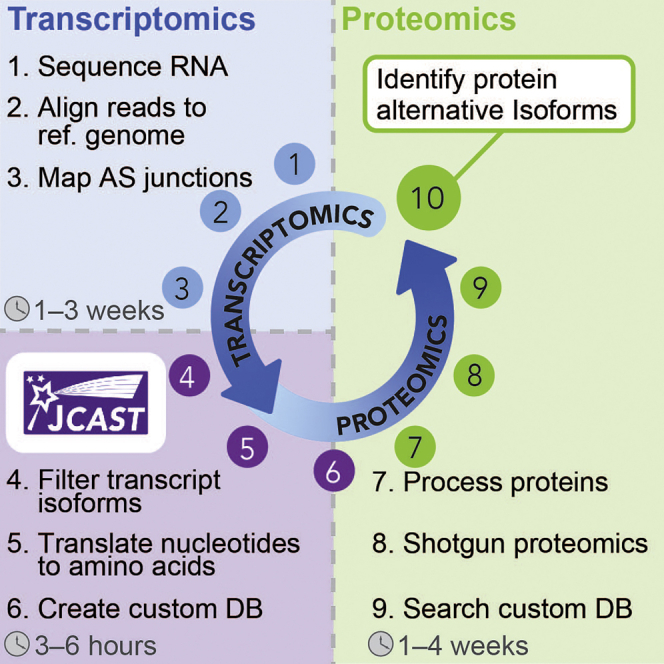
Highlights
-
•
Multi-omics workflow for identification of alternative protein isoforms
-
•
Applicable to publicly available or in-house generated RNA-seq and proteomics data
-
•
JCAST pipeline for creation of sample-specific protein sequence databases
-
•
Enables reanalysis of existing datasets to identify differentially regulated isoforms
Alternative splicing greatly expands the coding capacity of the human genome, but how many alternative transcripts are translated as proteins or carry functional importance remains unknown and awaits experimental verification. Here, we describe a protocol that combines transcriptomics (RNA-seq) and proteomics (mass spectrometry [MS]) analyses to identify alternative isoforms in proteomes. This workflow is applicable to custom-generated RNA-seq and MS data from matching samples, as well as the reanalysis of existing transcriptomics and proteomics datasets in public repositories.
Before You Begin
Preparation of Samples for RNA and Protein Extraction
Timing: ∼1–3 h
-
1.
RNA and proteins can be extracted from fresh cultured cells or frozen cell pellets.
Optional: add an RNA stabilization solution (e.g., RNAlater) to cell pellets prior to freezing.
Preparation of LC-MS/MS Instruments
-
2.
Calibrate the mass spectrometer with an appropriate mass calibration standard solution following manufacturer’s recommendation.
-
3.
Check LC and MS performance with bovine serum albumin protein digests to ensure optimal LC separation and MS detection.
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, Peptides, and Recombinant Proteins | ||
| Acetonitrile LC-MS Grade | VWR | Cat# JT9829 |
| Water LC-MS Grade | VWR | Cat# BDH83645.400 |
| Formic Acid LC-MS Grade | Thermo Fisher Scientific | Cat# 85178 |
| Iodoacetamide | Sigma-Aldrich | Cat# I1149 |
| Dithiothreitol | Sigma-Aldrich | Cat# D5545 |
| Trypsin MS Grade | Thermo Fisher Scientific | Cat# 90057 |
| Lys-C MS Grade | New England Biolabs | Cat# P8109S |
| RIPA | Thermo Fisher Scientific | Cat# 89901 |
| Halt Protease/Phosphatase Inhibitor | Thermo Fisher Scientific | Cat# 78442 |
| RNAlater Stabilization Solution | Thermo Fisher Scientific | Cat# AM7020 |
| TRIzol | Fisher | Cat# 15-596-026 |
| Ethanol Proof 195-200 | Fisher | Cat# 04-355-720 |
| Trypsin/Lys-C MS Grade | Thermo Fisher Scientific | Cat# A40007 |
| Pierce BSA Protein Digest, MS Grade | Thermo Fisher Scientific | Cat# 88341 |
| Pierce LTQ Velos ESI Positive Ion Calibration Solution | Thermo Fisher Scientific | Cat# 88323 |
| Ammonium Bicarbonate | Sigma-Aldrich | Cat# A6141 |
| Critical Commercial Assays | ||
| Pierce Rapid Gold BCA Protein Assay Kit | Thermo Fisher Scientific | Cat# A53225 |
| Quantitative Colorimetric Peptide Assay | Thermo Fisher Scientific | Cat# 23275 |
| Qubit RNA IQ Assay Kit | Thermo Fisher Scientific | Cat# Q33221 |
| Software and Algorithms | ||
| rMATS-Turbo v.0.1 | Shen et al., 2014 | rnaseq-mats.sourceforge.net |
| ProteoWizard msconvert v.3.0.11392 | Chambers et al., 2012 | http://proteowizard.sourceforge.net |
| MAXQuant v.1.6.10.43 | Tyanova et al., 2016 | https://www.maxquant.org/ |
| FastQC v.0.11.9 | Andrews, 2010 | https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ |
| JCAST v.0.2.8 | Lau et al., 2019 | https://github.com/ed-lau/jcast |
| STAR v.2.7.3 | Dobin et al., 2013 | https://github.com/alexdobin/STAR |
| Python v.3.7+ | Python Software | http://www.python.org |
| Pip v.20.2.1 | Python Packaging Authority | https://pip.pypa.io/en/stable/installing/ |
| Other | ||
| Direct-zol RNA Extraction Kit | ZYMO | Cat# R2072 |
| EASY-Spray HPLC Columns | Thermo Fisher Scientific | Cat# ES800A |
| Pierce Detergent Removal Spin Column, 0.5 mL | Thermo Fisher Scientific | Cat# 87777 |
| C18 Spin Columns | Thermo Fisher Scientific | Cat# 89870 |
| High pH Reversed-Phase Peptide Fractionation Kit | Thermo Fisher Scientific | Cat# 84868 |
| Protein LoBind tubes 2.0 mL | Eppendorf | Cat# 022431102 |
| Thermo Q Exactive HF Mass Spectrometer | Thermo Fisher Scientific | Cat# IQLAAEGAAPFALGMBFZ |
| EasynLC 1200 Nano LC | Thermo Fisher Scientific | Cat# LC140 |
| Omni Tissue Homogenizer (TH) | OMNI International | Cat# TH115-PCR5D |
| Benchtop Centrifuge with Temperature Control | Thermo Fisher Scientific | Cat# 75002441 |
| Nanodrop | Thermo Fisher Scientific | Cat# ND-2000 |
| Sonicator | Fisher Scientific | Cat# FB120110 |
| Qubit Fluorometer | Thermo Fisher Scientific | Cat# Q33238 |
Materials and Equipment
| LC Solvent A | Final Concentration | Amount |
|---|---|---|
| Formic Acid | 0.1% (v/v) | 1 mL |
| MS H2O | 999 mL | |
| Total | 1,000 mL |
| LC Solvent B | Final Concentration | Amount |
|---|---|---|
| Formic Acid | 0.1% (v/v) | 1 mL |
| Acetonitrile | 80% (v/v) | 800 mL |
| MS H2O | 199 mL | |
| Total | 1,000 mL |
| Ammonium Bicarbonate (ABC) (pH ~8.0) | Final Concentration | Amount |
|---|---|---|
| ABC | 100 mM | 79.06 mg |
| MS H2O | 10 mL | |
| Total | 10 mL |
| Iodoacetamide (IAA) | Final Concentration | Amount |
|---|---|---|
| IAA | 375 mM | 34.68 mg |
| MS H2O | 0.5 mL | |
| Total | 0.5 mL |
| Dithiothreitol (DTT) | Final Concentration | Amount |
|---|---|---|
| DTT | 60 mM | 46.28 mg |
| MS H2O | 5 mL | |
| Total | 5 mL |
| MS-grade Trypsin/Lys-C Mix Stock | Final Concentration | Amount |
|---|---|---|
| Trypsin/LysC mix | 1 μg/μL | 20 μg |
| MS H2O | 20 μL | |
| Total | 20 μL |
| RIPA Buffer with 1× Halt Protease Inhibitor | Final Concentration | Amount |
|---|---|---|
| Halt protease inhibitor cocktail (100×) | 1× | 10 μL |
| MS H2O | 990 μL | |
| Total | 1 mL |
CRITICAL: formic acid is highly irritating and corrosive to eyes, skins, and the respiratory system. Handle it under a chemical hood wearing gloves and protective eye goggles. ACN is flammable and should be handled with great caution.
Alternatives: alternatively, instead of using the trypsin/LysC mix, prepare individual stock (1 μg/μL) of trypsin and LysC as follows: dissolve 20 μg Trypsin or LysC in 20 μL LC-MS grade water. Store at −80°C.
Alternatives: alternative cell lysis buffer (e.g., M-PER Mammalian Protein Extraction Reagent) and protease inhibitor cocktails (e.g., Roche cOmplete Protease Inhibitor Cocktail) of preference can be used.
LC-MS/MS Preparation
MS Instrument Setup
-
•
For a typical MS Data Dependent Acquisition (DDA) experiment performed with a Thermo Fisher Q Exactive HF, set up instrument parameters as follows or modify according to instrument and/or sample type (Table 1):
Table 1.
MS Setup for MS Survey Scan and MS2 Scan
| MS Survey Scan | |
|---|---|
| Resolution | 60,000 |
| Mass range | 300–1,650 m/z |
| Maximum injection time | 20 ms |
| Automatic gain control target | 3e6 |
| MS2 Scan | |
| TopN | 15 |
| Resolution | 60,000 |
| Automatic gain control target | 2e5 |
| Maximum injection time | 110 ms |
| Isolation window | 1.4 m/z |
| Normalized collision energy (NCE) | 32 or stepped NCE of 27, 30, 32 |
Nano LC Setup
-
•
Equilibrate Nano LC column for 10 to 20 column volumes and set up Nano LC gradient to be run at a flow rate of 300 nL/min with the following gradient profile (Table 2):
-
•
Depending on MS instrument and sample type, inject ∼1 μg to 3 μg of digested peptides after C18 clean-up or from each high pH fraction to a C18 reversed-phase column (e.g., Thermo Scientific EASY-Spray HPLC Columns) with the LC autosampler or an injector.
Table 2.
Reversed-Phase Liquid Chromatography Gradient
| Minutes | % B |
|---|---|
| 0–105 | 0–40 |
| 105–110 | 40–70 |
| 110–115 | 70–100 |
| 115–120 | 100 |
Step-By-Step Method Details
Transcriptomics Analysis: RNA Extraction and Sequencing: Mammalian RNA Extraction from Frozen Cell Pellet
Alternatives: see “Retrieving Publicly Available RNA-seq Data”
The goal of this step is to extract high-quality RNA samples for downstream RNA sequencing.
-
1.
Place frozen cell pellets in sample tubes on dry ice and add 600 μL (for <5 × 106 cells) TRIzol directly to each frozen pellet. Transfer the sample tube on ice. Homogenize cells with a high-speed tissue homogenizer (e.g., OMNI TH) for 10 s, then cool down the samples on ice for 1 min. Repeat this step two additional times.
Note: Process one tube of cell pellets at a time. Homogenized samples in TRIzol can be kept on ice until all samples have been homogenized.
-
2.
Centrifuge at 16,000 × g for 15 min at 4°C to remove debris. Transfer the supernatant to a new 2.0 mL RNase-free tube.
-
3.
Measure the sample volume using a 1,000 μL pipette. Add an equal volume of ethanol (95%–100%) to the sample and mix thoroughly by pipetting up and down. Proceed to RNA extraction using a modified protocol with the Direct-zol RNA MiniPrep kit (steps 4–11). All reagents mentioned below are supplied in the Direct-zol RNA kit.
-
4.
Transfer up to 700 μL mixture into a Direct-zol spin column in a 2.0 mL collection tube. Centrifuge at 16,000 × g for 30 s at 20°C–22°C, discard the flow through. Transfer the remaining sample mixture into the spin column and repeat centrifugation.
-
5.In-column DNase I treatment:
-
a.Add 400 μL RNA Wash Buffer to the spin column and centrifuge at 16,000 × g for 30 s at 20°C–22°C.
-
b.In a new RNase-free tube, mix 75 μL DNA Digestion buffer with 5 μL DNase I (6 U/L; lyophilized DNase I is reconstituted in DNase/RNase - free water and stored at −20°C).
-
c.Add the mixture to the column and incubate at 20°C–22°C for 15 min.
-
a.
-
6.
Add 400 μL RNA PreWash Buffer to the column and centrifugate at 16,000 × g for 30 s at 20°C–22°C. Repeat this step two additional times.
-
7.
Add 700 μL RNA Wash Buffer to the column and centrifugate at 16,000 × g for 30 s at 20°C–22°C. Repeat this step two additional times.
-
8.
Centrifugate at 16,000 × g for 30 s to remove any remaining buffer in the column.
-
9.
Add 50 μL DNase/RNase-free water to the column. Sit the column at 20°C–22°C for 1 min. To elute RNA, centrifugate at 16,000 × g for 1 min at 20°C–22°C.
Note:
-
•
To improve RNA recovery, warm DNase/RNase-free water (up to 60°C) can be added to the column and allow to incubate for 3–5 min.
-
•
Use a smaller amount of water (≥ 35 μL) to elute RNA (if higher concentration of RNA is desired).
-
10.
Assess RNA quantity and quality by NanoDrop, Qubit, or Agilent Bioanalyzer. For RNA sequencing. See below for an example of typical RNA QC results using NanoDrop and Qubit IQ Integrity assay (with RNA extracted from two mouse tissues) (Table 3).
-
11.
Proceed to the next step “RNA Sequencing and Quality Control of Raw Sequencing Data”.
Pause Point: Snap freeze RNA samples in liquid nitrogen and store RNA at −80°C.
Table 3.
Examples of RNA QC Results
| Sample ID | NanoDrop A260/280 | NanoDrop A260/230 | Qubit RNA IQ |
|---|---|---|---|
| Sample 1 | 2.03 | 2.16 | 8.9 |
| Sample 2 | 2.02 | 2.15 | 9.0 |
RNA Sequencing and Quality Control of Raw Sequencing Data
-
12.
Perform short-read RNA sequencing, e.g., pair-ended mRNA sequencing with up to 150 bp read length on an Illumina Next-Seq platform.
-
13.
Quality assessment of raw sequencing data by using quality control (QC) software packages is recommended to ensure sequencing read quality. Example tools include FastQC (Andrews, 2010). Common quality metrics include sequence quality, adapter content, and overrepresented sequences/k-mers.
Retrieving Publicly Available RNA-Sequencing Data
As an alternative to generating custom RNA-seq data, it is possible to access and retrieve publicly available RNA-seq data for the purpose of generating custom protein sequence databases. RNA-seq data should be selected based on similarity or identity of sample tissue or cell type to the proteomics experiments counterparts in order to support sample-specific protein isoform discovery. Another emphasis should be on sequencing depth because of the low abundance of many alternative isoforms. High-quality RNA-seq data for different biological samples may be accessed from NCBI GEO or as part of the data generated by the ENCODE consortium. To download ENCODE cell and tissue data for custom database generation.
From https://www.encodeproject.org, input search terms of sample type through “Search ENCODE portal.”
Alternatives: sample types can be looked up via https://www.encodeproject.org/matrix/?type=Experiment&status=released
To retrieve datasets, follow the links to the sample and download the FASTQ files.
Follow the alignment and database generation steps below.
Generating Custom Databases of Alternative Protein Isoforms
Note: We provide a Python software Junction Centric Alternative Splicing Translator (JCAST; https://github.com/ed-lau/jcast), which is employed in the example workflow below to create sample-specific custom protein sequence databases for protein isoform identification. RNA-sequencing reads from biological replicates (e.g., n=3) of the same sample type are first mapped to a reference genome by an aligner to generate BAM files to be used as input files for rMATS (Shen et al., 2014) to identify splice junction nucleotides expressed in all replicates. JCAST then translates the junction nucleotide sequences into peptide sequences and recover full-length protein sequences to create sample-specific protein isoform databases. In the example below, we use STAR v.2.7.3 (Dobin et al., 2013) to map acquired RNA-sequencing reads to a reference genome. Other aligners including Tophat2 (Kim et al., 2013) and HISAT2 (Kim et al., 2019) can also be employed to map the RNA-sequencing reads for rMATS input.
Note: We provide minimal example commands below in order to execute the workflow on a Linux system (Ubuntu 18.04.4). Example or placeholder file names are provided for reference where applicable. Please refer to the documentations and publications from the software developers of STAR and rMATS for additional instructions and troubleshooting guides.
Alternatives: Besides JCAST, other software tools and workflows exist that can be used to translate RNA-sequencing reads into custom proteomics databases with different strengths and limitations. One example is alternative workflow world employed the software customProDB (Wang and Zhang, 2013) or other compatible tools to perform three-frame translation on assembled transcripts (e.g., from TopHat/StringTie to create custom FASTA databases).
Alignment of RNA-Sequencing Reads
The goal of this step is to map RNA-sequencing reads to a reference genome and generate BAM files containing alignment information.
-
14.
Download the genome assemblies from ensembl.org at ftp://ftp.ensembl.org/pub/. Select the most recent release folder (e.g., release-100; release date: 06/03/2020), navigate to the “fasta” folder and open the folder containing assemblies for the species of interest (we use Homo sapiens as an example in the following steps). Within the “homo_sapiens” folder, navigate to the “dna” folder and download the genome assembly by single clicking on the file named “Homo_sapiens.GRCh38.dna.primary_assembly.fa.gz.”
Note: Use primary assemblies rather than masked/soft masked genomes for alignment.
-
15.
Download the gene annotation GTF file from Ensembl. Within the “release-100” folder, open the “gtf” folder and then the “homo_sapiens” folder. Download the genome annotation file by single clicking on the file “Homo_sapiens.GRCh38.100.gtf.gz.”
-
16.Unzip downloaded assembly and annotation files.
- $ gunzip filepath/Homo_sapiens.GRCh38.dna.primary_assembly.fa.gz
- $ gunzip filepath/Homo_sapiens.GRCh38.100.gtf.gz
-
17.Download and install STAR v.2.7.3 or above.
- $ tar -xzf 2.7.3a.tar.gz
- $ cd STAR-2.7.3a/source
- $ make STAR
-
18.Build the STAR genome index for alignment.
-
$ STAR --runThreadN 8 --runMode genomeGenerate --genomeDir filepath/[STARindex file name] --genomeFastaFilesfilepath/Homo_sapiens.GRCh38.dna.primary_assembly.fa --sjdbGTFfilefilepath/Homo_sapiens.GRCh38.100.gtf --sjdbOverhang 149
-
Note: --runThreadN defines the number of threads to be used.
Note: --sjdbOverhang defines the length of the genomic sequence to be used in the construction of a splice junction database. The ideal value is ReadLength -1, e.g., for Illumina 2 × 150 bp paired-end reads, the value is 150 − 1 = 149. Alternatively, the default value of 100 works as well as the (ReadLength -1) value in most cases.
Note: Replace [STAR index file name] with the desired file name for the STAR index to be generated.
Note: Refer to the STAR documentations and publications for troubleshooting.
-
19.Align RNA- sequencing reads with STAR (e.g., paired-end reads R1.fastq.gz and R2.fastq.gz)
-
$ STAR --runThreadN 8 --genomeDir filepath/filename-of-STARindex --readFilesIn filepath/R1.fastq.gz filepath/R2.fastq.gz --readFilesCommandzcat --outSAMtype BAM SortedByCoordinate --outFileNamePrefix filepath/prefix
-
Note: --genomeDir defines the path to the genome indexes files generated in step 5.
Note: --readFilesIn specifies the read files to be mapped. Read files can be single-end reads or paired-end reads with R1 and R2 files separated by space. To map multiple samples, separate samples by comma, for example,
filepath/sample1.replicate1.fastq.gz,filepath/sample2.replicate1.fastq.gz
filepath/sample1.replicate2.fastq.gz,filepath/sample2.replicate2.fastq.gz.
Note: --readFilesCommand is required when input read files are compressed.
Refer to the STAR GitHub repository, documentations, and publications from the developer for additional instructions on installation and usage. (https://github.com/alexdobin/STAR).
Identification of Transcript Splice Junctions
The goal of this step is to identify splice junction nucleotide sequences that are expressed at mRNA level in the sample.
-
20.
Download the rMATS-turbo-0.1 docker image from https://sourceforge.net/projects/rnaseq-mats/files/MATS/rmats-turbo-0.1.tar.gz/download.
-
21.Install docker.
- $ sudo apt install docker.io
-
22.Unzip the compressed files.
- $ gunzip rmats-turbo-0.1.tar.gz
-
23.Install the rMATS-turbo-0.1 docker image.
- $ sudo docker load -i rmats-turbo-0.1.tar
-
24.Move the STAR output bam files and genome annotation gtf file into a designated folder (e.g., mydata). Specify the names of the STAR output BAM files in .txt files.
- $ cd mydata
- $ cat >b1.txt
- /data/Sample1-replicate1.bam,/data/Sample1-replicate2.bam
- Ctrl+Z
- $ cat >b2.txt
- /data/Sample1-replicate3.bam,/data/Sample1-replicate4.bam
- Ctrl+Z
Note: On some computer systems, Docker may lack permission to access the data directory (e.g., the folder “mydata” in our example) depending on security settings. Modify access permissions of files/directories with chrom as necessary or mount the host directory to a container directory with the “-v” option while using “docker run” as below (see also step 6; consult Docker documentation for details: https://docs.docker.com).
- $ docker run -v /host_directory:/container_directory run_commands
The created b1.txt and b2.txt files list the bam files of replicates from the same sample. In this protocol, we rely on the quantitative and statistical output of rMATS to identify the splice junctions that are consistently expressed in all replicates at appreciable levels and discard the other junctions.
-
25.Process the bam files from STAR outputs using rMATS to extract splice junction information.
-
$ sudo docker run -v ∼/mydata:/data rmats:turbo01 --b1 /data/b1.txt --b2 /data/b2.txt --gtf /data/Homo_sapiens.GRCh38.100.gtf --od /data/output -t paired --nthread 4 --readLength 150
- --anchorLength 1
-
Note: Change readLength value to the length of reads in your RNA-sequencing data.
Note: Refer to the rMATS documentations and publications for more information about usage of rMATS docker image at http://rnaseq-mats.sourceforge.net/rmatsdockerbeta/DockerImage-rMATS-turbo-0.1.pdfhttp://rnaseq-mats.sourceforge.net
Optional: Set up a virtual environment for rMATS in Python 2.7 if needed. Python 2.7 can be acquired from https://www.python.org. Instruction to setting up a virtual environment in Python 2.x using virtualenv can be found in the Python documentations: https://packaging.python.org/guides/installing-using-pip-and-virtual-environments/
In Silico Translation of Custom Protein Sequences
JCAST processes rMATS output files with options to filter out splice junctions with low or variable counts and then translates junction sequences into peptide and protein sequences.
-
26.
Install Python3.7+ (https://www.python.org) and pip (https://pip.pypa.io/en/stable/installing/) following developer instruction.
-
27.Install JCAST v.0.2.8 or a compatible version
- $ python3 -m pip install jcast
Note: JCAST can be acquired through pypi using pip or directly from Github. JCAST requires Python 3.7+ which can be acquired from https://www.python.org
Optional: To avoid potential conflicts in Python and dependency versions with other softwares, we recommend setting up a Python virtual environment for JCAST. For example, use the following command to set up a Python3.8 virtual environment
$ python3.8 -m venv ∼/venv
To use the virtual environment:
$ source ∼/venv/bin/activate
To install JCAST:
(venv) $ pip install jcast
Note: Further instruction for setting up a virtual environment in Python 3.x using venv can be found in the Python documentations: https://packaging.python.org/guides/installing-using-pip-and-virtual-environments/
To test whether JCAST is installed and print the help message for JCAST usage, type “jcast” in the Terminal command window:
- $ python3 -m jcast
- Or
(venv) $ jcast
The following message will show up in the command window:
usage: jcast [-h] [-o OUT] [-r READ] [-p PVALUE] rmats_folder gtf_file genome
Jcast retrieves splice junction information and translates into amino acid
positional arguments:
rmats_folder path to folder storing rMATS output
gtf_file path to ENSEMBL GTF file
genome path to Genome file
optional arguments:
-h, --help show this help message and exit
-o OUT, --out OUT name of the output files [default: psq_out]
-r READ, --read READ minimum read counts to consider [default: 1]
-p PVALUE, --pvalue PVALUE discard junctions with rMATS pvalue below this threshold [default: 0.01]
Note: JCAST provides the options to filter out splice junction sequences with low read counts by the -r argument and sequences with variable read counts by the -p argument.
Note: p is set at 0.01 by default, which means differentially expressed junctions (p < 0.01) between the biological replicates from the same sample group will be discarded. Only junctions expressed in all biological replicates will be kept and translated into peptide and protein sequences.
-
28.Process outputs from rMATs (RI.JC.txt, A5SS.JC.txt, A3SS.JC.txt, MXE.JC.txt, SE.JC.txt) and generate custom protein databases for protein identification.
-
(venv) $ jcast path/to/rmats_folder/ path/to/Homo_sapiens.GRCh38.100.gtfpath/to/Homo_sapiens.GRCh38.dna.primary_assembly.fa -o OUT
-
Note: JCAST requires three inputs. The first is the path to the folder of the splice junction files from rMATS output. The second is the path to the genome annotation file, and the third is the path to the genome FASTA file.
Note: rMATS generates two output files for each splicing event, JC.txt and JCEC.txt. JC.txt includes sequences that span splice junctions of two adjacent exons and is what we use as JCAST input. JCEC.txt contains sequences which span splice junctions and locates within alternative exons.
Note: The output of JCAST is a number of protein sequence database (FASTA) files named according to their content. These files include canonical sequences in the psq_canonical.fasta file, non-canonical/alternative sequences in one of four translation tiers in the psq_T1.fasta, psq_T2.fasta, psq_T3.fasta, and psq_T4.fasta files, as well as orphan sequence fragments that are not compatible with the Swissprot canonical sequence at the N- and/or C- terminal ends. Refer to the JCAST documentation for details on the translation tiers. The FASTA files can be combined manually using a text editor to yield the custom protein sequence databases used in database search for the shotgun proteomics workflows below.
Optional: To test JCAST performance, download a small dataset containing rMATS input files, human genome chromosome 15 annotation file and assembly file from https://github.com/ed-lau/jcast/tree/master/tests/data. Run JCAST:
(venv)$ jcast path/to/rmats_folder/
path/to/Homo_sapiens.GRCh38.89.chromosome.15.gtf
path/to/Homo_sapiens.GRCh38.dna.chromosome.15.fa -o OUT
With this test dataset JCAST will create protein sequence databases including two protein isoform sequences for the gene PKM. One is the canonical sequence which can be found in the psq_canonical.fasta file (>sp|P14618|KPYM_HUMAN Pyruvate kinase PKM), corresponding to the UniProt P14618 (KPYM_HUMAN) canonical sequence. The other non-canonical PKM isoform can be found in the psq_T1. fasta file (>sp|P14618|KPYM_HUMAN|E NSG00000067225|MXE2) and differs with the canonical sequence at amino acids 389–426 IYHLQLFEELRRLAPITSDPTEATAVGAVEASFKCCSG. The alternative peptide sequence was generated by a mutually exclusive splicing (MXE) event at the mRNA transcript.
Optional: if several samples (i.e., a time course) need to be analyzed, this entire process may be batched using a shell script that loops through individual rMATS output folders.
Mammalian Protein Extraction from Frozen Cell Pellets
This goal of this step is to extract and solubilize proteins from cellular samples for MS experiments.
-
29.
Place frozen cell pellets in sample tubes on dry ice. Add 1 mL of cold RIPA buffer containing 1× Halt Protease Inhibitor Cocktail into one sample tube with frozen cell pellets (∼5 × 106 cells).
Note: The RIPA buffer and protease inhibitor mixture should be freshly prepared and kept on ice.
Optional: RNA and protein can be extracted simultaneously from the same sample. Extractions can be performed using acidic guanidinium-thiocyanate-phenol (TRIzol; Life Technologies) followed by chloroform extraction of RNA in the aqueous phase and precipitation of proteins in the organic phase, or using solid-phase columns and reagents such as the AllPrep DNA/RNA/Protein Mini Kit (QIAGEN) following manufacture’s manual. Please see Limitations section for additional discussion on experimental design.
-
30.
Vortex for 10 s.
-
31.
With a handheld Omni TH homogenizer, homogenize cells on ice at high speed for 10 s. Sit the cell pellets in sample tubes on ice for 1 min to avoid overheating samples by homogenization.
-
32.
Repeat step 3 for two additional times for a total of three rounds of homogenization.
Note: Process one tube of cell pellets at a time. Homogenized samples in the RIPA buffer can be kept on ice until all samples have been homogenized.
-
33.
Clean the homogenizer probe between different samples by rinsing with 50% LC-MS grade isopropanol or ethanol followed by LC-MS grade H2O for three times.
-
34.
With a handheld sonicator (we use Fisher Model 120), sonicate homogenized samples at 40% amplitude for 1 s, pause for 5 s. Repeat step 6 for a total of 15 cycles.
-
35.
Centrifuge samples at 5,000 × g for 1 min at 4°C, and vortex for 10 s.
-
36.
Repeat steps 6 and 7 for two additional times for a total of three rounds of sonication.
-
37.
Centrifuge samples at 14,000 × g for 15 min at 4°C. Collect supernatants and measure protein concentration with the BCA protein assay kit following manufacturer instruction.
-
38.
Proceed to the next step “Protein Digestion using Trypsin and Lys-C,” or snap freeze protein samples in liquid nitrogen and store at −80°C.
Protein Processing and Shotgun Proteomics Analysis
This goal of this step is to reduce, alkylate, and proteolyze proteins into peptides for MS experiments.
-
39.
Remove detergent from protein samples using the Detergent Removal Spin Column (Pierce) following manufacturer instruction.
-
40.
Quantify protein concentration with the BCA Protein Assay Kit (Thermo).
-
41.
Measure 100 μg protein and adjust sample volume to 100 μL in a protein low-bind Eppendorf tube with ammonium bicarbonate (100 mM).
-
42.
Add 10 μL of 60 mM DTT into the sample. Wrap the sample tube with parafilm to prevent samples from drying out or accidental spillage. Vortex for 10 s and quickly spin on a mini spin centrifuge. Incubate at 55°C for 30 min with 600 rpm shaking.
-
43.
Quickly spin the sample on a mini spin centrifuge. Allow the sample to cool down to 20°C–22°C.
-
44.
Add 5 μL of 375 mM IAA to the sample. Vortex for 10 s and quickly spin on a mini centrifuge. Incubate in the dark at 20°C–22°C for 30 min with optional 600 rpm shaking.
-
45.
Quickly spin the sample on a mini centrifuge. Add 2 μg of MS-grade trypsin/Lys-C Protease Mix to the sample and wrap the tube with parafilm to prevent the sample from drying. Incubate at 37°C with optional 600 rpm shaking for 16–20 h.
Note: The ratio of proteases to protein can range from 1:100–1:20 (w/w) depending on the digestion efficiency on the samples.
Alternatives: Instead of using the trypsin/Lys-C mix, a sequential digestion can be performed in step 45: first, add 1 μg of MS-grade Lys-C to the sample and incubate the sample at 37°C for 3 h with 600 rpm shaking; next, add 1 μg of MS-grade trypsin into the sample and incubate the sample at 37°C with 600 rpm shaking for 16–20 h.
-
46.
Quantify peptide amount with the Pierce Quantitative Colorimetric Peptide Assay.
-
47.
Proceed to the next step “Peptide Clean-Up for Downstream Mass Spectrometry Analysis.”
Peptide Clean-Up for Downstream Mass Spectrometry Analysis
This goal of this step is to clean up the peptides to remove interfering contaminants (e.g., salts) for MS analysis.
Option 1
Clean up samples with the Pierce C18 mini spin columns for MS analysis. Dry eluted peptides in a vacuum dryer and resuspend peptides in 0.1% formic acid (in LC-MS grade water). Quantify peptides concentration using a quantitative peptide assay.
Option 2
Process samples with the Pierce High pH Reversed-Phase Peptide Fractionation Kit. This kit will clean up peptides as well as perform an offline fractionation of peptides. This is necessary if two-dimensional- (2D) LC separation is intended. Dry eluted peptides in a vacuum dryer and resuspend peptides in 0.1% formic acid (in LC-MS grade water). Quantify peptides concentration using a quantitative peptide assay.
Note: Due to the potentially low abundance of protein alternative isoforms, we recommend the use of 2D-LC separation prior to MS acquisition to increase proteome coverage.
Mass Spectrometry and Liquid Chromatography
Retrieving Publicly Available Shotgun Proteomics Data
As an alternative to analysis of newly generated MS data, the custom protein sequence databases output by JCAST can be used to re-analyze existing data including publicly available proteomics data from persistent repositories. For example, it is possible to access and retrieve publicly available shotgun proteomics data from ProteomeXchange/PRIDE as an alternative approach to generating experimental data for protein alternative isoform identification. Proteomics data should be selected based on their similarity in tissue type and experimental condition to the RNA-seq data used to generate custom protein databases.
Note: Access PRIDE via https://www.ebi.ac.uk/pride/; Input search terms for datasets of interest (e.g., “cardiac”) into the “Search” field on the front page.; Follow links to datasets and download the .raw files.
Protein Database Search
The goal of this step is to search the acquired MS spectra against the custom isoform databases to identify expressed protein isoforms in the samples.
-
48.
Download and install msconvert v.3.0.11392 (Chambers et al., 2012) (Tools - ProteoWizardproteowizard.sourceforge.net › tools) on a Windows or Linux computer.
-
49.Convert vendor specific .raw files into .mzML files using msconvert with the following command in the Windows Command Prompt (Windows):
- filepath\output folder\”filepath\msconvert.exe” “filepath\MS.raw” --filter “peakPicking vendor”
-
50.Peptide and Protein identification by MaxQuant v.1.6.10.43 (Tyanova et al., 2016) with its built-in search engine Andromeda.
-
a.Download and install MaxQuant software from https://www.maxquant.org/ following developer instructions.
-
b.To load the custom proteome database generated by JCAST, go to the “Global Parameters” session and choose the “Sequences” tab. Click the “Add” button to designate the file path to the FASTA file. Once the upload is complete, the FASTA file will be displayed in the window. An example screen capture is shown below where the file Human_Heart_JCAST.fasta is uploaded in MaxQuant (Figure 1).
-
a.
Note: The exact user interface may differ based on operating systems or the version of MaxQuant used, and may change in future MaxQuant updates. Please refer to the latest MaxQuant documentations for detailed instructions.
-
c.Typical parameter settings are shown in the table below (Table 4). Use default values for parameters not specified here.
-
d.MaxQuant stores search results as txt files containing information about identified peptides and proteins in the folder “\combined\txt\.” A detailed description of each output file can be found in the “tables” pdf file. To collect isoform-specific unique peptides, open “peptides.txt” file and check the values in the column “Unique (Proteins)”, if the value is “yes” then this peptide is unique to a single protein sequence in the FASTA protein database; if the value is “no” then this peptide is a razor peptide shared by multiple protein sequences. The MS2 scan information for all peptides can be found in the txt file “msms.txt”. Identification confidence can be determined by the Posterior Error Probability (PEP) values and Andromeda score with lower PEP values and higher Andromeda scores indicating higher confidence.
-
c.
Alternatives: Besides MaxQuant, the custom sequence databases are compatible with other popular proteomics database search engines including Thermo ProteomeDiscoverer.
Figure 1.

Loading a Custom Database through the MaxQuant User Interface
Screenshot showing a custom protein sequence database generated by JCAST (Human_Heart_JCAST.fasta) being loaded to MaxQuant for database search. Databases are loaded by clicking the “Add” under the “Global Parameters” > “Sequences” tabs. Additional options, including Identifier rule, Description rule, and Taxonomy can be specified (see MaxQuant documentation for details).
Table 4.
MS Spectrum Searching Parameters in MaxQuant
| MaxQuant Parameter | |
|---|---|
| Digestion enzyme | LysC and trypsin |
| Maximal missed cleavage | 2 |
| Fixed modification | carbamidomethylation of cysteine |
| Variable modification | N-terminal protein acetylation; oxidation of methionine |
| Peptide length range | 7-25 aa |
| Precursor mass tolerance | ±4.5 ppm |
| MS/MS ions mass tolerance | ±20 ppm |
| False discovery rate (FDR) for peptide-spectrum match (PSM) and protein identification | 0.01 |
| Peptide for protein quantification | unique (minimal number =1) |
Expected Outcomes
This protocol generates custom protein sequence databases from RNA-sequencing data containing sample-specific information on gene and protein isoform expression. The generated custom protein sequence database is then used to support the identification of proteins and protein alternative isoforms in mass spectrometry-based proteomics data. When combined with a quantitative shotgun proteomics workflow (e.g., employing isobaric labels), this protocol allows large-scale discovery and quantification of isoform expression changes in different samples at a proteome level, including previously documented alternative protein isoforms and novel protein isoform candidates. The results can further be coupled to downstream targeted MS applications for the targeted detection and quantification of isoform peptides across samples.
Figure 2 shows an example peptide-spectrum match using RNA-seq guided protein sequence database. A deep short-read RNA-seq (100 M) data set was generated from human iPSC-derived cardiomyocyte, identifying an MXE event for the PDLIM5 gene. The translated database was used to search against a TMT-labeled shotgun proteomics dataset of various human iPSC-cardiomyocyte time points (Lau et al., 2019), and supported the identification of an alternative PDLIM5 isoform through the isoform-specific peptide sequence SGPHPPATPQVLTIGSQVATLSK (PEP 1.6e–3) (top), which correspond to the uncharacterized isoform 6 of PDLIM5 on UniProt. When the spectrum was searched against the canonical SwissProt human database, it was identified to another sequence IQSNYDIAIAELHVQKSK belonging to the KIF2B protein, but with fewer matching peaks and at notably lower confidence (PEP 0.93) (bottom).
Figure 2.

Identification of Non-canonical Protein Isoforms
A peptide-spectrum match is shown showing an experimental spectrum matched to a non-canonical peptide sequence in a custom database, leading to the identification of a PDLIM5 isoform (top). Searching the same spectrum against a canonical Swissprot protein database without the non-canonical protein sequence led to a low-quality match to a different peptide.
Quantification and Statistical Analysis
Many contemporary proteomics workflows employ target-decoy databases and post-search modeling to help filter peptide identification and ascertain false discovery rates (FDR). Because the inclusion of alternative protein isoform sequences can potentially expand the database search space, these methods should be taken into consideration during database search using the custom sequence databases. In particular, an appropriate decoy database that accounts for alternative sequences should be employed. A stringent FDR threshold should be chosen (e.g., q ≤ 0.01 or based on PEP), and other distinguishing information may be used to adjudicate the likelihood of alternative sequence identification, including whether the alternative sequence has been known to exist in similar samples from past experiments, the existence of sister peptides from the same isoform, and the expected retention time of the sequence.
In addition to statistical approaches to determine false discovery, peptide spectrum matches for protein isoform sequences should be reviewed to consider possible alternative explanations to the origin of the identifying spectrum, which can include single amino acid variants of the canonical protein, known or unknown post-translational modifications causing mass shifts, or exogenous peptide sequences arising from another genome or transcriptome (Nesvizhskii, 2014). Where applicable, orthogonal means to verify potential novel sequences should be sought, which may include immunoblot analysis using various antibodies known to target specific amino acid segment epitopes, targeted MS in conjunction with chemically synthesized peptides, or in vitro expression of sequences of interest.
Limitations
For the acquisition of original RNA-sequencing and MS datasets, this protocol assumes RNA and proteins to be extracted from identical samples, which may require a higher quantity of initial samples available over conventional proteomics experiments. An alternative design would be to perform stepwise extraction such as using TRIzol-chloroform extraction or commercially available silica columns that extract RNA and proteins sequentially from one aliquot of samples. An advantage of this alternative approach would be a decrease in total sample amount requirement as well as sample variability, such as from variations between biological replicates or between different tissue region sampling. A potential disadvantage is the protein precipitation step from sequential extraction protocols may decrease protein yield due to the difficulty of resolubilizing precipitated pellets, or otherwise exclude some proteins based on their physicochemical properties. The RNA and protein extraction protocols, quality, and yield will need to be optimized for the specific biospecimens analyzed or experimental design.
This protocol identifies alternative isoform and junction specific peptides using the output of short-read RNA-sequencing experiments. Due to the inherent nature of short-read RNA sequencing and mass spectrometry-based bottom-up proteomics, both of which only directly analyze fragments of a transcript or protein, the results do not directly inform on the existence of full-length alternative protein isoforms. For instance, only full-length protein isoforms containing some particular combinations of alternative junctions may exist in a given sample. These limitations may be overcome using long-read sequencing and top-down proteomics as these technologies continue to mature. A more in-depth discussion may be found in the literature (Hardwick et al., 2019, Kovaka et al., 2019, Tiambeng et al., 2019, Tran et al., 2011).
The total complexities of proteoforms in different proteomes continue to be defined. Besides alternative splicing variants, the configurations of protein molecules encoded in a gene can be influenced by single amino acid variants within a population, somatic mutations, post-translational cleavages, and chemical and enzymatic modifications to proteins. These considerable sources of protein variants are not the target of this protocol. Other workflows and protocols are available that address some of these other emerging aspects of proteome complexity.
Troubleshooting
Problem
Low yield of RNA and/or protein extraction.
Potential Solution
-
•
Increase the starting materials (e.g., higher number of cells).
-
•
Adjust the amount of TRIzol (for RNA extraction) and RIPA buffer (for protein extraction) to avoid incomplete cell lysis due to the insufficient amount of TRIzol.
-
•
Optimize speed and cycles of the homogenization step for the sample.
Problem
RNA and/or protein degradation.
Potential Solution
-
•
Make sure all supplies and reagents are RNAase free.
-
•
Make sure to add protease inhibitor cocktail into RIPA buffer prior to cell lysis and always keep samples cold throughout the extraction procedure.
-
•
Keep RNA samples cold post extraction to avoid degradation by residual RNAases in the sample.
-
•
Avoid repeated freeze-thaw cycles of purified RNA/protein.
Problem
Extracted RNA with Low A260/280 and A260/230 ratios (indicative of presence of DNA and contaminants with absorbance at 280 or 230nm such as proteins and phenol, guanidine, and buffer components).
Potential Solution
-
•
Add additional wash steps prior to RNA elution.
-
•
Use silica-based spin columns for additional clean-up (e.g., Monarch RNA Cleanup Kits; NEB).
Problem
Very few alternative sequences are created in the database.
Potential Solution
-
•
Increase the read depth of RNA sequencing.
-
•
Adjust the --read flag to set a lower threshold if necessary to permit more junctions to be translated.
Problem
rMATS docker image running errors.
Potential Solution
Make sure the versions of Python and pip are compatible with rMATS.
Problem
rMATS (non-docker image version) installation and running errors.
Potential Solution
-
•
Check if all required dependencies are pre-installed successfully.
-
•
Make sure the versions of Python and pip are compatible with rMATS.
Problem
MaxQuant database search unable to finish.
Potential Solution
-
•
Make sure there is enough storage space for MaxQuant to store output files and temporary files.
-
•
Number of threads used for processing needs to be ⋦number of logical cores and be aware that each thread requires ⋧2 GB of RAM.
-
•
Please refer to the MaxQuant documentations or publications for additional troubleshooting instructions.
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Maggie Pui Yu Lam, PhD (maggie.lam@cuanschutz.edu).
Materials Availability
This protocol did not generate new unique reagents.
Data and Code Availability
JCAST codes and detailed instruction are deposited in the GitHub at https://github.com/ed-lau/jcast.
Acknowledgments
This study was supported in part by National Institutes of Health (NIH); National Heart, Lung, and Blood Institute (NHLBI) awards R00-HL127302, R01-HL141278, and R21-HL150456 to M.P.Y.L. and R00-HL144829 to E.L.; NIH National Research Service Award (NRSA) Postdoctoral Fellowship F32-HL149191 to Y.H.; the University of Colorado Postdoctoral Fellowship in Cardiovascular Research T32-HL007822 to Y.H.; the University of Colorado Consortium for Fibrosis Research and Translation Pilot Grant to M.P.Y.L.; and the University of Colorado Undergraduate Research Opportunity Grant and Mini Grant to J.M.W.
Author Contributions
Y.H., E.L., and M.P.Y.L. conceived the project and drafted the manuscript. Y.H., J.M.W., E.L., and M.P.Y.L. edited and finalized the manuscript.
Declaration of Interests
The authors declare no competing interests.
Contributor Information
Yu Han, Email: yu.han@cuanschutz.edu.
Maggie Pui Yu Lam, Email: maggie.lam@cuanschutz.edu.
References
- Andrews S. FastQC: a quality control tool for high throughput sequence data. 2010. https://www.bioinformatics.babraham.ac.uk/projects/fastqc/
- Chambers M.C., Maclean B., Burke R., Amodei D., Ruderman D.L., Neumann S., Gatto L., Fischer B., Pratt B., Egertson J. A cross-platform toolkit for mass spectrometry and proteomics. Nat. Biotechnol. 2012;30:918–920. doi: 10.1038/nbt.2377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardwick S.A., Joglekar A., Flicek P., Frankish A., Tilgner H.U. Getting the Entire Message: Progress in Isoform Sequencing. Front. Genet. 2019;10:709. doi: 10.3389/fgene.2019.00709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D., Pertea G., Trapnell C., Pimentel H., Kelley R., Salzberg S.L. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D., Paggi J.M., Park C., Bennett C., Salzberg S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019;37:907–915. doi: 10.1038/s41587-019-0201-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovaka S., Zimin A.V., Pertea G.M., Razaghi R., Salzberg S.L., Pertea M. Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome Biol. 2019;20:278. doi: 10.1186/s13059-019-1910-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau E., Han Y., Williams D.R., Thomas C.T., Shrestha R., Wu J.C., Lam M.P.Y. Splice-Junction-Based Mapping of Alternative Isoforms in the Human Proteome. Cell Rep. 2019;29:3751–3765.e5. doi: 10.1016/j.celrep.2019.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesvizhskii A.I. Proteogenomics: concepts, applications and computational strategies. Nat. Methods. 2014;11:1114–1125. doi: 10.1038/nmeth.3144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen S., Park J.W., Lu Z., Lin L., Henry M.D., Wu Y.N., Zhou Q., Xing Y. rMATS: robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proc. Natl. Acad. Sci. U S A. 2014;111:E5593–E5601. doi: 10.1073/pnas.1419161111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiambeng T.N., Tucholski T., Wu Z., Zhu Y., Mitchell S.D., Roberts D.S., Jin Y., Ge Y. Analysis of cardiac troponin proteoforms by top-down mass spectrometry. Methods Enzymol. 2019;626:347–374. doi: 10.1016/bs.mie.2019.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran J.C., Zamdborg L., Ahlf D.R., Lee J.E., Catherman A.D., Durbin K.R., Tipton J.D., Vellaichamy A., Kellie J.F., Li M. Mapping intact protein isoforms in discovery mode using top-down proteomics. Nature. 2011;480:254–258. doi: 10.1038/nature10575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyanova S., Temu T., Cox J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 2016;11:2301–2319. doi: 10.1038/nprot.2016.136. [DOI] [PubMed] [Google Scholar]
- Wang X., Zhang B. customProDB: an R package to generate customized protein databases from RNA-Seq data for proteomics search. Bioinformatics. 2013;29:3235–3237. doi: 10.1093/bioinformatics/btt543. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
JCAST codes and detailed instruction are deposited in the GitHub at https://github.com/ed-lau/jcast.