

Summary
Lentiviral vectors are an ideal gene-delivery system for large gene-editing tools, such as the clustered regularly interspaced short palindromic repeat (CRISPR)-Cas9 system, due to their high packaging capacity and broad tropism. Here, we present a calcium phosphate-based protocol for lentiviral production and concentration for in vitro and in vivo use. This revised procedure has been optimized to ensure high viral titers and transduction efficiency and is scalable to meet specific production needs.
Graphical Abstract
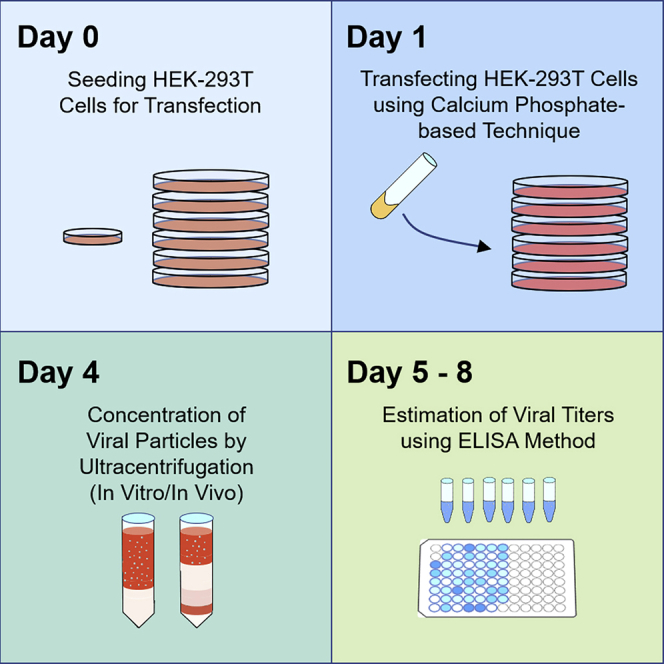
Highlights
-
•
Lentiviruses are ideal for gene delivery due to packaging capacity and broad tropism
-
•
We present a calcium phosphate-based protocol for lentiviral production
-
•
This protocol has been optimized to ensure high titers and transduction efficiency
-
•
This protocol is easily scalable to meet specific production and experimental needs
Lentiviral vectors are an ideal gene-delivery system for large gene-editing tools, such as the clustered regularly interspaced short palindromic repeat (CRISPR)-Cas9 system, due to their high packaging capacity and broad tropism. Here, we present a calcium phosphate-based protocol for lentiviral production and concentration for in vitro and in vivo use. This revised procedure has been optimized to ensure high viral titers and transduction efficiency and is scalable to meet specific production needs.
Before You Begin
Reagent and Material Check
Timing: ∼0.5–4 h
-
1.
Prepare cell culture media both with and without fetal bovine serum (FBS) beforehand and store at 20°C–22°C. Ensure there are enough solutions and reagents needed for the protocol.
-
2.
Purchase and prepare necessary packaging plasmid (psPAX2; AddGene plasmid #12260), envelope plasmid (pMD2.G; AddGene plasmid #12259), and Rev plasmid (pRSV-Rev; AddGene plasmid #12253). Prepare lentiviral vector cassette carrying a transgene-of-interest
Note: We recommend using a second generation packaging system, which excludes all four accessory proteins, Vpu, Vpr, Vif, and Nef, but includes the regulatory proteins, Tat and Rev. To maximize expression of viral mRNA, heterologous promoters, such as cytomegalovirus (CMV) and the Rous sarcoma virus (RSV) promoters, were successfully incorporated into the packaging cassette in place of the parental long terminal repeats (LTRs) Naldini L,et al 1996 PNAS. Importantly, this modification greatly reduced the risk of replication-competent viruses (RCVs), as packaging and envelope cassettes of the vectors are expressed separately from two different plasmids delivering the respective proteins in trans. In addition, non-LTR promoters drive expression of the unspliced RNA in a Tat- independent manner, which further enhances vector safety. Paradoxically, leaving Tat within the packaging plasmid results in higher production titers. In contrast, the Rev gene product is involved in the export of unspliced and incompletely spliced mRNAs, making it indispensable for productive packaging. Interestingly, we found that supplementing with additional Rev from pRSV-Rev increased production titers (data not published). The psPAX2 packaging plasmid supplies the Gag and Pol genes which carry structural and enzymatic proteins of the virus, respectively. The pMD2.G plasmid supplies the heterologous (pseudo) envelope gene, derived from the vesicular stomatitis virus protein G (VSV-G).
-
3.
At least one day before transfection, check to make sure pH of 2× BES-buffered solution (BBS) solution is 6.95. If not, prepare new BBS solution.
Note: The following protocol uses a standard hemocytometer to count cells.
Note: All tissue culture plates are incubated at 37°C with 5% CO2 in a standard tissue culture incubator.
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Experimental Models: Cell Lines | ||
| Human embryonic kidney 293T (HEK293T) cells | ATCC | CRL-3216 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Dulbecco’s Modified Eagle Medium (DMEM 1×) | Gibco | Cat#11965-092 |
| Cosmic Calf Serum (Fetal Bovine Serum) | Hyclone | Cat#SH30087.01 |
| Antibiotic-antimycotic solution, 100× | Sigma Aldrich | Cat#A5955-100ML |
| Sodium pyruvate | Sigma Aldrich | Cat#S8636-100ML |
| Non-Essential Amino Acid (NEAA) | Hyclone | Cat#SH30087.04 |
| L-Glutamine | Sigma Aldrich | Cat#G8540 |
| Trypsin-EDTA solution | Gibco | Cat#25300054 |
| BES (N, N-bis (2-hydroxyethyl)-2- amino-ethanesulfonic acid) | Sigma Aldrich | Cat#B9879-BES |
| Gelatin from porcine skin | Sigma Aldrich | Cat#G1890-100G |
| 1× PBS | Corning | Cat#21-040-CM |
| Sucrose | Sigma Aldrich | Cat#S0389 |
| CaCl2 (Calcium Chloride Dihydrate) | Sigma Aldrich | Cat#C3881 |
| NaCl (Sodium Chloride) | Fisher Scientific | Cat#S640 |
| Na2HPO4 (Sodium Phosphate Dibasic) | Sigma Aldrich | Cat#S5136 |
| Tween-20 | Sigma Aldrich | Cat# P7949 |
| HIV-1 standardsa | NIH AIDS Research and Reference Reagent Program | Cat#SP968F |
| Normal mouse serum, Sterile, 500 mLa | Equitech-Bio | Cat#SM30-0500 |
| Goat serum, Sterile, 10 mLa | Sigma Aldrich | Cat#G9023 |
| TMB microwell peroxidase substratea | SeraCare KPL SureBlue | Cat#5120-0075 |
| Antibodies | ||
| Monoclonal anti-p24 antibodya | NIH AIDS Research and Reference Reagent Program | Cat#3537 |
| Polyclonal rabbit anti-p24 antibodya | NIH AIDS Research and Reference Reagent Program | Cat#SP451T |
| Goat anti-rabbit horseradish peroxidase IgGa | Sigma Aldrich | Cat#12-348 |
| Recombinant DNA | ||
| psPAX2 | Addgene | Cat#12260 |
| pMD2.G | Addgene | Cat#12259 |
| pRSV-Rev | Dull et al., 1998 | Addgene Cat#12253 |
| Other | ||
| Optima XPN-80 Ultracentrifuge | Beckman Coulter | Cat#A99839 |
| Allegra 25R tabletop centrifuge | Beckman Coulter | Cat#369434 |
| SW32Ti swinging-bucket rotor | Beckman Coulter | Cat#369650 |
| Conical bottom ultracentrifugation tubes | Seton Scientific | Cat#5067 |
| Conical tube adapters | Seton Scientific | Cat#PN 4230 |
| xMark Microplate Absorbance plate reader | Bio-Rad | Cat#1681150 |
| 15 mL conical centrifuge tubes | Corning | Cat#430791 |
| 50 mL conical centrifuge tubes | Corning | Cat#430291 |
| 1.7 mL microcentrifuge tubes | Corning | Cat#CLS3621 |
| High-binding 96-well plates | Corning | Cat#3366 |
| 150 mm TC-Treated Cell Culture dishes with 20 mm Grid | Corning | Cat#353025 |
| 100 mm TC-Treated Culture Dish | Corning | Cat#430167 |
| 0.22 μM filter unit, 1 L | Corning | Cat#430513 |
| 0.45 μM filter unit, 500 mL | Corning | Cat#430773 |
| Serological pipettes, 5 mL | Corning | Cat#4487 |
| Serological pipettes, 10 mL | Corning | Cat#4488 |
| Serological pipettes, 25 mL | Corning | Cat#4489 |
| Vortex | N/A | N/A |
| Hemocytometer with cover slips | Cole-Parmer | Cat#UX-79001-00 |
P24 ELISA Reagents
Materials and Equipment
HEK293T Cell Media
| Reagent | Final Concentration | Amount |
|---|---|---|
| Dulbecco’s Modified Eagle Medium (DMEM 1×) | N/A | 500 mL (1 bottle) |
| Fetal bovine serum (FBS)a | 10% | 50 mL |
| Antibiotic-antimycotic solution, 100× | 1× | 5 mL |
| 100 mM Sodium pyruvate | 1 mM | 5 mL |
| Non-Essential Amino Acid (NEAA), 100× | 1× | 5 mL |
| 200 mM L-Glutamine | 2 mM | 5 mL |
Store at −20°C. Warm to 20°C–22°C before use.
Two different types of cell media need to be prepared beforehand, one with fetal bovine serum (FBS) and one without. FBS will be omitted from the above table for the media not containing FBS.
Transfection Reagent: 2× BES-buffered solution (BBS)
| Reagent | Final Concentration | Amount |
|---|---|---|
| BES (N, N-bis (2-hydroxyethyl)-2- amino-ethanesulfonic acid) | 0.5 M | 106.63 g |
| NaCl (Sodium Chloride) | 2.5 M | 146.1 g |
| Na2HPO4 (Sodium Phosphate Dibasic) | 150 mM | 21.3 g |
| dd-H2O | N/A | Add up to 1 L |
Dissolve and titrate to pH 6.95 with 1 M NaOH. Filter using 0.45 μM filter unit. Store at −20°C. Warm to 20°C–22°C before use.
Transfection Reagent: 1 M CaCl2
| Reagent | Final Concentration | Amount |
|---|---|---|
| 1 M CaCl2 (Calcium Chloride Dihydrate) | 1 M | 110.98 g |
| dd-H2O | N/A | Add up to 1 L |
Filter using 0.45 μM filter unit. Store at 4°C. Warm to 20°C–22°C before use.
Other Solutions
| Name | Reagents |
|---|---|
| 20% Sucrose | 20% sucrose dissolved in 1× PBS, filtered using 0.22 μM filter unit |
| 30% Sucrose | 30% sucrose dissolved in DMEM, filtered using 0.22 μM filter unit |
| 60% Sucrose | 60% sucrose dissolved in DMEM, filtered using 0.45 μM filter unit |
| 70% Sucrose | 70% sucrose dissolved in 1× PBS, filtered using 0.45 μM filter unit |
| PBS-T Solution | Sterile PBS with 0.05% Tween-20 |
| Sample Diluent (SD) Solution | DMEM supplemented with 0.2% Tween 20 and 1% BSA |
| Primary Antibody Diluent (PD) Buffer | DMEM supplemented with 10% FBS, 0.025% BSA, and 2% normal mouse serum (NMS) |
| Secondary Antibody Diluent | DMEM supplemented with 5% normal goat serum, 2% normal mouse serum, 0.25% BSA, and 0.01% Tween-20 |
Store all solutions at 4°C. Warm to 20°C–22°C before use.
Step-By-Step Method Details
Seeding HEK293T Cells for Transfection
Prior to transfection, the HEK293T cells must be seeded.
-
1.
Start new HEK293T cell culture. Seed low passage HEK293T cells into a 100 mm tissue culture plate. Grow cells at 37°C with 5% CO2 in a standard tissue culture incubator.
Note: Low passage cells are used to ensure quality and adequate cell growth speed. It is recommended to not use cells that have been passaged more than 15 times, or less than 2 times, or if growth has slowed.
-
2.Once cells reach 90%–95% confluent growth, prepare and collect cells for reseeding.
-
a.Aspirate media from the confluent 100 mm tissue culture plate and gently rinse the cells with ∼3.0 mL of sterile 1× PBS.
-
b.Incubate cells with 1.0 mL of dissociation reagent (e.g., Trypsin-EDTA) for 3–5 min until cells have detached from each other and from the bottom of the plate.
-
c.Add 8 mL of media prepared with FBS to inactivate the dissociation reagent.
-
d.Triturate the cells against the bottom of the plate 10–15 times to create single cell suspension with a cell density of approximately 5 × 107 cells/mL.
-
a.
Note: The following measurements and calculations of the protocol are for a single 150 mm tissue culture plates. Scale accordingly to experimental needs.
Optional: To enhance cell adherence to the tissue culture plate, pre-coat tissue culture plates by adding 5 mL of 0.2% gelatin per plate. Spread the gelatin evenly across the surface of the plate, incubate at 20°C–22°C for 10 min, and then siphon off the liquid.
-
3.Day 0: Reseed HEK293T cells into 150 mm plates for production.
-
a.Add 22.5 mL of warm (37°C) FBS containing media to each 150 mm plate.
-
b.Seed 2.5 mL of cells from step 2 into each plate (total ∼4 × 106 cells/plate).
-
c.Incubate the plates 12–18 h or until 70%–80% confluence is reached.
-
a.
Note: For concentrated lentiviral vectors, up to six 150 mm plates can be used in a single production run, with capacity being limited by ultracentrifuge space. One 100 mm plate can seed approximately four 150 mm plates, and one 150 mm plate can produce ≥2.5 × 107 vg/mL for non-concentrated lentivirus and ≥2.5 × 109 vg/mL for concentrated lentivirus.
Transfecting HEK293T Cells Using Calcium Phosphate-Based Technique
Once the HEK293T cells in the 150 mm tissue culture plates have reached 70%–80% confluence, they are ready for transfection.
CRITICAL: The calcium phosphate transfection (Day 1) should be performed in the afternoon, and it is crucial to replace the media the following morning (Day 2) as it affects cell viability.
-
4.
Day 1: Aspirate the old media from the 150 mm plates and gently replace with 20 mL of freshly prepared media without FBS by pipetting the media against the sidewall of the plate. The lack of serum stunts cell growth and enhances uptake of the transfection mixture.
-
5.
Prepare transfection mixture in a 15-mL conical tube following the volumes for each reagent listed in Table 1.
-
6.
Add 2× BBS Solution. Set tabletop vortex to ∼4–5 speed. While vortexing the 15-mL conical tube, slowly add 1.25 mL of 2× BBS solution dropwise to the plasmid mixture (total transfection mixture volume is 2.5 mL). Once completed, incubate for 30 min at 20°C–22°C.
-
7.Add transfection mixture to cells.
-
a.Following 30 min incubation, immediately add the transfection mixture dropwise to 150 mm tissue culture plate.
-
b.Swirl plates gently and incubate for 1 h.
-
c.Check size of CaPO4 precipitates in transfected cells under light microscope.
-
d.Add 2.5 mL (10%) pure FBS to the plate and incubate 12–18 h.
-
a.
Note: The calcium phosphate precipitates should be small for ideal transfection.
Note: Some labs use 3% CO2 incubators to stabilize media pH. However, we do not observe any difference in transfection efficiency between 3% and 5% CO2.
-
8.Day 2: 12–18 h post-transfection replace media with fresh FBS containing media to remove CaCl2 and 2× BBS solution.
-
a.Observe cells to ensure that they are near 100% confluency with little to no cell death.
-
b.Gently aspirate old media.
-
c.Add 25 mL prepared media with FBS, and continue incubating for an additional 48 h.
-
a.
Note: If using more than four 150 mm tissue culture plates for production, adjust volume of media per plate to ∼20–22 mL (refer to step 19 Note of protocol for rationale).
Table 1.
Volume of Plasmids and Chemicals for Lentiviral Transfection Mixture
| Reagent | Amount (μg) | Volume (μL) |
|---|---|---|
| Expression cassette of Interest (X μM) | 37.5 | 37.5/X |
| psPAX2 (Y μM) | 25 | 25/Y |
| pMD2.G (Z μM) | 12.5 | 12.5/Z |
| pRSV-Rev (A μM) | 6.25 | 6.25/A |
| 1 M CaCl2 | n/a | 312.5 |
| Sterile dd-H2O | n/a | Add up to total volume |
| Total | n/a | 1250 |
Harvesting Virus
48 h after replacing media, the virus is ready to be harvested.
-
9.
Day 4: Without disturbing the transfected cells, carefully collect the supernatant from the 150 mm tissue culture plate into a new, sterile 50-mL conical tube.
-
10.
Centrifuge the 50-mL conical tube at 400–450 × g for 5 min using a tabletop centrifuge to separate any detached cells from the supernatant. Filter the supernatant through a 0.45 μm vacuum filter unit to remove large cellular debris.
Pause Point: The filtered supernatant can be stored at 4°C for up to 4 days prior to the concentration of viral particles.
-
11.
If not concentrating viral particles, prepare a 1:10 dilution of the samples for subsequent estimation of viral titers as shown in Table 2 using ELISA method in step 35. ELISA samples can be stored at −20°C for an extended period of time.
Table 2.
1:10 Dilution Sample Preparation of Non-concentrated Viral Supernatant to Test for Viral Titer Using ELISA
| Reagent | Volume (μL) |
|---|---|
| Sample | 10 |
| Triton X-100 | 10 |
| Sterile dd-H2O | 80 |
| Total | 100 |
Concentration of Viral Particles by Ultracentrifugation (In Vitro)
Using ultracentrifugation and a sucrose filter, the lentiviral particles are concentrated for in vitro use.
-
12.
Add 4 mL of 20% sucrose dissolved in 1× PBS to the bottom of the conical ultracentrifuge tube. Without disturbing the sucrose, carefully load the filtered supernatant on top of sucrose layer dropwise as shown in Figure 1.
Note: Each conical ultracentrifugation tube has a volume capacity of up to 30 mL. When using four or more 150 mm tissue culture plates during transfection, use all six ultracentrifugation tubes per spin to process the full volume of the filtered viral supernatant. If producing more than four 150 mm plates, adjust the changed media with serum volume (from step 8) to ∼20–22 mL/plate to accommodate the six ultracentrifuge tube limit.
-
13.
Balance the ultracentrifuge tubes to within a difference of 0.01 g using sterile 1× PBS solution.
-
14.
Spin down the tubes in the ultracentrifuge for 2 h at 70,000 × g at 17°C.
Note: To prevent disruption of the sucrose layer during acceleration and deceleration of the centrifuge spin, set rotor to slowly accelerate to 10 × g during the first 3 min of the spin, and then slowly decelerate from 10 × g to 0 × g during last 3 min of the spin.
-
15.Once the spin is complete, gently extract the conical ultracentrifuge tubes and isolate the pellet.
-
a.Aspirate all media and sucrose. Be careful not to disturb the pellet.
-
b.Use a sterile paper towel to carefully wipe the sides of the tubes, without touching the pellet.
-
c.Set the tubes inverted on sterile paper towel to dry at 20°C–22°C for 10 min.
-
a.
Note: The lentivirus pellets should be visible as a small translucent dot at the bottom of the conical ultracentrifuge tubes.
-
16.
Resuspend the pellet with cold 1× PBS solution using half the total desired volume and transfer to a sterile microfuge tube. Repeat to get desired final volume and store at −80°C (e.g., If a total volume of 100 μL of 1× PBS solution is used, first resuspend with 50 μL, then wash with another 50 μL and combine).
Note: The final viral mixture should appear clear, however, the mixture may appear cloudy due to cellular impurity remnants. Typically, 25 μL of 1× PBS solution per 150 mm tissue culture plate is sufficient to resuspend and wash the pellet.
-
17.
Prepare a 1:100 dilution of the samples for estimation of viral titers as shown in Table 3 using ELISA Method in step 35. Store ELISA samples at −20°C.
Figure 1.

Graphical Depiction of Sucrose Layer for Concentration of Lentiviral Vectors for In Vitro
Table 3.
1:100 Dilution Sample Preparation of Concentrated Viral Supernatant to Test for Viral Titer Using ELISA
| Reagent | Volume (μL) |
|---|---|
| Sample | 1 |
| Triton X-100 | 10 |
| Sterile dd-H2O | 89 |
| Total | 100 |
Concentration of Viral Particles by Ultracentrifugation (In Vivo)
Using two ultracentrifuge spins, a sucrose gradient filter, and a sucrose cushion step, the lentiviral particles are concentrated for in vivo use.
-
18.Create a sucrose gradient in the following order in the conical ultracentrifuge tube, which is also shown in Figure 2.
-
a.Add dropwise 0.5 mL 70% sucrose.
-
b.Add dropwise 0.5 mL 60% sucrose.
-
c.Add dropwise 1 mL 30% sucrose.
-
d.Add dropwise 2 mL 20% sucrose.
-
a.
-
19.
Being careful not to disturb the sucrose gradient, load the filtered supernatant on top of sucrose gradient dropwise.
Note: Each conical ultracentrifugation tube has a volume capacity of up to 30 mL. When using four or more 150 mm tissue culture plates during transfection, use all six ultracentrifugation tubes per spin to process the full volume of the filtered viral supernatant. If using more than four 150 mm plates, adjust the changed media with serum volume (from step 8) to ∼20–22 mL/plate to accommodate within six ultracentrifuge tubes.
-
20.
Balance the ultracentrifuge tubes to within a difference of 0.01 g using sterile 1× PBS solution.
-
21.
First spin. Spin down the tubes in the ultracentrifuge for 2 h at 70,000 × g at 17°C.
Note: To prevent disruption of the sucrose layer during acceleration and deceleration of the centrifuge spin, set rotor to slowly accelerate to 10 × g during the first 3 min of the spin, and then slowly decelerate from 10 × g to 0 × g during last 3 min of the spin.
Note: After spin down the viral particle should be concentrated between the 30% and 60% sucrose layers.
-
22.
Once the first spin is complete, gently extract the conical ultracentrifuge tubes and aspirate all the media and most of the 20% sucrose layer. DO NOT touch the 30% sucrose layer.
-
23.
Use a P1000 pipette to gently extract the 30% and 60% sucrose layers and collect into a new 50-mL conical tube.
-
24.
Add 15 mL of sterile cold 1× PBS solution to the 50-mL conical tube for each ultracentrifuge tube collected and resuspend well. (e.g., if the 30%–60% sucrose layers were collected from four ultracentrifuge tubes, resuspend in 60 mL of cold 1× PBS solution.)
-
25.
Add 4 ML of 20% sucrose to the bottom of a new conical ultracentrifuge tube. Being careful not to disturb the sucrose layer, load the resuspended viral-PBS mixture from step 24 on top of the sucrose layer dropwise.
-
26.
Balance the ultracentrifuge tubes to within a difference of 0.01 g using sterile 1× PBS solution.
-
27.
Second spin. Spin down the tubes in the ultracentrifuge for 2 h at 70,000 × g at 17°C.
-
28.Once the second spin is complete, gently extract the conical ultracentrifuge tubes and isolate the pellet.
-
a.Aspirate all media and sucrose. Be careful not to disturb the pellet.
-
b.Use a sterile paper towel to carefully wipe the sides of the tubes.
-
c.Set the tubes inverted on sterile paper towel to dry at 20°C–22°C for 10 min.
-
a.
Note: The lentivirus pellets should be visible as a small translucent dot at the bottom of the conical ultracentrifuge tubes.
-
29.
Resuspend the pellet with cold 1× PBS solution using half the total desired volume and transfer to a sterile microfuge tube. Repeat to get desired final volume and store at −80°C. (e.g., If a total volume of 100 μL of 1× PBS solution is used, first resuspend with 50 μL, then wash with another 50 μL and combine.)
Note: The final viral mixture should appear clear. Typically, 25 μL of 1× PBS solution per 150 mm tissue culture plate is sufficient.
-
30.
Prepare a 1:100 dilution of the samples for estimation of viral titers as shown in Table 3 using ELISA method in step 35 Store ELISA samples at −20°C.
Figure 2.

Graphical Depiction of Sucrose Gradient for Purification of Lentiviral Vectors for In Vivo
Estimation of Viral Titers Using ELISA method
Estimation of viral titers is performed using the p24-enzyme-linked immunosorbent assay (ELISA) method. This protocol follows the instructions for the NIH AIDS Vaccine Program for HIV-1 p24 Antigen Capture Assay with slight modifications.
-
31.
Coat the 96-well Costar Assay plate using 80 μL of monoclonal anti-p24 antibody diluted 1:600 in 1× PBS solution per well. Incubate 12–18 h at 4°C.
-
32.
The next day, wash the 96-well plate three times with 200 μL/well of 0.05% PBS-T solution.
-
33.
To avoid non-specific binding, block the plate with 200 μL/well of 1% BSA in 1× PBS solution. Incubate for 1 h at 20°C–22°C.
-
34.
Prepare HIV-1 standards using a 2-fold serial dilution as shown in Table 4.
-
35.Dilute prepared samples from harvesting (step 11) or concentration (step 17 or 30) as shown in Table 5.
-
a.Dilute non-concentrated lentiviral samples in SD solution to establish 1:500 and 1:2,500 dilutions.
-
b.Dilute concentrated lentiviral samples in SD solution to obtain 1:10,000, 1:50,000, and 1:250,000 dilutions.
-
a.
-
36.
Following incubation with blocking solution, wash the 96-well plate three times with 200 μL/well of PBS-T solution. Apply 100 μL/well of controls (one empty well and one with SD buffer only), standards, and samples on the plate in duplicates as shown in Figure 3 and incubate 12–18 h at 4°C.
-
37.
The next day, dump the samples and wash the plate six times with 200 μL/well of PBS-T solution.
-
38.
Add 100 μL/well of polyclonal rabbit anti-p24 antibody diluted 1:600 in PD Buffer. Incubate at 37°C for 4 h.
-
39.
Dump the polyclonal rabbit anti-p24 antibody and wash the plate six times with 200 μL/well of PBS-T solution.
-
40.
Add 100 μL/well of goat anti-rabbit horseradish peroxidase IgG diluted 1:5,000 in secondary solution. Incubate at 37°C for 1 h.
-
41.
Dump the goat anti-rabbit horseradish peroxidase IgG and wash the plate six times with 200 μL/well PBS-T solution.
-
42.
Add 100 μL/well of SureBlue TMB peroxidase substrate. Incubate at 20°C–22°C for 10–30 min.
Note: A gradient of blue should appear across wells A to F containing standards, reflective of sample concentration, with tube A being the most intense blue and tube F being the most diffuse.
-
43.
Add 100 μL/well of 1 N HCL solution to stop the reaction. Once HCl is added, the wells will turn yellow.
-
44.
Measure sample absorbance at 450 nm wavelength and 5 s mix time using plate reader.
Table 4.
Preparing HIV-1 Standards Using a 2-Fold Serial Dilution
| Tube | Final Concentration (ng/mL) |
|---|---|
| A | 5 |
| B | 2.5 |
| C | 1.25 |
| D | 0.625 |
| E | 0.3125 |
| F | 0.156 |
Table 5.
Diluting Prepared Samples Collected from Transfection
| 1:10,000 Dilution | 1:50,000 Dilution | 1:250,000 Dilution | |
|---|---|---|---|
| Concentrated Vector 1:100 | Tube 1 | Tube 2 | Tube 3 |
| 495 μL SD solution | 400 μL SD solution | 400 μL SD solution | |
| 5 μL diluted sample | 100 μL from Tube 1 | 100 μL from Tube 2 | |
| Non-concentrated Vector 1:10 | Tube 1a | Tube 2a | \ |
| 490 μL SD solution | 400 μL SD solution | \ | |
| 10 μL diluted sample | 100 μL from Tube 1a | \ |
Figure 3.

Graphical Example of 96-Well Plate (8 × 12) with Each Box of the Table Representing a Well of the Plate
The above example displays the position of the controls, standards, and four diluted concentrated samples in duplicates.
Expected Outcomes
Successful lentiviral production should yield physical titers ≥2.5 × 107 vg/mL for non-concentrated vectors, and ≥2.5 × 109 vg/mL for concentrated vectors. Functional titers will vary depending on the expression cassette packaged. It was found that using this protocol, the functional viral titer for Sp1-LVs carrying CRISPR/Cas9 transgenes was around 1 × 1,010 TU/mL per ∼5 × 107 producer cells (Vijayraghavan and Kantor, 2017) and (Tagliafierro et al., 2019).
Quantification and Statistical Analysis
To estimate viral titers using the absorbance reader from the ELISA method, use the following equation:
∗1.0 ng/mL = 10,000 vg/mL
Note: Dilution factor refers to the dilution of the sample, such as 1:10,000, 1:50,000, and 1:250,000 dilutions. If the absorbance reader accounts for dilution factor, omit this consideration.
Note: For reliable results, check to make sure the coefficient of determination (r2) is above 0.96 for an accurate curve fitting of concentration vs. optical density (OD) values.
Note: Check the concentration of tubes A to F, where concentration of tube F must be lower than tubes A to E. Concentration of tube F must also be higher than the controls (empty well and SD solution). Only rely on sample value results that fall within the range of standards concentrations (tubes A through F).
Estimation of Viral Titers by Counting Fluorescent-Reporter Expressing Cells
Estimation of viral titers can also be obtained for samples expressing a fluorescent reporter, such as green fluorescent protein (GFP), by transducing cells with the viral prep and counting the number of cells which express the fluorescent reporter.
-
1.
Determine the optimal multiplicity of infection (MOI) to use for transduction by performing a pilot experiment testing a range of different MOIs (1–10), with increasing MOIs resulting in a higher transduction efficiency. The MOI represents the relationship between the number of infectious units and the number of cells to be transduced.
Note: For example, an MOI of 10 = (1.0 × 106 vg/mL × 1.0 mL)/1.0 × 105 cells
-
2.
Prior to transduction, seed a 6-well plate with approximately 3–4 × 105 cells per well. Once the cells reach >90% confluency (usually within 24 h), transduce with the virus at pre-determined MOIs.
-
3.
Incubate plate at 37°C with 5% CO2 in a standard tissue culture incubator and monitor cells at regular intervals for 1–7 days for changes in GFP signal.
-
4.
Count the number of GFP-positive cells with a fluorescent microscope (PLAN 4X objective, 0.1 NA, 40× magnification) fitted with a GFP filter set (excitation wavelength-470 nm, emission wavelength-525 nm), using naïve (un-transduced) cells to set the population of GFP-negative and positive cells.
-
5.
Estimate the final titer by adjusting for the dilution factor and the volume, using the following formula:
Note: N = number of GFP-positive cells, D = dilution factor, M = magnification factor (usually 20×), V = volume of virus used for transduction. For example, for 20 GFP-positive cells (N) counted at a dilution of 10−4 (1:10,000) (D) in a 10 μL sample (V) at 20× magnification (M) would result in a functional titer of (20 × 104) × (20) × (10) × (100∗) = 4 × 108 TU/mL. (∗to adjust to per mL).
Limitations
Lentiviral vectors are a common gene-delivery vehicle used for a wide range of experimental procedures. With the increasing reach in precision medicine and high-precision genome editing, lentiviral vectors are advantageous for delivering large programmable nucleases, such as zinc-finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs), and CRISPR-Cas systems. Despite advantages such as low immunogenicity and large packaging capacity, lentiviral vectors can produce unwanted insertional mutagenesis effects due to genome integration (Schaffer et al., 2008). Alternatives in gene delivery vehicles include adeno-associated virus (AAV), which are easier to produce and generally have a higher viral titer. However, recent advances in lentivirus engineering have demonstrated that removal of the integrase gene to generate integrase-deficient lentivirus (IDLVs) can achieve similar viral efficiency as its counterpart integrase-competent lentivirus (ICLVs), while not integrating the delivered gene into the genome (Ortinski et al., 2017).
Troubleshooting
Problem 1
Transfecting HEK293T Cells using Calcium Phosphate-based Technique:
Cloudy mixture after adding 2× BBS solution vs. non-cloudy mixture (step 6).
Potential Solution
Repeat the procedure and prepare a new transfection plasmid mixture. The transfection mixture must be clear prior to its addition onto the cells in order to obtain small CaPO4 precipitates, which are crucial for effective transfection. If 2× BBS solution is added rapidly into the plasmid mixture, cloudiness will appear immediately. We have found that vortexing the 15-mL conical tube at ∼4–5 speed, while slowly dropwise adding the 1.25 mL of 2× BBS solution, helps 2× BBS solution mix better with the transfection mixture. Slanting the 15-mL conical tube and letting each added drop of 2× BBS solution slide down along the inner wall of the conical tube also helps maximizing mixture of the reagent in the transfection mixture.
Always check the transfection mixture before applying it to the tissue culture plate. If cloudiness appears during the 30 min 20°C–22°C incubation period after the addition of 2× BBS solution, then it is likely fresh 2× BBS (pH = 6.95) is needed.
Problem 2
Transfecting HEK293T Cells using Calcium Phosphate-based Technique:
Large calcium phosphate precipitates (step 7c).
Potential Solution
Rapid addition of 2× BBS solution or cloudy mixtures will result in large calcium phosphate precipitates. This is not ideal for lentiviral production and will result in lower production efficiency. Adding the 2× BBS solution slowly in a dropwise manner and checking the pH of the 2× BBS solution before the experiment will help prevent the error.
Problem 3
Harvesting virus:
Cloudy media with many cell deaths (step 9).
Potential Solution
Forgetting to change media 12–18 h after the addition of the transfection mixture and the serum will result in cell death due to the toxicity of 2× BBS solution and 1 M CaCl2. The cloudiness is caused by the leftover 1 M CaCl2 from the transfection mixture. Be gentle when collecting the media to prevent disturbing the transfected cellular layer. Centrifuge the 50-mL conical tube of collected media for a longer period of time to separate all the dead cells from the viral supernatant. This will prevent the excess dead cells clogging the 0.45 μm vacuum filter unit during filtration. The viral titer will be lower than expected due to excess cell death. If final product does not meet the necessary viral titer needed, repeat experiment.
Problem 4
Concentration of Viral Particles by Ultracentrifugation (In Vitro/In Vivo):
Disrupted supernatant/sucrose layer or sucrose gradient (step 12, 18, or 19).
Potential Solution
Load each sucrose layer or the viral supernatant dropwise into the conical ultracentrifuge tube. Slanting the conical ultracentrifuge tube and letting each drop slide down along the inner wall of the conical tube also helps prevent mixing of the layers.
At the same time, check acceleration and deceleration speed of the ultracentrifuge, set rotor to slowly accelerate to 10 × g during the first 3 min of the spin, and then slowly decelerate from 10 × g to 0 × g during last 3 min of the spin.
Problem 5
Concentration of Viral Particles by Ultracentrifugation (In Vitro/In Vivo):
Spillage of viral supernatant during ultracentrifugation (step 15, 22, or 28).
Potential Solution
Leave at least 10% headspace to prevent loss of samples. Make sure to clean and check equipment if spillage occurs to prevent equipment damage.
Problem 6
Estimation of Viral Titers using ELISA method:
Low viral titers (step 44).
Potential Solution
Titers lower than the range of 2.5 × 109 TU/mL per ∼5 × 107 producer cells indicate error in the production process. (1) Check if the producer cells have only gone through a low number of passages. It is advised not to use cells that have undergone 15 or more passages, or if cell growth has slowed down. (2) Check the cell culture media components. Ensure that FBS was included in the usual cell culture media, but not in the media prior to transfection. The lack of serum in the media stunts cell growth and enables better uptake of the transfection mixture. (3) Check the HEK producer cell line used in the experiment. It has been shown that there is a 3-fold difference in viral production yield in HEK293T cells compared to HEK293TF cells (Vijayraghavan and Kantor, 2017). (4) Make sure that the cells were transfected at 70%–80% confluency. Low cell density results in premature cell death due to toxicity of lentiviral vectors, while high cell densities results in a drop in production efficiency. (5) Make sure transfection mixture was not cloudy and that the pH of 2× BBS solution added was 6.95. Transfection efficiency is highly dependent on the pH of 2× BBS solution.
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to, and will be fulfilled by, the Lead Contact, Boris Kantor (boris.kantor@duke.edu).
Materials Availability
The lentiviral vectors generated in this study will be made available for customization upon request, some of the components may require a Materials Transfer Agreement.
Data and Code Availability
No new datasets or code were generated in the preparation of this manuscript.
Acknowledgments
We would like to thank the Department of Neurobiology, Duke University School of Medicine. We also thank members of the Duke Viral Vector Core for comments and advice on the manuscript. This research was supported by Seelos Therapeutics, INC through Sponsored Research Agreement Project 2933505; SPS#: 260286 to B.K.
Author Contributions
Conceptualization, W.D., L.B., and B.K.; Investigation, W.D., L.B., and B.K.; Writing – Original Draft, W.D.; Writing – Review & Editing, W.D., L.B., and B.K.
Declaration of Interests
Boris Kantor is a founder of CLAIRIgene and a member of its scientific advisory board.
Contributor Information
Logan Y. Brown, Email: logan.brown@duke.edu.
Wendy Dong, Email: wendy.dong@duke.edu.
Boris Kantor, Email: boris.kantor@duke.edu.
References
- Dull T., Zufferey R., Kelly M., Mandel R.J., Nguyen M., Trono D., Naldini L. A third-generation lentivirus vector with a conditional packaging system. J. Virol. 1998;72:8463–8471. doi: 10.1128/jvi.72.11.8463-8471.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortinski P.I., O’Donovan B., Dong X., Kantor B. Integrase-deficient lentiviral vector as an all-in-one platform for highly efficient CRISPR/Cas9-mediated gene editing. Mol. Ther. Methods Clin. Dev. 2017;5:153–164. doi: 10.1016/j.omtm.2017.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaffer D.V., Koerber J.T., Lim K. Molecular engineering of viral gene delivery vehicles. Annu. Rev. Biomed. Eng. 2008;10:169–194. doi: 10.1146/annurev.bioeng.10.061807.160514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijayraghavan S., Kantor B. A protocol for the production of integrase-deficient lentiviral vectors for CRISPR/Cas9-mediated gene knockout in dividing cells. J. Vis. Exp. 2017;130:e56915. doi: 10.3791/56915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tagliafierro L., Ilich E., Moncalvo M., Gu J., Sriskanda A., Grenier C., Murphy S.K., Chiba-Falek O., Kantor B. Lentiviral vector platform for the efficient delivery of epigenome-editing tools into human induced pluripotent stem cell-derived disease models. J. Vis. Exp. 2019:e59241. doi: 10.3791/59241. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No new datasets or code were generated in the preparation of this manuscript.