

Summary
Recent advances in high-resolution multiparametric flow cytometry enable ever deeper analysis of human lymphocyte subsets that require rigorous methodology development and optimization. Here, we detail methods to characterize glycosylated Sialyl-LewisX (SLeX)- or cutaneous lymphocyte-associated antigen (CLA)-expressing CD4+ T cells using two separate multiparametric flow cytometry panels enabling the identification of memory subsets, Th subsets, and expression of diverse activation markers and chemokine receptors. The proposed protocol allows optimal resolution of the measured parameters while minimizing background in a 25-parameter experiment.
For complete details on the use and execution of this protocol, please refer to Colomb et al. (2020).
Graphical Abstract
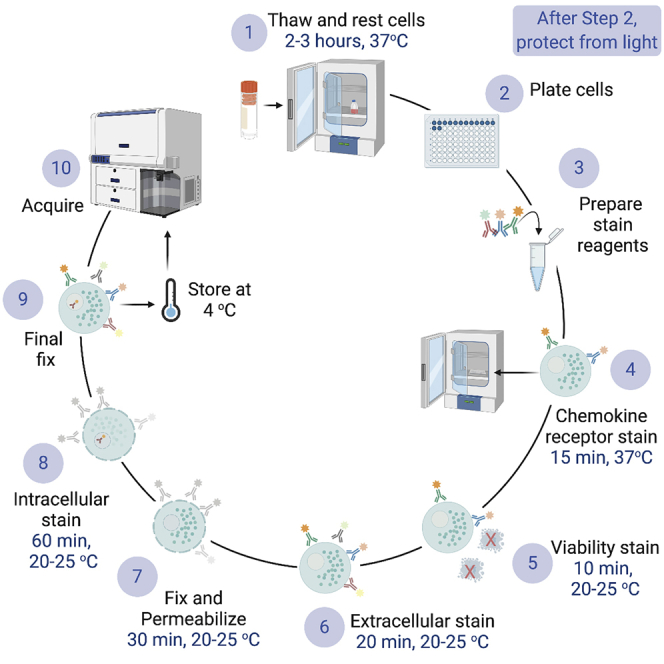
Highlights
-
•
25-parameter panels for glycosylated CD4+ T cell deep immune profiling
-
•
Characterization of memory subsets and Th profiles on CD4+ T cells
-
•
Quantification of expression levels of many activation markers and chemokine receptors
-
•
Optimal resolution and minimal background
Recent advances in high-resolution multiparametric flow cytometry enable ever deeper analysis of human lymphocyte subsets that require rigorous methodology development and optimization. Here, we detail methods to characterize glycosylated Sialyl-LewisX (SLeX)- or cutaneous lymphocyte-associated antigen (CLA)-expressing CD4+ T cells using two separate multiparametric flow cytometry panels enabling the identification of memory subsets, Th subsets, and expression of diverse activation markers and chemokine receptors. The proposed protocol allows optimal resolution of the measured parameters while minimizing background in a 25-parameter experiment.
Before You Begin
This protocol details the procedure for staining two separate panels to characterize in detail the phenotype of CD4+ (or CD8+ T cells) expressing SLeX or CLA. For SLeX detection, sequential staining for chemokine receptors, viability exclusion, extracellular markers, and intracellular markers is performed using combined antibody cocktails from Tables 2, 3, and 5. For CLA characterization, a combination of antibody cocktails detailed in Tables 2, 4, and 5 is indicated.
Note: this protocol does not explore co-expression of these markers, but experienced users can modify accordingly to adjust for their research interest.
CRITICAL: Before starting, make sure that your BD FACS Symphony A5 flow cytometer has the correct configuration to detect all parameters included in this panel. The detailed configuration can be found under Materials and Equipment.
Table 2.
Chemokine Receptor Extracellular Antibody Cocktail
| Marker | Fluorochrome | Comp Bead | 1X μL/test | 10X (+1) μL/test | 20X (+3) μL/test |
|---|---|---|---|---|---|
| CXCR4 | PE Cy5 | mouse | 2.5 | 30 | 57.5 |
| CXCR3 | AF700 | mouse | 2 | 24 | 46 |
| CCR7 | APC-Cy7 | mouse | 2 | 24 | 46 |
| CCR6 | BV421 | mouse | 1 | 12 | 23 |
| CCR4 | PE-CF594 | mouse | 1 | 12 | 23 |
| CCR5 | BV650 | mouse | 0.6 | 7.2 | 13.8 |
| Sum of antibodies | 9.1 | 109.2 | 209.3 | ||
| PBS | 35.9 | 430.8 | 825.7 | ||
| Final volume | 45 | 540 | 1,035 | ||
Table 3.
Extracellular Antibody Cocktail with SLeX
| Marker | Fluorochrome | Comp Bead | 1X μL/test | 10X (+1) μL/test | 20X (+3) μL/test |
|---|---|---|---|---|---|
| CD69 | PE | mouse | 10 | 110 | 230 |
| CD27 | BV570 | mouse | 2 | 22 | 46 |
| CD127 | BV480 | mouse | 2 | 22 | 46 |
| PD-1 | BV785 | mouse | 2 | 22 | 46 |
| CD38 | BUV661 | mouse | 1.5 | 16.5 | 34.5 |
| CD95 | BV711 | mouse | 1 | 11 | 23 |
| CXCR5 | BB515 | rat | 0.7 | 7.7 | 16.1 |
| CD4 | BB700 | mouse | 0.6 | 6.6 | 13.8 |
| SLeX (CD15s) | AF647 | mouse | 0.5 | 5.5 | 11.5 |
| CD19 | BV510 | mouse | 0.5 | 5.5 | 11.5 |
| CD14 | BV510 | mouse | 0.5 | 5.5 | 11.5 |
| CD45RA | BUV563 | mouse | 0.5 | 5.5 | 11.5 |
| CD25 | BUV737 | mouse | 0.5 | 5.5 | 11.5 |
| CD8 | BUV496 | mouse | 0.5 | 5.5 | 11.5 |
| CD3 | BUV805 | mouse | 0.5 | 5.5 | 11.5 |
| HLA-DR | BV605 | mouse | 0.4 | 4.4 | 9.2 |
| Sum of antibodies | 23.7 | 260.7 | 545.1 | ||
| Staining buffer | 26.3 | 289.3 | 604.9 | ||
| Final volume | 50 | 550 | 1,150 | ||
Table 5.
Example Antibody Cocktails for FMO Preparation
| Marker | Fluorochrome | FMO CD69 | FMO PD-1 | FMO CD38 | FMO CLA | FMO CD25 | FMO HLA-DR |
|---|---|---|---|---|---|---|---|
| CD69 | PE | - | 10 | 10 | 10 | 10 | 10 |
| CD27 | BV570 | 2 | 2 | 2 | 2 | 2 | 2 |
| CD127 | BV480 | 2 | 2 | 2 | 2 | 2 | 2 |
| PD-1 | BV785 | 2 | - | 2 | 2 | 2 | 2 |
| CD38 | BUV661 | 1.5 | 1.5 | - | 1.5 | 1.5 | 1.5 |
| CD95 | BV711 | 1 | 1 | 1 | 1 | 1 | 1 |
| CXCR5 | BB515 | 0.7 | 0.7 | 0.7 | 0.7 | 0.7 | 0.7 |
| CD4 | BB700 | 0.6 | 0.6 | 0.6 | 0.6 | 0.6 | 0.6 |
| CLA | AF647 | 0.5 | 0.5 | 0.5 | - | 0.5 | 0.5 |
| CD19 | BV510 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 |
| CD14 | BV510 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 |
| CD45RA | BUV563 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 |
| CD25 | BUV737 | 0.5 | 0.5 | 0.5 | 0.5 | - | 0.5 |
| CD8 | BUV496 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 |
| CD3 | BUV805 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 |
| HLA-DR | BV605 | 0.4 | 0.4 | 0.4 | 0.4 | 0.4 | - |
| Sum of antibodies | 13.7 | 21.7 | 22.2 | 23.2 | 23.2 | 23.3 | |
| Staining buffer | 36.3 | 28.3 | 27.8 | 26.8 | 26.8 | 26.7 | |
| Final volume | 50 | 50 | 50 | 50 | 50 | 50 | |
Table 4.
Extracellular Antibody Cocktail with CLA
| Marker | Fluorochrome | Comp Bead | 1X μL/test | 10X (+1) μL/test | 20X (+3) μL/test |
|---|---|---|---|---|---|
| CD69 | PE | mouse | 10 | 110 | 230 |
| CD27 | BV570 | mouse | 2 | 22 | 46 |
| CD127 | BV480 | mouse | 2 | 22 | 46 |
| PD-1 | BV785 | mouse | 2 | 22 | 46 |
| CD38 | BUV661 | mouse | 1.5 | 16.5 | 34.5 |
| CD95 | BV711 | mouse | 1 | 11 | 23 |
| CXCR5 | BB515 | rat | 0.7 | 7.7 | 16.1 |
| CD4 | BB700 | mouse | 0.6 | 6.6 | 13.8 |
| CLA | AF647 | mouse | 0.5 | 5.5 | 11.5 |
| CD19 | BV510 | mouse | 0.5 | 5.5 | 11.5 |
| CD14 | BV510 | mouse | 0.5 | 5.5 | 11.5 |
| CD45RA | BUV563 | mouse | 0.5 | 5.5 | 11.5 |
| CD25 | BUV737 | mouse | 0.5 | 5.5 | 11.5 |
| CD8 | BUV496 | mouse | 0.5 | 5.5 | 11.5 |
| CD3 | BUV805 | mouse | 0.5 | 5.5 | 11.5 |
| HLA-DR | BV605 | mouse | 0.4 | 4.4 | 9.2 |
| Sum of antibodies | 23.7 | 260.7 | 545.1 | ||
| Staining buffer | 26.3 | 289.3 | 604.9 | ||
| Final volume | 50 | 550 | 1,150 | ||
Antibody Titration
Timing: 10 min for cell plating, ~1 h per antibody stain
Note: This should be done in advance, on a different day before the main experiment is performed.
The protocol below details the titration for a single antibody as an example. Titration can be performed for each antibody separately, or multiplexed to decrease the total cell requirement. However, fluorophores with high spectral overlap should optimally be titrated separately. Note that staining time and temperature are critical variables, and conditions must remain consistent for titrations and the ultimate cell staining protocol.
-
1.Prepare ~10–15 × 106 freshly isolated PBMC or thaw cryopreserved PBMC cryopreserved PBMC (1–2 × 106 cells/condition for seven conditions).
-
a.Resuspend samples at 10 × 106 cells/mL in 1× PBS. For cryopreserved PBMC, thaw and wash as indicated in major steps 1–8, under Cryopreserved PBMC Sample Thawing and resting.
-
a.
-
2.
Label two V-bottom 96-well plate for serial dilutions: 1:20, 1:40, 1:80, 1:160, 1:320, 1:640, and NC for negative control (Figure 1). Mark one as “titration plate,” and the other one as “dilution plate.”
Note: These are suggested dilutions optimized for the antibodies used in this protocol. A wider span of dilutions can be tested to reach optimal titration for certain antibodies as needed.
-
3.Prepare antibody dilutions in the dilution plate. This step can be performed before or during the 10 min incubation of the Live/Dead Fixable Aqua Dead Cell Stain.Note: Cells will be stained in a 100 μL final volume, by adding equal volumes of cells and antibody dilution. Therefore, antibody dilutions are prepared in double concentration (2×).
-
a.Label a 1.5 mL microcentrifuge tube as “staining buffer.” Prepare 1 mL of a 1:2 FACS buffer with BD Brilliant Stain buffer solution in a microcentrifuge tube.
-
b.Aliquot 90 μL of staining buffer into wells A1 and A7.
-
c.Aliquot 50 μL of staining buffer into wells A2-A6.
-
d.Add 10 μL of the antibody into well A1 (1:10 dilution, 2X). Mix well with pipette, being mindful of not creating bubbles.
-
e.Transfer 50 μL of the volume in well A1 into well A2 to perform a 1:2 dilution. Mix well with pipette, being mindful of not creating bubbles.
-
f.Continue performing serial dilutions: A2 → A3 → A4 → A5 → A6.
-
g.A7 should only contain staining buffer and will be used as the unstained negative control.
-
h.Set the dilution plate aside in the dark.
-
a.
-
4.Prepare titration plate.
-
a.Plate 1 × 106 cells/well (100 μL/well) into the titration plate in wells A1-A7.
-
b.Centrifuge plate 5 min at 400 × g. Remove the supernatant carefully using pipette by leaning the pipette tip at the edge of the well cone as shown in Figure 2.
-
a.
-
5.
Resuspend cells in 45 μL of 1× PBS. Mix well with pipette.
-
6.Perform viability stain using Live/Dead Fixable Aqua Dead Cell Stain.
-
a.Thaw one vial from the stock, prepared by adding 25 μL of DMSO into the lyophilized vial. A previously prepared vial can be used (See Notes at the end of this step).
-
b.In a 500 μL microcentrifuge tube, prepare a 1:60 dilution of Live/Dead Fixable Aqua Dead Cell Stain in 1× PBS. Mix well with pipette.
-
▪This dilution should be freshly prepared at the time of the experiment.
-
▪
-
c.Add 5 μL of the Live/Dead Fixable Aqua Dead Cell Stain dilution to each well. Mix well with pipette, being mindful of not creating bubbles.
-
a.
Note: At this point, the final volume is 50 μL/well.
-
d.Incubate at 20°C–25°C in the dark for 10 min.
-
d.
Note: The Live/Dead Fixable Aqua Dead Cell Stain can be performed in PBS, Dulbecco’s PBS, and Hanks’ Balanced Salt Solution, without proteins such as bovine serum albumin or serum as per manufacturer’s instructions. The use of protein-enriched buffers will affect the resolution of the stain.
Note: Manufacturer’s instructions indicate the preparation of one vial of Live/Dead Fixable Aqua Dead Cell Stain with 50 μL of DMSO, and used fresh at 1 μL/test for 1 × 106 cells. The suggested incubation conditions are 30 min at 4°C. The staining conditions and titrations indicated in step 6 have been optimized for this protocol. Live/Dead Fixable Aqua Dead Cell Stain should also be titrated appropriately.
Note: One vial of Live/Dead Fixable Aqua Dead Cell Stain diluted with 25 μL of DMSO can be used for approximately one month and throughout multiple freeze-thaw cycles, as long as it is kept in the dark and stored at −20°C immediately after use.
-
7.Stain cells.
-
a.Transfer 50 μL of A1 from the dilutions plate into A1 of the titration plate. Mix well with pipette.Note: At this point, the final volume is 100 μL per well (45 μL of cell suspension + 5 μL of Live/Dead Fixable Aqua Dead Cell Stain + 50 μL of the antibody diluted in staining).Note: The final stain dilution is now 1:20, as the antibody was diluted once more when adding 50 μL of the antibody dilution to the 50 μL volume of cell suspension in the well.
-
b.Transfer 50 μL of A2-8 from the dilutions plate to their corresponding well in the titration plate. Mix well with pipette.
-
c.Incubate for 20 min at 20°C–25°C.Note: For antibodies used for chemokine receptor labeling, place the plate inside the incubator at 37°C 5% CO2, for 15 min.Note: The detailed antibodies for chemokine receptors used in these panels are listed on Table 2.
-
d.Add 100 μL/well of FACS buffer. Centrifuge for 5 min at 400 × g.
-
e.Remove supernatant using pipette by leaning the pipette tip at the edge of the well cone, as indicated previously.
-
f.Fix with 200 μL/well of PBS with 1% paraformaldehyde. Mix well with pipette.
-
g.Transfer volume into 1.2 mL microtiter tubes or 5 mL FACS tubes.
-
a.
-
8.
Prepare compensation controls using the corresponding anti-mouse or anti-rat beads, and amine reactive beads for dead cell stain (see Notes at the end of this section).
-
9.
Store at 4°C until acquisition. Collect an equivalent number of events of the parent population of interest.
-
10.
Select titration based on the detection signal plateau of the marker, considering percentage and separation of positive population, median fluorescence intensity (MFI) and staining index (Maecker and Trotter, 2008; McLaughlin et al., 2008). An example of CD4 BB700 antibody titration is shown in Figure 3. In this example, a titer of 1:160 (rounded to 0.6 μL/test) was selected based on separation of the positive population.
Note: Prepare compensation control beads. Use ArC™ Amine Reactive compensation beads for the Live/Dead Fixable Aqua Dead Cell Stain, and the corresponding BD Anti-Mouse or Anti-Rat Ig compensation beads. Prepare a universal negative control for each type of bead used.
Note: In this protocol, we recommend the use of beads to prepare compensation controls. Although some labs prefer the use of cells, achieving a distinct or bright enough level of expression for certain markers with cells may be challenging, resulting in uncompensated events throughout the analysis. Further details are described in step 20 under “Preparing Antibody Cocktails and Staining Reagents” and Figure 4. Furthermore, the high number of parameters would call for a large number of cells for this purpose.
Figure 1.

Example Labeling of a 96-Well Plate for Preparing Dilutions and Titrations
Numbers indicate dilution 1:X. NC, negative control.
Figure 2.

Example Placement of Pipette Tip for Removing Supernatant in a V-Bottom 96-Well Plate
Figure 3.

CD4+ BB700 Titration on Frozen PBMC
All samples were acquired on a FACS Symphony A5 cytometer, collecting 100,000 events within the viable CD3+ cell gate.
(A) Representative examples of staining of viable CD3+ using serial dilutions for CD4 as indicated in the protocol. Cells are gated on lymphocytes (identified by morphology based on FSC-A and SSC-A), selected by viability exclusion and gated within CD3+ cells. Total CD4+ (teal) and CD4− (black) cells were gated. Numbers inside the gates indicate the percentage of cells within the parent population.
(B) MFI of CD4+ population on stained samples (solid lines) and negative control population (NC, dotted line).
(C)Graph showing the percentage of CD4+ cells (left Y axis, circles) and MFI of the CD4+ population (right Y axis, squares) throughout the tested dilutions.
(D) Stain index calculated as MFI of CD4+ population – MFI of CD4− population, divided by 2× the standard deviation of the CD4− population ((MFIpos-MFIneg)/2σneg).
(E) Equivalence of dilutions and μL/test.
Figure 4.

Importance of Using the Correct Beads for Compensation Controls
Example generated from cryopreserved PBMC stimulated in vitro with anti-CD3 (clone UCHT1, Bio-Rad, Hercules, CA) and CD49d/CD28 co-stimulatory antibodies (clones L293 and L25, BD Biosciences) for 6 h at 37°C, 5% CO2. Following 1 h of stimulation, monensin (0.7 μg/mL final concentration; BD Biosciences) and brefeldin A (1 μg/mL final concentration; Sigma-Aldrich, St. Louis, MO) were added, and cells were incubated for 5 h more. Cells were stained to quantify cytokine production. The example shows the expression of TNF-α (TNF-α PE Cy7 clone Mab11, eBioscience) and IFN-γ (IFN-γ Alexa Fluor 700, Invitrogen) in memory CD8+ T cells.
(A) Compensation control prepared with Plus beads.
(B) Representative example of stimulated memory CD8+ T cells. In A and B, the orange selection indicates the events above median of the compensation peak, which would not be compensated correctly.
(C) Compensation control prepared with normal beads.
(D) Representative example of stimulated memory CD8+ T cells. In C and D, the purple selection indicates the events above the compensation peak, which would not be compensated correctly.
Note: For certain antibodies, it is recommended to include lineage markers to help characterize the population of interest, as shown in Figure 3 where CD3 was included to clearly identify CD4+ T cells.
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-human CCR4 PECF594 (clone 1G1) | BD | Cat# 565391 |
| Anti-human CCR5 BV650 (clone 3A9) | BD | Cat# 564999 |
| Anti-human CCR6 BV421 (clone 11A9) | BD | Cat# 562515 |
| Anti-human CD127 BV480 (clone HIL-7R-M21) | BD | Cat# 566101 |
| Anti-human CD25 BUV737 (clone 2A3) | BD | Cat# 564385 |
| Anti-human CD3 BUV805 (clone UCHT1) | BD | Cat# 565515 |
| Anti-human CD38 BUV661 (clone HIT2) | BD | Cat# 565069 |
| Anti-human CD4 BB700 (clone SK3) | BD | Cat# 566392 |
| Anti-human CD45RA BUV563 (clone HI100) | BD | Cat# 565702 |
| Anti-human CD69 PE (clone FN50 ) | BD | Cat# 555531 |
| Anti-human CD8 BUV496 (clone RPA-T8 ) | BD | Cat# 564804 |
| Anti-human CXCR4 PE-Cy5 (clone 12G5) | BD | Cat# 555975 |
| Anti-human CXCR5 BB515 (clone RF8B2) | BD | Cat# 564624 |
| Anti-human HLA-DR BV605 (clone G46-6) | BD | Cat# 562845 |
| Anti-human Ki67 BUV395 (clone B56) | BD | Cat# 564071 |
| Anti-human SLeX (CD15s) AF647 (clone CSLEX1) | BD | Cat# 563526 |
| Anti-human CCR7 APC-Cy7 (clone G043H7) | Biolegend | Cat# 353211 |
| Anti-human CD14 BV510 (clone M5E2) | Biolegend | Cat# 301842 |
| Anti-human CD19 BV510 (clone HIB19) | Biolegend | Cat# 302242 |
| Anti-human CD27 BV570 (clone O323) | Biolegend | Cat# 302825 |
| Anti-human CD95 BV711 (clone DX2) | Biolegend | Cat# 305644 |
| Anti-human CLA AF647 (clone HECA-452) | Biolegend | Cat# 321309 |
| Anti-human CXCR3 AF700 (clone G025H7) | Biolegend | Cat# 353731 |
| Anti-human PD-1 BV785 (clone EH12.2H7) | Biolegend | Cat# 302827 |
| Anti-human Foxp3 PE-Cy7 (clone 236A/E7) | eBioscience | Cat# 25-4777-42 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| RPMI 1640 | Corning | Cat# 10-040-CM |
| Dulbecco's Phosphate-Buffered Saline (D-PBS) | Corning | Cat# 21-031-CM |
| Penicillin/streptomycin (10,000 U Pen/mL; 10,000 μg Strep/mL) | Lonza | Cat# 17-602E |
| Fetal Bovine serum | GeminiBio | Cat# 100-500 |
| Bovine Serum Albumin (BSA) | GeminiBio | Cat# 700-100P |
| L-Glutamine (200 mM solution) | Corning | Cat# 25-005-CI |
| Paraformaldehyde 10% in aqueous solution, EM Grade | Electron Microscopy Sciences | Cat# 15714-S |
| Sodium Azide | Fisher Scientific | Cat # S227I-500 |
| Critical Commercial Assays | ||
| Brilliant Stain buffer | BD | Cat# 563794 |
| Foxp3/Transcription Factor Staining Buffer Set | eBioscience | Cat# 00-5523-00 |
| LIVE/DEAD™ Fixable Aqua Dead Cell Stain Kit | Invitrogen | Cat# L34957 |
| DNase I recombinant, RNase-free (10,000 U) | Roche | Cat# 4716728001 |
| ArC™ Amine Reactive Compensation Bead Kit | Invitrogen | Cat# A10346 |
| Anti-Mouse Ig, κ/Negative Control Compensation Particles Set | BD | Cat# 552843 |
| Anti-Rat Ig, κ/Negative Control Compensation Particles Set | BD | Cat# 552844 |
| Countess™ cell counting chamber slides | Invitrogen | Cat# C10228 |
| Trypan blue stain (0.4%) for use with the Countess™ automated cell counter | Invitrogen | Cat# T10282 |
| Software and Algorithms | ||
| FlowJo software (Tree Star) | FlowJo LLC | Version 10.7.1 |
| FACSDiva software | BD | Version 8.5 |
| Cell Accense | CellAccense.com | Version 0.5-beta |
| Other | ||
| 1.2 mL polypropylene micro titer tubes | Fisherbrand | Cat# 02-681-377 |
| 96 V-bottom well microtest Plate with Lid | Sarstedt | Cat# 82-1583-001 |
| Falcon™ round-bottom, disposable, polystyrene 5 mL FACS tubes | Corning | Cat# 350254 |
| Falcon™ 15 mL conical centrifuge tubes | Corning | Cat# 14-959-49B |
| 1.5 mL microcentrifuge tubes | Eppendorf | Cat# 022364111 |
| 0.5 mL microcentrifuge tubes | Eppendorf | Cat# 022363671 |
| BD FACS Symphony A5 Flow cytometer SORP | BD | Ref# 660964 |
| Countess II™ automated cell counter | Invitrogen | Cat# AMQAX1000 |
| Cell culture CO2 Incubator | Sanyo | Cat# MCO-18AIC |
| Centrifuge (with rotor for plates) | Beckman Coulter | Cat# Avanti J-15R |
| Multichannel and single channel micropipettes | Gilson | Multiple references |
BV, Brilliant Violet; BUV, Brilliant Ultraviolet; BB, brilliant blue; AF, Alexa Fluor.
Alternatives: A hemocytometer may be used if an automated cell counter is not available.
Materials and Equipment
Human Samples
Human samples used in figures were obtained from the Human Immunology Core facility of the University of Pennsylvania, which collects mononuclear cells from aphaeresis donors and distributes them to investigators under a blanket IRB protocol (Protocol #705906).
Flow Cytometer
The panels used in this protocol were designed to be used on a BD FACS Symphony A5 flow cytometer with the following configuration: 3 channels for the blue 488 nm laser (forward and side scatter, Brilliant Blue 515, Brilliant Blue 700), 3 channels for the red 640 nm laser (Alexa Fluor 647, Alexa Fluor 700, APC-Cy7), 6 channels for the ultraviolet 355 nm laser (Brilliant Ultraviolet 395, Brilliant Ultraviolet 496, Brilliant Ultraviolet 563, Brilliant Ultraviolet 661, Brilliant Ultraviolet 737, Brilliant Ultraviolet 805), 8 channels for the violet laser 405 nm (Brilliant Violet 405, Brilliant Violet 480, Brilliant Violet 510, Brilliant Violet 570, Brilliant Violet 605, Brilliant Violet 650, Brilliant Violet 750, Brilliant Violet 785) and 4 channels for the yellow green 561 nm laser (PE, PE-CF594, PE-Cy5, PE-Cy7).
FACS Buffer
FACS buffer used for washes and for preparation of staining buffer is prepared by making a dilution of 0.1% sodium azide and 1% bovine serum albumin in 1X PBS.
Extracellular Staining Buffer
Prepare the necessary volume of a 1:1 mix of FACS buffer and BD Brilliant Stain buffer to dilute extracellular antibody mix.
Complete R-10 Medium
Medium used for thawing and culturing cells is prepared by supplementing RPMI 1640 with 10% fetal bovine serum, 2 mM L-glutamine, 100 U/mL penicillin, and 100 mg/mL streptomycin.
Live/Dead Fixable Aqua Dead Cell Stain Kit
To prepare stock solution, thaw aqua fluorescent reactive dye (component A) and DMSO (component B) included in the kit. Add 25 μL of DMSO into the aqua fluorescent reactive dye and mix well with pipette to make the stock solution. Store at −20°C.
Note: The 1:60 dilution for Live/Dead Fixable Aqua Dead Cell Stain should be freshly prepared before performing the experiment.
Step-By-Step Method Details
This protocol can be performed in one day or broken down into 2–3 days based on the number of samples and instrument availability (Table 1). Thawing, plating, and acquisition time will depend on the number of samples stained.
Table 1.
Time Course for Experiment Development
| Day | Duration | Process |
|---|---|---|
| Option 1: Day 0 | ~0.5 h | cell thawing and counting |
| ~18 h | overnight resting | |
| Option 2: Day 1 | ~0.5 h | cell thawing and counting |
| 3 h | cell resting | |
| Day 1 | ~1–1.5 h | preparing antibody cocktail and staining reagents |
| ~20 min | cell counting and plating | |
| 1 h | extracellular staining | |
| 2 h | fixation/permeabilization and intracellular staining | |
| Option 1: Day 1 Option 2: Day 2 |
~20 min | experiment setup |
| ~20–30 min | compensation | |
| 3–7 min/sample | acquisition | |
| ~10 min | experiment clean up and shut down |
This protocol can be used for freshly isolated or cryopreserved PBMC. Antibodies should be titrated in fresh or cryopreserved cells, respectively, to ensure adequate resolution of the markers as detailed in the Antibody Titration section.
Cryopreserved PBMC Thawing and Resting
This protocol details optimized steps for PBMC samples stored in liquid nitrogen to yield >90% viability and ensure high quality resolution. When using cryopreserved samples, cells should be thawed and rested with DNAse I to remove debris from dead cells and reduce background signal. The following steps should be taken for each individual sample.
-
1.
Prepare a waste container with ~5 mL of 10% bleach.
-
2.Aliquot 13 mL of warm complete R10 medium into an adequately labeled 15 mL conical tube, and place in water bath at 37°C.
-
a.If multiple aliquots are being used, adjust the volume of complete R10 medium added to the 15 mL conical tube to avoid overfilling it. Approximately 1 mL of frozen volume per cryotube is assumed.
-
a.
-
3.
Thaw cryopreserved PBMC sample in the water bath for 30–60 s (for ~1 mL of frozen volume) until just melted.
-
4.
Quickly decant into the 15 mL conical tube with warm medium. Carefully pipette out the remaining volume from the cryotube using a P1000 micropipette.
-
5.Mix well without creating bubbles. Count cells and record total number of viable cells and % viability.
-
a.In a 1.5 mL microcentrifuge tube, prepare a 1:1 mixture of 10 μL of cell suspension and 10 μL of trypan blue.
-
b.Load 10 μL of the 1:1 mix into a cell counting slide.
-
c.Insert slide into the automated cell counter and count.
-
a.
-
6.
Centrifuge for 10 min at 400 × g.
-
7.
Decant and resuspend at 2 × 106/mL in complete R10 medium with 1 μL/mL of DNAse I.
-
8.
Transfer the cell suspension to a culture flask adequate for the volume. Rest in the incubator at 37°C 5% CO2 for 2–3 h on the same day of the stain, or ~18 h to stain the following day.
Pause Point: Cells can be rested for ~18 h to perform the cell staining the next day (Table 1).
Note: All centrifugations should be performed at 20°C–25°C.
Note: Based on manufacturer’s instructions (Corning), suggested volumes to be used in culture flasks are: 2–7.5 mL for a 25 cm2 flask, 15–22.5 mL for a 75 cm2 flask, and 30–45 mL for a 150 cm2 flask. These volumes were calculated based on 0.2 to 0.3 mL medium for each square centimeter of culture vessel growth surface area.
Alternatives: Cell staining can be performed in polystyrene 5 mL FACS tubes. The adjusted protocol for tubes is detailed under section “Quick protocol for FACS tubes.”
Preparing Antibody Cocktails and Staining Reagents
This section indicates how to prepare the antibody cocktails in the appropriate staining buffers for the corresponding steps. If the staining is performed on the same day of sample thawing, the antibody cocktail preparation can be done during the incubation for cell resting. Examples are shown for 1, 10, and 20 tests.
One single stain compensation control will be prepared for each parameter measured in this protocol (full panel detailed on Table 7), including their corresponding negative controls (mouse, rat, “plus;” detailed in step 20 at the end of this section). These panels require single stain compensation controls for 22 shared parameters, plus SLeX and CLA for each corresponding panel, and unstained negative controls.
For clear data analysis, placement of gates can be based on fluorescence minus one (FMO controls), particularly for markers with continuous expression, such as activation markers (i.e., Ki67, CD38, HLA-DR, etc.), or for the main markers of interest SLeX and CLA. The need to use FMOs in every single experiment or throughout different samples has been detailed elsewhere (McGowan et al., 2015). FMOs may be used for initial panel setup and further selected based on user’s experience.
Note: While handling fluorochrome marked antibodies, all steps should be performed at 20°C–25°C in the dark.
Note: Foxp3/Transcription Factor Staining Buffer Set contains three components: Permeabilization buffer 10X, Fixation/Permeabilization concentrate and Fixation/Permeabilization diluent. The preparation and use of each component is detailed as needed in the corresponding step.
-
9.
Remove from the refrigerator all antibodies, FACS buffer, BD Brilliant Stain buffer and Foxp3/Transcription Factor Staining Buffer Set and place inside the hood.
-
10.
Remove Live/Dead Fixable Aqua Dead Cell Stain stock solution from the freezer and place inside the hood.
-
11.Determine the number of necessary tests. Prepare an excess of 1–2 samples (for <11 samples) or 3–5 more (for >10 samples).
-
a.Live/Dead Fixable Aqua Dead Cell Stain stock solution diluted 1:60 → 5 μL/test.
-
b.Staining buffer: 1:2 mix of FACS buffer and BD Brilliant Stain buffer → 26.3 μL/test.
-
c.1X eBioscience wash buffer: 94.5 μL/test for antibody dilution + 200 μL/test for washes → 294.5 μL/test.
-
a.
Note: Remember to account for FMOs.
-
12.In an appropriately labeled 1.5 mL microcentrifuge tube, or a 15 mL conical tube (depending on the necessary volume), prepare the chemokine receptor cocktail following the volumes indicated in Table 2.
-
a.Start by adding PBS.
-
b.Continue by adding antibodies in order of decreasing volumes.
-
c.Once finished, vortex for 5 s and do a quick spin to collect the volume at the bottom of the tube.
-
d.Set the chemokine receptor antibody cocktail aside in the dark inside the hood.
-
a.
-
13.In an appropriately labeled 0.5 mL microcentrifuge tube, prepare the 1:60 dilution of the stock solution of Live/Dead Fixable Aqua Dead Cell Stain in PBS.
-
a.Make sure that the Live/Dead Fixable Aqua Dead Cell Stain stock solution is fully thawed before pipetting the necessary volume.
-
b.Place the stock solution back in the freezer immediately.
-
c.Vortex for 5 s and follow by a quick spin to collect the volume at the bottom of the tube.
-
d.Set the Live/Dead Fixable Aqua Dead Cell Stain dilution aside in the dark inside the hood.
-
a.
- 14.
-
15.In an appropriately labeled 1.5 mL microcentrifuge tube, or a 15 mL conical tube (depending on the necessary volume), prepare the extracellular antibody cocktail following the volumes indicated in Table 3 for SLeX staining, or Table 4 for CLA staining.
-
a.Start by adding staining buffer.
-
b.Continue by adding antibodies in order of decreasing volumes.
-
c.Once finished, vortex for 5 s and follow by a quick spin to collect the volume at the bottom of the tube.
-
d.Set the extracellular antibody cocktails aside in the dark inside the hood.
-
a.
-
16.
In appropriately labeled 1.5 mL microcentrifuge tubes, prepare the antibody cocktails for the relevant FMOs. Examples of cocktails from preparing FMOs for the antibody cocktail including CLA are shown in Table 5. FMOs suggested for activation markers and CLA. While critical for initial characterization of the panel, users can evaluate the need to include these in every subsequent experiment depending on the consistency of the signal for each marker.
-
17.In an appropriately labeled 1.5 mL microcentrifuge tube, or a 15 mL conical tube (depending on the necessary volume), prepare fresh Foxp3 fixation/permeabilization working solution by adding 1 part of Foxp3 Fixation/Permeabilization concentrate and 3 parts Foxp3 Fixation/Permeabilization diluent (1:4 dilution, calculate 100 μL/test). These reagents are components of the Foxp3/Transcription Factor Staining Buffer Set.
-
a.Mix well with pipette without making bubbles.
-
b.Set aside in the hood.
-
a.
-
18.Prepare eBioscience 1X permeabilization buffer to be used for washes and intracellular antibody cocktail preparation (Table 6). The eBioscience 1X permeabilization buffer is a component of the Foxp3/Transcription Factor Staining Buffer Set.
-
a.Label a 15 mL conical tube “1X permeabilization buffer”.
-
b.Prepare the necessary volume of a 1:10 dilution of eBioscience 10X Permeabilization buffer in ddH2O.
-
c.Mix well with pipette without making bubbles.
-
d.Set aside in the hood.
-
a.
-
19.In an appropriately labeled 1.5 mL microcentrifuge tube or a 15 mL conical tube (depending on the necessary volume), prepare the intracellular antibody cocktail following the volumes indicated in Table 6.
-
a.Start by adding 1X permeabilization buffer.
-
b.Continue by adding antibodies in order of decreasing volumes.
-
c.Once finished, vortex for 5 s and follow by a quick spin to collect the volume at the bottom of the tube.
-
d.Set the intracellular antibody cocktail aside in the dark inside the hood.
-
a.
Optional: The intracellular antibody cocktail can be prepared during the fixation/permeabilization incubation step (30 min) following the extracellular staining.
-
20.Prepare compensation controls.
-
a.Label a 5 mL FACS tube for each corresponding antibody and fluorochrome.
-
b.Label one tube as negative for mouse antibodies.
-
c.Label one tube as negative for rat antibodies.
-
d.Add one drop of Anti-Rat Ig κ beads to the CXCR5 BB515 and CLA AF647 compensation tubes.
-
e.Add one drop of Anti-Mouse Ig or Anti-Rat Ig Negative Control beads to the corresponding negative control tubes.
-
f.Add one drop of Anti-Mouse Ig κ beads to all remaining tubes.
-
g.Add the corresponding antibody and mix well with pipette.
-
i.Add 0.5 μL of the antibody if the titer is 1:200 (0.5 μL/test) or higher.
-
ii.Add the corresponding antibody volume if the dilution is lower than 1:200 ( <0.5 μL/test).
-
i.
-
h.Incubate in the dark at 20°C–25°C.
-
a.
Note: Compensation tubes can be prepared at any time and incubated for ~10 min. In this protocol, we indicate to prepare them initially and incubate for the duration of the experiment.
Note: The use of Plus beads (BDTM CompBead Plus Particles, anti-mouse Ig κ Cat# 560497 or anti-rat Ig κ Cat# 560499) may be necessary for certain parameters based on the flow cytometer configuration. An example of the use of Plus beads for compensation is shown in Figure 4. The positive peak of the compensation control must be brighter than the brightest population. Given the brightness of the anti-human IFN- γ AF700 antibody, the use of Plus beads is required. As depicted in this example, the peak for the Plus beads in the AF700 channel fell above 104 (Figure 4A), and all events would be correctly compensated (Figure 4B). The uncompensated area is indicated by an orange box. When using a normal compensation bead, the peak fell below 104 (Figure 4C). All events below the peak would be correctly compensated, but every event above the peak would not (Figure 4D, purple selection indicated uncompensated area). Note that the example provided was generated from a different experiment, as the panel detailed in this protocol does not call for the use of Plus beads for compensation. When using Plus beads in a compensation control, the corresponding unstained negative control for Plus beads should be prepared and acquired as well.
-
21.
Prepare the appropriate volume of a dilution of 1% paraformaldehyde in PBS. Calculate 200–300 μL/sample.
Note: Account for all type of samples: unstained controls, FMO controls, compensation controls, and experimental samples.
Note: 1% paraformaldehyde dilution is only necessary to perform final fixation of infectious samples. Further details on step 37 under "Final fixation and acquisition."
Optional: FMOs for chemokine receptors or intracellular markers can be prepared in the same fashion as exemplified in Table 5 for the extracellular antibody cocktail. Adjust the volumes from Tables 2 and 3 (for the SLeX panel) or 6 accordingly.
Note: The use of FMOs in different steps of the analyses is discussed under “Expected Outcomes.”
Table 6.
Intracellular Antibody Cocktail
| Marker | Fluorochrome | Comp Bead | 1X μL/test | 10X (+1) μL/test | 20X (+3) μL/test |
|---|---|---|---|---|---|
| Foxp3 | PE-Cy7 | mouse | 5 | 55 | 115 |
| Ki67 | BUV395 | mouse | 0.5 | 5.5 | 11.5 |
| Sum of antibodies | 5.5 | 60.5 | 126.5 | ||
| 1X Permeabilization Buffer | 94.5 | 1,039.5 | 2,173.5 | ||
| Final volume | 100 | 1,100 | 2,300 | ||
Cell Counting and Plating for Staining
After resting, cells should be counted and plated at a similar number per test to ensure homogeneity in the stain. For a small number of samples, the stain can be performed in 5 mL polystyrene FACS tubes. The adjusted volumes for tubes are described in the “Brief Protocol for FACS tubes” section.
Note: All steps are performed at 20°C–25°C.
-
22.
Label a 15 mL conical tube with the corresponding sample ID.
-
23.
Label a 96-well V-bottom plate with the sample IDs in the desired order.
-
24.After incubation for resting on complete R10 medium with DNAse I, transfer the sample into the corresponding 15 mL conical tube and count.
-
a.Complete the volume to 15 mL with PBS as an initial washing step.
-
b.Detailed counting detailed on step 5 under “Cryopreserved PBMC thawing and resting.”
-
c.Record number of viable cells and viability.
-
a.
-
25.
Centrifuge tube for 10 min at 400 × g.
-
26.
Decant and resuspend in PBS at 30 × 106 cells/mL, yielding a concentration of 3 × 106 in 100 μL.
Note: The cell number can be adjusted from 1–5 × 106 cells/well (taking into account the volume used for antibody titration). When performing the experiment on multiple samples, it is important to maintain a similar staining volume for all samples to ensure homogeneity in the stain.
-
27.
Transfer 100 μL of the cell suspension (in PBS) into the corresponding well in the 96-well V-bottom plate (3 × 106 cells/well).
-
28.Centrifuge plate for 5 min at 400 × g.
-
a.Remove the supernatant carefully using pipette by leaning the pipette tip at the edge of the well cone; as shown in step 4 of the “Antibody titration” section.
-
a.
Extracellular Staining
In this section of the protocol, cells are stained sequentially for chemokine receptor expression, viability, and extracellular markers. These stains require different buffers, as well as incubation temperatures and times to achieve optimal resolution.
-
29.Immediately after removing the supernatant, resuspend the cells with 45 μL/test of the chemokine receptor extracellular antibody cocktail (prepared in PBS as indicated in Table 2, under Preparing Antibody Cocktails and Staining Reagents).
-
a.Mix well to fully resuspend the cell pellet, being mindful not to create bubbles.
-
b.Cover the plate with the lid and place the plate inside the incubator at 37°C 5% CO2, for 15 min.
-
a.
-
30.Remove the plate from the incubator and, without washing, add 5 μL/test of the 1:60 dilution of the Live/Dead Fixable Aqua Dead Cell Stain.
-
a.Mix well with pipette, being mindful not to create bubbles.
-
b.Cover the plate with the lid and incubate in the hood for 10 min at 20°C–25°C.
-
c.At this point, each well contains a volume of ~50 μL: 45 μL of chemokine receptor extracellular antibody cocktail and 5 μL of Live/Dead Fixable Aqua Dead Cell Stain dilution.
-
a.
-
31.Without washing, add 50 μL/test of the corresponding extracellular antibody cocktail (prepared in staining buffer as indicated in Tables 3 and 4, under Preparing Antibody Cocktails and Staining Reagents).
-
a.Mix well with pipette, being mindful not to create bubbles.
-
b.Cover the plate with the lid and incubate in the hood for 20 min at 20°C–25°C.
-
c.At this point, each well contains a final staining volume of ~100 μL.
-
a.
-
32.Wash with 100 μL/well of FACS buffer.
-
a.Centrifuge for 5 min at 400 × g.
-
b.Carefully remove supernatant with the pipette tip as previously indicated.
-
a.
Note: If staining a large number of samples, the antibody mixes can be transferred into a reservoir and added to the cells using a multichannel pipette, being mindful not to create bubbles while mixing.
Fixation/Permeabilization and Intracellular Staining
In this section of the protocol, the cells will be fixed and permeabilized to allow for intracellular stain of Ki67 and Foxp3.
-
33.Immediately after removing the supernatant from the wash, resuspend the cells with 100 μL/well of the Foxp3 fixation/permeabilization working solution (prepared as indicated in step 16, under Preparing Antibody Cocktails and Staining Reagents).
-
a.Mix well to fully resuspend the cell pellet, being mindful of not creating bubbles.
-
b.Cover the plate with the lid and incubate for 30 min at 20°C–25°C in the dark.
-
a.
Optional: The intracellular antibody cocktail can be prepared during this incubation following Table 6, under Preparing Antibody Cocktails and Staining Reagents.
-
34.Wash with 100 μL/well of 1X permeabilization buffer.
-
a.Centrifuge for 5 min at 800 × g.
-
b.Carefully remove supernatant with the pipette tip.
-
a.
Note: Cells may be less visible after fixation/permeabilization.
-
35.Resuspend with 100 μL/well of the intracellular antibody cocktail (prepared in 1× permeabilization buffer as indicated in Table 6, under Preparing Antibody Cocktails and Staining Reagents in steps 17 and 18).
-
a.Mix well to fully resuspend the cell pellet, being mindful of not creating bubbles.
-
b.Cover the plate with the lid and incubate for 1 h at 20°C–25°C in the dark.
-
a.
-
36.Wash with 100 μL/well of 1× permeabilization buffer.
-
a.Centrifuge for 5 min at 800 × g.
-
b.Carefully remove supernatant with the pipette tip.
-
a.
Final Fixation and Acquisition
In this section of the protocol, the cells and compensation controls are prepared for acquisition.
Note: Depending on the type of sample, the concentration of paraformaldehyde may need to be adjusted to comply with biosafety protocols.
-
37.
Immediately after removing the supernatant from the wash, resuspend the cells in 200 μL of 1% paraformaldehyde in PBS. Mix well to fully resuspend the cell pellet, being mindful of not creating bubbles.
Alternatives: At this point, cells will have already been fixed before performing the intracellular stain. For non-infectious samples, cells can be resuspended in 200 μL of PBS instead of PFA for acquisition. As this protocol was optimized to be used in HIV-infected samples, an extra fixation step is recommended for biosafety purposes.
-
38.
Transfer the volume into an appropriately labeled 1.2 mL microtiter tube or a 5 mL FACS tube as preferred.
-
39.
Add 200–300 μL of a freshly made solution of 1% paraformaldehyde in PBS to all compensation controls.
-
40.
Store samples and compensation tubes in the dark at 4°C until acquisition.
-
41.
Acquire using a FACS Symphony A5 cytometer within 24 h of staining.
Note: Although the most fixed/permeabilized samples will remain intact for up to 7 days if kept in the dark at 4°C, best results are obtained if acquired in the first 48 h, being 24 h the optimal time to acquisition. Scatter properties can change with extended storage in PFA.
Note:Table 7 includes the full list of antibodies and dilutions used for the panels described in this protocol.
Table 7.
Flow Cytometry Panel
| Laser | Filter | Fluorochrome | Antigen | Manufacturer | Clone | Dilutiona |
|---|---|---|---|---|---|---|
| 488 nm | 515/20 | BB515 | CXCR5 | BD | RF8B2 | 140 |
| 710/50 | BB700 | CD4 | BD | SK3 | 160 | |
| 640 nm | 660/20 | AF647 | CLAb | Biolegend | HECA-452 | 200 |
| 660/20 | AF647 | SLeX (CD15S)b | BD | CSLEX1 | 200 | |
| 730/45 | AF700 | CXCR3 | Biolegend | G025H7 | 50 | |
| 780/60 | APC-Cy7 | CCR7 | Biolegend | G043H7 | 50 | |
| 355 nm | 379/28 | BUV395 | Ki67 | BD | B56 | 200 |
| 515/30 | BUV496 | CD8 | BD | RPA-T8 | 200 | |
| 586/15 | BUV563 | CD45RA | BD | HI100 | 200 | |
| 660/20 | BUV661 | CD38 | BD | HIT2 | 66.7 | |
| 740/35 | BUV737 | CD25 | BD | 2A3 | 200 | |
| 820/60 | BUV805 | CD3 | BD | UCHT1 | 200 | |
| 405 nm | 450/50 | BV421 | CCR6 | BD | 11A9 | 100 |
| 470/15 | BV480 | CD127 | BD | HIL-7R-M21 | 50 | |
| 515/20 | Aqua Blue | Viability | Invitrogen | n/a | 20 | |
| 515/20 | BV510 | CD14 | Biolegend | M5E2 | 200 | |
| 515/20 | BV510 | CD19 | Biolegend | HIB19 | 200 | |
| 586/15 | BV570 | CD27 | Biolegend | O323 | 50 | |
| 610/20 | BV605 | HLA-DR | BD | G46-6 | 100 | |
| 660/20 | BV650 | CCR5 | BD | 3A9 | 166.7 | |
| 710/50 | BV711 | CD95 | Biolegend | DX2 | 100 | |
| 780/60 | BV785 | PD-1 | Biolegend | EH12.2H7 | 50 | |
| 561 nm | 586/15 | PE | CD69 | BD | FN50 | 10 |
| 610/20 | PE-CF594 | CCR4 | BD | 1G1 | 100 | |
| 660/40 | PE-Cy5 | CXCR4 | BD | 12G5 | 40 | |
| 780/60 | PE-Cy7 | FoxP3 | eBioscience | 236A/E7 | 20 |
n/a, not applicable; BB, brilliant blue; AF, Alexa Fluor; APC, allophycocyanin; BUV; Brilliant Ultraviolet; BV, Brilliant Violet; PE, phycoerythrin.
Dilutions are calculated by titration of all antibodies. In some cases, antibody volumes/dilution factors are rounded up to facilitate pipetting (for example, CD38 BUV661 is used at a 1.5 μL/test or a 1:66.7 dilution).
This protocol indicates the staining of CLA AF647 and SLeX AF647 in two independent panels, not simultaneously in the same antibody cocktail.
Brief Protocol for FACS Tubes
This section indicates how to perform the staining procedure described in steps 1–41 in 5 mL polystyrene FACS tubes for a small number of samples. Refer to the fully described protocol above for details. Authors recommend using FACS tubes for under 8–10 samples to optimize reagents and duration of procedure.
-
42.Determine the number of necessary tests. Prepare an excess of 1–2 samples (for <11 samples) or 3–5 more (for >10 samples).
-
a.Live/Dead Fixable Aqua Dead Cell Stain stock solution diluted 1:60 → 5 μL/test
-
b.Staining buffer: 1:1 mix of FACS buffer and BD Brilliant Stain buffer → 26.7 μL/test
-
c.1X eBioscience wash buffer: 94.5 μL/test for antibody dilution + 2 mL/tube for washes → 2,094.5 μL/test
-
a.
-
43.
In an appropriately labeled 1.5 mL microcentrifuge tube, prepare the chemokine receptor cocktail following the volumes indicated in Table 2.
-
44.
In an appropriately labeled 0.5 mL microcentrifuge tube, prepare the 1:60 dilution of the stock solution of Live/Dead Fixable Aqua Dead Cell Stain in PBS.
-
45.
Prepare the adequate amount of staining buffer by mixing 1:1 volume of FACS buffer and BD Brilliant Stain buffer, as indicated in Table 3 for SLeX staining, or Table 4 for CLA staining.
-
46.
Prepare the extracellular antibody cocktail following the volumes indicated in Table 3 for SLeX staining, or Table 4 to for CLA staining.
-
47.
Determine the volume of Foxp3 fixation/permeabilization working solution needed (250 μL/sample) and prepare by adding 1 part of Foxp3 Fixation/Permeabilization concentrate and 3 parts Foxp3 Fixation/ Permeabilization diluent (these reagents are components of the Foxp3/Transcription Factor Staining Buffer Set).
-
48.Determine the necessary volume of eBioscience 1× permeabilization buffer.
-
a.94.5 μL/test as indicated in Table 6
-
b.4 mL/test for washing
-
a.
-
49.
Prepare eBioscience 1X permeabilization buffer by making a 1:10 dilution of eBioscience 10× Permeabilization buffer in ddH2O (this reagent is a component of the Foxp3/Transcription Factor Staining Buffer Set).
-
50.
Prepare the intracellular antibody cocktail in 1× permeabilization buffer following the volumes indicated in Table 6.
-
51.
Prepare compensation controls.
-
52.
Prepare 1% paraformaldehyde in PBS. Calculate 200–300 μL/sample.
-
53.
Label 5 mL polystyrene tubes with the corresponding sample ID.
-
54.
Aliquot 3 × 106 of the corresponding sample in each tube.
-
55.
Add 2 mL of PBS, and centrifuge for 5 min at 400 × g. Decant the supernatant into a container with the right disinfectant. Dry any remaining drops at the end of the tube in a paper towel, while the tube is still being held upside down to prevent disturbance of the pellet by backflow of the drops. Discard the towel appropriately.
-
56.
Immediately after removing the supernatant, resuspend the cells with 45 μL/test of the chemokine receptor extracellular antibody cocktail (prepared in PBS as indicated in Table 2, under Preparing Antibody Cocktails and Staining Reagents in step 2). Mix well and place inside the incubator at 37°C, 5% CO2, for 15 min.
-
57.
Without washing, add 5 μL/tube of the 1:60 dilution of the Live/Dead Fixable Aqua Dead Cell Stain. Mix well and incubate in the hood for 10 min at 20°C–25°C.
-
58.
Without washing, add 50 μL/tube of the corresponding extracellular antibody cocktail (prepared in staining buffer as indicated in Table 3 and Table 4, under Preparing Antibody Cocktails and Staining Reagents in steps 4 and 5). Mix well and incubate in the hood for 20 min at 20°C–25°C.
-
59.
Wash with 2 mL/tube of FACS buffer. Centrifuge for 5 min at 400 × g. Carefully decant supernatant. Dry any remaining drops at the end of the tube in a paper towel. Discard the towel appropriately.
-
60.
Immediately after removing the supernatant from the wash, resuspend the cells with 250 μL/tube of the Foxp3 fixation/permeabilization working solution (prepared as indicated in step 6, under Preparing Antibody Cocktails and Staining Reagents).Mix well and incubate for 30 min at 20°C–25°C in the dark.
-
61.
Wash with 2 mL/tube of 1X permeabilization buffer. Centrifuge for 5 min at 800 × g. Carefully decant supernatant. Dry any remaining drops at the end of the tube in a paper towel. Discard the towel appropriately.
-
62.
Resuspend with 100 μL/tube of the intracellular antibody cocktail (prepared in 1× permeabilization buffer as indicated in Table 6, step 9 under Preparing Antibody Cocktails and Staining Reagents). Mix well and incubate for 1 h at 20°C–25°C in the dark.
-
63.
Wash with 2 mL/tube of 1X permeabilization buffer. Centrifuge for 5 min at 800 × g. Carefully decant supernatant. Dry any remaining drops at the end of the tube in a paper towel. Discard the towel appropriately.
-
64.
Immediately after removing the supernatant from the wash, resuspend the cells in 200 μL of 1% paraformaldehyde in PBS.
Note: Cells can be resuspended in PBS for non-infectious samples. See Notes above in the fully detailed protocol.
-
65.
Store samples and compensation tubes in the dark at 4°C until acquisition.
-
66.
Acquire using a FACS Symphony A5 cytometer within 24 h of staining.
Expected Outcomes
At the end of this protocol, fcs files will be generated with markers which will allow characterization of CD4+ (and CD8+) T cells expressing CLA or SLeX. The markers included in this panel will also permit characterization of T cell memory subsets, Th subsets for CD4+ T cells, and quantify the levels of activation (CD25, CD38, HLA-DR) and cell cycling (Ki67), and the expression of CXCR4 and CCR5. Depending on the gating approach, and whether combination/Boolean gates are used, this panel will generate >400 quantifiable parameters including frequencies and level of expression (MFI) within different cell subsets. Data generated using this protocol has been recently published (Colomb et al., 2020).
The recommended gating strategy for the analysis of memory subsets is shown in Figure 5. Firstly, the most stable period of acquisition should be selected to yield optimal resolution of the data. Single cells are then selected, and lymphocytes are identified based on their morphology on forward (FSC-A) and side scatter (SSC-A). This protocol includes the Live/Dead Fixable Aqua Dead Cell Stain, CD14 BV510 and CD19 BV510 to be read in the same channel (violet laser, ~515 nm LP). As dead cells, monocytes and B cells will be excluded from this analysis, these are all removed by using a “Dump” channel. CD3+ dump- cells are selected, and CD8− CD4+ cells are then gated subgated from them (Figure 5A). For experienced users, we recommend using CD27 in the Y axis to help identify the cutoff for the negative/positive gate for SLeX and CLA. In the case of CLA+ cells, analyses showed two distinct populations expressing low and high levels of CLA (named CLAlow and CLAhi, for simplicity). Otherwise, an FMO control can be used to clearly determine the positive and negative gates (Figure 5B).
Figure 5.

Gating Strategy for Identification of CD4+ T Cells Expression of SLeX and CLA, and Memory Subset Characterization within CD4+ T Cells
Representative example of two possible gating strategies used for analysis of a cryopreserved PBMC sample, stained simultaneously with the two described panels.
(A) General gating strategy to characterize total CD4+ T cells. First, the most stable acquisition time is selected. Single lymphocytes were then gated using FSC and SSC. Dead cells, monocytes, and B cells were excluded using a dump channel and by gating on CD3+ T cells. Total CD8− CD4+ cells were then selected. The initial characterization of CD4+ T cells is common for both stains.
(B) Identification of SLeX+ in Stain 1 (using the antibody cocktail described on Table 3) or CLA+ cells (black gate), CLAhi (teal gate), and CLAlow (orange gate) in Stain 2 (using the antibody cocktail described in Table 4). The negative (gray) and positive (black) gates were determined using an FMO control.
(C) Characterization of CD4+ T cell memory subsets using CD27, CD45RA, CCR7, and CD95.
Different approaches can be used to analyze the phenotype of glycosylated CD4+ T cells depending on the research question: 1) Gating of SLeX+/CLA+/low/hi cells within total CD4+ T cells, followed by identification of memory subsets using CD45RA, CD27, CCR7, and CD95. 2) Identifying memory subsets in total CD4+ cells, and then characterizing the frequencies of SLeX+/CLA+/low/hi within said subsets. For this analysis, memory subsets were defined as: naive (TN: CD45RA+ CCR7+ CD27+ CD95-), stem cell memory (TSCM: CD45RA+ CCR7+ CD27+ CD95+), RA+ effector memory (TEMRA: CD45RA+ CCR7- CD27+), transitional memory (TTM: CD45RA- CCR7- CD27+), central memory (TCM: CD45RA- CCR7+ CD27+), and effector memory (TEM: CD45RA- CCR7- CD27-). A detailed gating strategy is shown in Figure 5C.
As a next step in the phenotyping of SLeX+/CLA+/low/hi CD4+ T cells, gating based on PD-1 and chemokine receptor expression within memory cells will allow to characterize Th subsets (Figure 6). As indicated above, two approaches can be followed for this analysis: 1) Gate on memory CD4+ T cells, followed by selection of SLeX- or CLA- expressing cells and then characterization of Th subsets. 2) Alternatively, Th subsets can be identified in memory CD4+ T cells, followed by the measurement of SLeX or CLA expression within them. In this analysis, the gating strategy identified: circulating follicular T cells (cTfh, PD-1+ CXCR5+) and CXCR3+ cTfh, regulatory T cells (Tregs, CD127low CD25+ Foxp3+ non-cTfh), CXCR5+ non-Tfh/non-Tregs, Th1 cells (CXCR5- CCR6- CXCR3+ CCR4- non-Tfh/non-Tregs), Th2 cells (CXCR5- CCR6- CXCR3- CCR4+ non-Tfh/non-Tregs), Th17+1 cells (CXCR5- CCR6+ CXCR3+ CCR4- non-Tfh/non-Tregs) and Th17 cells (CXCR5- CCR6+ CXCR3- CCR4+ non-Tfh/non-Tregs).
Figure 6.

Example of Gating Strategy for the Characterization of Th Subsets in Memory CD4+ T Cells
Representative example of analyses in a cryopreserved PBMC sample. Memory CD4+ T cells are first selected by excluding naive cells (CCR7+ CD45RA+ CD4+ T cells). Inside memory CD4+ T cells, SLeX+ were selected from Stain 1, using the antibody cocktail described on Table 3. CLA+ cells (black gate), CLAhi (teal gate), and CLAlow (orange gate) were selected from Stain 2, using the antibody cocktail described in Table 4. cTfh cells are identified using PD-1 and CXCR5. CD127low CD25+ cells were selected within non-cTfh cells, and Foxp3+ cells were then selected to identify Tregs. Non-cTfh/Tregs cells were selected based on CXCR5 and CCR6 expression. Th1 and Th2 cells were identified within the CXCR5- CCR6- gate, while Th17 and Th17+1 were characterized ins the CXCR5- CCR6+ gate using CXCR3. The frequency of CXCR3+ Tfh can also be measured with this panel.
To continue with the phenotypic characterization of SLeX+/CLA+/low/hi cells CD4+ T cells, a similar approach can be used. Cells expressing activation markers (such as CD38, HLA-DR, Ki67, CD25, CD69, PD-1), as well as other chemokine receptors not used to characterize Th subsets (such as CXCR4 and CCR5) can be selected, followed by analysis of CLA/SLex expression within those (example of analysis with HLA-DR shown in Figure 7A). Conversely, cells which express SLeX+/CLA+/low/hi can be identified first, and then quantify specific markers within those populations (example of analysis with HLA-DR is shown in Figure 7B). To set up gates for activation markers and chemokine receptors, using the naive cell gate or plotting against CD27 is recommended. Different approaches can be used to define the positive and negative gates for each marker of interest. The standard approach is to use an FMO for the specific marker, as shown for HLA-DR in Figure 7C. Experienced users could explore co-expression of the marker of interest with others, as exemplified with CD38 and HLA-DR (known to be co-expressed in chronic HIV infection (Appay and Sauce, 2008; Kestens et al., 1992)), and use quadrant gates to determine positive/negative populations (Figure 7D). Alternatively, experienced users can also use a population known to lack the expression of the marker (or express it at very low levels) as a reference to set the cutoff when compared to the population of interest. An example of this type of biological gating is shown in Figure 7E, comparing the expression of HLA-DR in naive vs. memory CD4+ T cells. Note the use of CD27 on the Y axis to facilitate the identification of the positive/negative gates, as suggested above. The need to use FMOs for all markers and all samples has been published elsewhere (McGowan et al., 2015).
Figure 7.

Example of Analysis of Activation Markers in CLA+/hi/low CD4+ T Cells
Representative dot plots gated on total CD4+ T cells as shown in Figure 5, stained on cryopreserved PBMC from an HIV-infected individual under antiretroviral treatment (using the antibody cocktail detailed in Table 4).
(A) Gating on HLA-DR+ CD4+ T cells, followed by selection of subsets based on expression of CLA. Number inside the gate indicates the percentage of HLA-DR+ CD4+ T cells.
(B) Gating of subsets based on expression of CLA, followed by an overlay plot showing the selection of HLA-DR+ cells within each subset. Corresponding frequencies are shown to the right of the plots.
(C) FMO used to determine the HLA-DR+ gate.
(D) Example of biological gating based on CD4+ T cell co-expression of CD38 and HLA-DR.
(E) Example of biological gating using expression of CD27 and HLA-DR, and naive cells as a reference to determine positive and negative populations. Total memory CD4+ T cells were selected as shown in Figure 6.
For further analyses, the expression of activation markers and other chemokine receptors can be quantified in Th subsets expressing SLeX or CLA. Conversely, the Th phenotype can be characterized within activated cells expressing SLeX or CLA. The number of cells stained and events acquired may need to be increased depending on how many levels of gating are intended in the analyses.
Alternatively, unbiased approaches can be used to perform broad exploration of the data. To do so, after analyzing in FlowJo, a new .fcs file was generated from the CD4+ T cell gate. This file was then analyzed using Cell Accense (without downsampling) to produce t-SNE plots and highlight the expression of different markers. An example of co-localization of all other parameters included in this panel within CD4+ T cells, in a stain using CLA is shown in Figure 8. Similar analyses can be performed by gating on CD8+ T cells, or within total CD3+ T cells for a broader overview of the data.
Figure 8.

Example of Phenotypic Characterization of CD4+ T Cells in a CLA Stained Sample
Dimensionality reduction analysis performed from 65,982 cells in 19 dimensions. t-SNE visualization of parameters measured by flow cytometry on a CLA stained (using the antibody mix detailed in Table 4) sample of cryopreserved PBMC. This analysis was performed on an independent .fcs file generated from the CD4+ T cell gate, and excluding parameters used to define this population (time, FSC, SSC, viability, CD14, CD19, CD3, CD4, and CD8).
Limitations
This protocol assumes the availability of a 30-parameter flow cytometer which has been properly optimized for the detection of all fluorochromes included in this panel (Giesecke et al., 2017; Perfetto et al., 2012), as well as biosafety cabinets and fume hood for handling of potentially hazardous materials (such as sodium azide and paraformaldehyde). This protocol may take longer depending on the number of samples used, and may require more than one person to perform it, especially at the cryopreserved sample thawing step. Both sample preparation and sample acquisition are time consuming. As we recommend the preparation of fresh reagents before performing each experiment, this introduces the possibility of inter-experiment variation mainly through master mix preparation.
This protocol has been standardized to be used for immunophenotyping of human PBMC, either fresh or cryopreserved, isolated using Ficoll density-gradient centrifugation. Although not specifically annotated here, this protocol can be used for staining tissue samples provided the cells are in good condition. Titration of the antibodies for the specific cell suspension will be required to achieve optimal resolution.
Finally, given the complexity of these high parameter panels, this protocol requires mid to advanced knowledge on multiparametric flow cytometry. For proper data analysis, the researcher must know how to appropriately acquire compensation controls, and then manually adjust automatically calculated compensation matrices to yield optimal results.
Troubleshooting
Problem 1
Poor cell viability after thawing.
Potential Solutions
-
1.
Fast thawing is critical to avoid cell death due to DMSO exposure. Make sure that the samples are thawed and washed quickly.
-
2.
Add a 2nd wash after step 6 under Cryopreserved PBMC Thawing and Resting to ensure complete removal of DMSO.
Problem 2
Cannot detect a positive signal while acquiring a compensation control.
Potential Solutions
-
1.
Make sure that the correct bead was used for preparing the compensation control. For example, the CXCR5 BB515 antibody used in this protocol was made using rats. If the compensation control was prepared using anti-mouse beads, the stain will not work.
-
2.
Although this protocol was designed to use BD Compensation Particles, eBioscience OneComp eBeads (Cat# 01-111-42), UltraComp eBeads (Cat# 01-2222-42) and UltraComp Plus eBeads (01-333-42) can be used as an alternative. These beads are reactive to antibodies made in mouse and rat.
Problem 3
Cannot detect intracellular stain.
Potential Solutions
-
1.
Ensure that the intracellular stain (Table 6) is prepared in eBioscience 1× permeabilization buffer. The permeabilization process continues during the 1-h incubation of the intracellular staining. If the intracellular antibody cocktail is prepared in regular staining buffer, membrane permeabilization will be suboptimal and will affect the resolution of intracellular markers.
-
2.
Make sure that the Foxp3/Transcription Factor Staining Buffer Set and antibodies have not passed the expiration date. Old reagents will produce suboptimal results. If obtaining newer reagents is not possible, re-titrate the antibodies to yield optimal staining.
Problem 4
Very low events recovered upon acquisition.
Potential Solutions
-
1.
This protocol includes multiple washes and fixation/permeabilization steps, which can make the pellets fragile. Ensure that fixation/permeabilization does not go past the recommended incubation time.
-
2.
If using V-bottom plates, make sure that the supernatant is removed carefully as shown in Figure 2, without mistakenly affecting the pellet. If using tubes, make sure that the pellet is not lost when decanting supernatant from the washes.
-
3.
Start with a larger cell number. By doing so, make sure the antibodies are being titrated using a similar cell number.
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, betts@pennmedicine.upenn.edu Leticia Kuri-Cervantes, kuri@pennmedicine.upenn.edu.
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
There is no dataset/code associated with the paper.
Acknowledgments
We thank the Human Immunology Core at the University of Pennsylvania for assistance with sample processing for these studies and NIH P30-CA016520 and P30-AI0450080 for infrastructure support. The studies were supported by NIH UM1AI126620, R01-AI118694, and P01-AI131338 to M.R.B. Graphical abstract and Figures 1 and 2 were created using BioRender.com
Author Contributions
Conceptualization and Methodology, L.K.-C. and M.R.B. Formal Analysis, Visualization, Data Curation, L.K.-C. Writing – Original Draft, L.K.-C. Figure Preparation, L.K.-C. and M.B.P. Writing – Review & Editing: L.K.-C., M.B.P., and M.R.B. Supervision, Resources, and Funding Acquisition, M.R.B.
Declaration of Interests
The authors declare no competing interests.
References
- Appay V., Sauce D. Immune activation and inflammation in HIV-1 infection: causes and consequences. J. Pathol. 2008;214:231–241. doi: 10.1002/path.2276. [DOI] [PubMed] [Google Scholar]
- Colomb F., Giron L.B., Kuri-Cervantes L., Adeniji O.S., Ma T., Dweep H., Battivelli E., Verdin E., Palmer C.S., Tateno H. Sialyl-Lewis(X) glycoantigen is enriched on cells with persistent HIV transcription during therapy. Cell Rep. 2020;32:107991. doi: 10.1016/j.celrep.2020.107991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming K.K., Hubel A. Cryopreservation of hematopoietic and non-hematopoietic stem cells. Transfus. Apher. Sci. 2006;34:309–315. doi: 10.1016/j.transci.2005.11.012. [DOI] [PubMed] [Google Scholar]
- Giesecke C., Feher K., von Volkmann K., Kirsch J., Radbruch A., Kaiser T. Determination of background, signal-to-noise, and dynamic range of a flow cytometer: a novel practical method for instrument characterization and standardization. Cytometry A. 2017;91:1104–1114. doi: 10.1002/cyto.a.23250. [DOI] [PubMed] [Google Scholar]
- Kestens L., Vanham G., Gigase P., Young G., Hannet I., Vanlangendonck F., Hulstaert F., Bach B.A. Expression of activation antigens, HLA-DR and CD38, on CD8 lymphocytes during HIV-1 infection. AIDS. 1992;6:793–797. doi: 10.1097/00002030-199208000-00004. [DOI] [PubMed] [Google Scholar]
- Liseth K., Abrahamsen J.F., Bjorsvik S., Grottebo K., Bruserud O. The viability of cryopreserved PBPC depends on the DMSO concentration and the concentration of nucleated cells in the graft. Cytotherapy. 2005;7:328–333. doi: 10.1080/14653240500238251. [DOI] [PubMed] [Google Scholar]
- Maecker H., Trotter J. Selecting reagents for multicolor flow cytometry with BD™ LSR II and BD FACSCanto™ systems. Nature Methods. 2008;5:an6–an7. [Google Scholar]
- McGowan I., Anton P.A., Elliott J., Cranston R.D., Duffill K., Althouse A.D., Hawkins K.L., De Rosa S.C. Exploring the feasibility of multi-site flow cytometric processing of gut associated lymphoid tissue with centralized data analysis for multi-site clinical trials. PLoS One. 2015;10:e0126454. doi: 10.1371/journal.pone.0126454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin B.E., Baumgarth N., Bigos M., Roederer M., De Rosa S.C., Altman J.D., Nixon D.F., Ottinger J., Oxford C., Evans T.G. Nine-color flow cytometry for accurate measurement of T cell subsets and cytokine responses. Part I: Panel design by an empiric approach. Cytometry A. 2008;73:400–410. doi: 10.1002/cyto.a.20555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perfetto S.P., Ambrozak D., Nguyen R., Chattopadhyay P.K., Roederer M. Quality assurance for polychromatic flow cytometry using a suite of calibration beads. Nat. Protoc. 2012;7:2067–2079. doi: 10.1038/nprot.2012.126. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
There is no dataset/code associated with the paper.