

Summary
Tumors have complex microenvironments that provide not only biochemical but also mechanical cues to the cells within. In particular, tumor stiffness has been shown to regulate tumor growth, invasion, and metastasis. Understanding how the mechanical microenvironment controls cell behavior could be key to unraveling new therapeutic approaches. Here, we describe a protocol to prepare primary and metastatic human colorectal cancer samples and measure tumor stiffness at the tissue level using an atomic force microscope in spectroscopy mode.
For complete details on the use and execution of this protocol, please refer to Shen et al. (2020).
Subject Areas: Biophysics, Atomic Force Microscopy (AFM), Cancer
Graphical Abstract
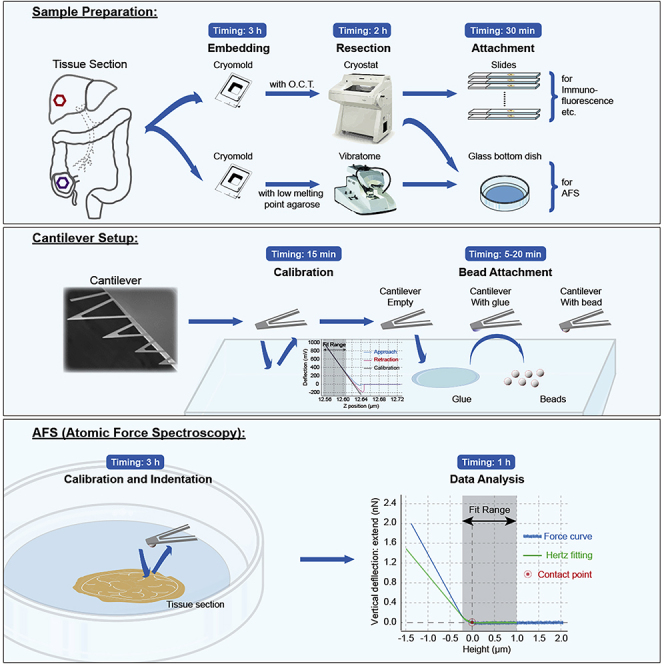
Highlights
-
•
An optimized protocol for sectioning and measuring tissue stiffness in human samples
-
•
Acquisition of mechanical properties of a tissue slice at micrometer scale using an AFM
-
•
Preservation of serial sections to correlate mechanics with other tissue signatures
Tumors have complex microenvironments that provide not only biochemical but also mechanical cues to the cells within. In particular, tumor stiffness has been shown to regulate tumor growth, invasion, and metastasis. Understanding how the mechanical microenvironment controls cell behavior could be key to unraveling new therapeutic approaches. Here, we describe a protocol to prepare primary and metastatic human colorectal cancer samples and measure tumor stiffness at the tissue level using an atomic force microscope in spectroscopy mode.
Before You Begin
Tumor stiffness measurements can be performed on fresh or snap frozen samples. Fresh tissues represent the tumor microenvironment better but require the experimentalist to measure tissue mechanics on the same day as the surgery takes place, which can be challenging. We have shown that the difference in tissue stiffness between colorectal cancer primary tumors (pTU) and liver metastases (LM) is not affected by snap freezing and thawing (Figure 1), as previously reported for mammary gland tissues (Lopez et al., 2011). This could be due to the fact that they are highly desmoplastic tissues and the ECM is more resistant to fracture by the crystals that form during freezing (Boettcher et al., 2014). For other tissues and tumor types we recommend a similar assessment as this validates the use of biobank samples.
Note: The four protocols in the Before You Begin section can be made prior to the day of measurements.
Figure 1.

Effect of Snap Freezing and Rapid Thawing on Tissue Stiffness Measurements
(A) Quantification of tissue stiffness in fresh primary tumor (Fresh pTU) and liver metastases tissue (Fresh LM) (n = 6 Fresh pTU/4 Fresh LM; p = 0.0017; t = 4.627, df = 8).
(B) Quantification of tissue stiffness in Rapid Freeze-Rapid Thaw primary tumor (Cryo pTU) and liver metastases (Cryo LM) (n = 6 Cryo pTU/4 Cryo LM; p < 0.0001; t = 8.235, df = 8).
(C) Pearson correlation between Fresh pTU and Cryo pTU (n = 6; r = 0.9255; p = 0.0081).
(D) Pearson correlation between Fresh LM and Cryo LM (n = 4; r = 0.9686; p = 0.0314). Displayed dots are the average of 3 measurements. Figure modified from Shen et al. (2020).
Snap Freezing of Tissue Samples and Preparation for Cryosectioning
Timing: 3 h
Upon collection of surgery specimens from the operating room (OR) remove large necrotic areas before processing if possible. A quality control of the tissue with evaluation of necrosis and immunochemistry to identify stromal areas will be performed in steps 24 and 25 of the main protocol. Snap frozen biobank samples (collected in the OR, directly immersed into liquid nitrogen and stored at −80°C) can also be processed as follows.
-
1.
Cut fresh tissue into an easy to handle block (we used 3 × 3 × 3 mm) with razor blade or scalpel or use a similarly sized snap frozen sample.
-
2.
Place tissue block in the middle of a disposable cryomold and embed in Optimal Cutting Temperature (OCT) compound.
-
3.
Place cryomold into container with isopropanol chilled to −20°C for 30 min, keeping the liquid below the edge of the mold so it does not touch the sample directly.
-
4.
Move the cryomold into isopropanol chilled to −80°C for 2 h, again keeping the liquid below the edge of the mold.
-
5.
Store at −80°C or in liquid nitrogen.
Bead Gluing to Cantilevers
Cantilevers with spherical indenters are used to map the stiffness of soft materials such as living cells or tissues. Using a smooth spherical surface in the cantilever tip reduces the indentation depth and localized pressure and provides an average measurement of local elasticity (Rosenbluth et al., 2006). These cantilevers can be purchased or they can be produced in the lab for a fraction of the price by gluing a silica bead to a flat-tipped cantilever (Figure 2A).
Figure 2.

Gluing a Silica Bead to a Flat-Tipped Cantilever
(A) Schematic of the protocol.
(B) The cantilever is placed perpendicular to a glue patch and approaches on the edge.
(C) The cantilever is moved to an area with isolated beads, placed on top of one and approaches on it.
(D and E) The final cantilever with a bead attached. Scale bar, 50 μm.
For both pTU and LM we used flat-tipped cantilevers with a nominal spring constant of 0.03 N/m and attached a silica bead of 10 μm diameter using 2-component epoxy glue that is only workable for 5 min following the protocol provided by our AFM manufacturer (JPK Instruments - Bruker, 2014). These are parameters needed to obtain tissue stiffness using the classical Hertz model for a spherical indenter (Hertz, 1882):
where, F is the loading force, Esample is the Young’s modulus, νsample is the cell’s Poisson ratio (which is usually close to 0.5 (Harris and Charras, 2011), as for any incompressible material), r is the radius of the indenting bead and δ is the indentation depth.
Note: The stiffness of the cantilever should match that of the sample so it is sensitive enough to measure the desired sample while being on the linear range of the photodetector. For other tissue types test which cantilever stiffness provides the best force resolution.
Note: It is difficult to attach a bead with a diameter of less than 2 μm without enveloping it in glue. For such small indenters consider purchasing cantilevers from a commercial source.
-
1.
Determine the actual spring constant of each cantilever by thermal calibration in air.
-
2.
Place silica beads in an ethanol suspension or directly dry in one half of an ethanol-cleaned slide using a pipette tip. This will generate a mixture of clusters and individual spheres on the surface, from which the latter can be easily selected.
-
3.
On the other half of the slide distribute a thin and flat patch of glue using a pipette tip and place on the AFM stage.
-
4.
Choose a part of the glue patch where the edge is perpendicular to the cantilever direction so glue is only placed in the tip and not all over the length of the cantilever, which could change its spring constant. Place the cantilever so that the tip only overlaps with the glue by a few microns (Figure 2B).
-
5.
Approach the edge of the glue patch with the cantilever tip by approaching the cantilever to the surface to transfer the glue to the tip region of the cantilever.
Optional: If glue is observed on the sides of the cantilever probably too much is attached to the tip. To prevent the glue from embedding the bead, deposit some back onto the glass by approaching the glue-covered cantilever on bare glass. This can be repeated several times if needed.
CRITICAL: The viscosity, surface tension, and hardening time of the glue are critical, especially for soft cantilevers that can sink into the glue. Some practice is initially needed to use quick hardening glues that reduce the risk of glue enveloping the bead. Alternatively, UV- curable glues have been used by other labs in the field.
-
6.
Position the cantilever tip on top of the desired isolated bead. Again, approach the surface to “catch” the bead. Remain in contact for 20 s and retract the cantilever away from the surface (Figure 2C).
-
7.
At low magnifications (<60×), successful bead gluing can be assessed after cantilever retraction: the bead will no longer be on the slide (Figures 2D and 2E).
-
8.
The cantilever with attached bead is ready for use or can be carefully stored until use in a cantilever box.
Note: If the gluing of the first bead fails, move to another bead and try again. As the glue will take around 5 min to harden several tries can be performed when working quickly.
Coating of AFM-Compatible Glass Bottom Dishes with Poly-L-lysine
Attachment of tissue slices to AFM-compatible 3.5 cm glass bottom dishes is critical for measurements. For fresh tissue measurements coat dishes with poly-L-lysine.
-
1.
Place 0.2 mL of 0.01% w/v poly-L-lysine on AFM-compatible glass bottom dishes.
-
2.
Incubate 10 - 16 h at 37°C.
-
3.
Remove liquid and place under a tissue culture hood until dry (around 30 min).
Note: Coated dishes can be stored for up to a year at 20°C–25°C.
Prepare 1% Pluronic Solution
Pluronic will prevent the cantilever from attaching to the sample upon contact. That is a pre-requisite to use the Hertz model to fit the data.
-
1.
As Pluronic has low solubility at 20°C–25°C prepare a solution by incubating 1% Pluronic in PBS at 37°C for 30 min.
-
2.
Sterile filter it to get rid of nucleating crystals.
Note: Solution can be stored at 20°C–25°C and used for months.
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Collagen I | Abcam | Cat#ab34710; RRID:AB_731684 |
| αSMA | Sigma-Aldrich | Cat#C6198; RRID:AB_476856 |
| Alexa Fluor 488-conjugated donkey anti-rabbit | Jackson ImmunoResearch | Cat#711-545-152; RRID:AB_2313584 |
| Alexa Fluor 647-conjugated donkey anti-mouse | Jackson ImmunoResearch | Cat#715-605-151; RRID:AB_2340863 |
| Other | ||
| Cryostat, CM3050S | Leica | N/A |
| Vibratome, VT1200S | Leica | N/A |
| CellHesion200 | Bruker | N/A |
| Fluorescent microscope, Nikon Ti | Nikon | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Mayer’s hemalum solution (for H&E staining) | Millipore/Sigma-Aldrich | Cat#1.09249.0500 |
| Eosin Y (for H&E staining) | Millipore/Sigma-Aldrich | Cat#1.15935.0100 |
| Dulbecco’s phosphate-buffered saline (D-PBS) | Gibco | Cat#14040133 |
| DAPI | Invitrogen | Cat#D1306; RRID:AB_2629482 |
| OCT Compound | Tissue-Tek | Cat#4583 |
| Triton X-100 | Sigma | Cat#T8787 |
| cOmplete Mini EDTA-free | Roche | Cat#11836170001 |
| Cryomold (15 mm × 15 mm × 5 mm) | Tissue-Tek | Cat#4566 |
| Cover Slips, Circles, diameter 10 mm | Thermo Scientific | Cat#12893804 |
| Poly-L-lysine solution | Sigma-Aldrich | Cat#P4707 |
| Flat-tipped cantilever, MLCT | Bruker | N/A |
| Silica bead, 10 μm | Microparticles | SiO2-F-SC100 |
| Agarose, low gelling temperature | Sigma | A9414 |
| Commercial cantilever with 10 μm spherical tip | Novascan Tech | PT.GS |
| Commercial cantilever with 10 μm spherical tip | NanoAndMore | Sqube |
| 2-component epoxy glue | Araldite, Switzerland | Rapid |
| 2-component epoxy glue | UHU, Germany | 3996 |
| Glass bottom dishes, CellHesion 200 compatible, FluoroDish Cell Culture Dishm 35 mm, 23 mm well | WPI | FD35-100 |
| Pluronic | Sigma | P2443 |
| Software and Algorithms | ||
| NIH ImageJ | NIH | N/A |
| R | N/A | https://www.r-project.org |
| Pheatmap function | Raivo Kolde | N/A |
| CellHesion 200 | JPK | N/A |
| Prism 7.0 software | GraphPad | N/A |
Step-By-Step Method Details
Fresh Tissue Sample Preparation for Sectioning
Note: As patient derived samples are precious we suggest novice users practice on mouse samples.
Fresh tissue keeps on degrading, so measurements have to be performed within 8 h after surgical resection. Once validated for tissue of interest (Figure 1) frozen samples can also be used.
-
1.
Collect fresh tissue samples from the pTU or LM from the OR in D-PBS.
-
2.
Cut 3 × 3 × 3 mm tissue blocks with razor blade or scalpel.
-
3.
For each tissue block prepare 1.5 mL of 2% low melting point agarose in D-PBS and warm it to 37°C for 10 min.
Note: 3%–4% agarose might be used to further stabilize very soft tissues prior to cutting (Wang et al., 2017).
Note: In tissues with large pores such as lung or bone the agarose can infiltrates the tissue block and one risks measuring the mechanical properties of agarose instead of those of the tissue of interest. Such samples will require an optimization of the sectioning protocol. For example, by pre-inflating mouse lungs before embedding in agarose (Liu and Tschumperlin, 2011).
-
4.
Remove D-PBS from the tissue block with cotton pad.
-
5.
Embed tissue block into pre-warmed agarose within a disposable cryomold (Figure 3).
-
6.
Place cryomold into bath of D-PBS at 4°C for 15–30 min, keeping the liquid below the edge of the mold so it does not touch the sample directly. The agarose will gently stabilize the tumor tissue structures. If needed, the agarose-stabilized tissue can be stored at 4°C in a fridge till cutting (steps 7–20) and measuring on the same day (steps 28–39).
Figure 3.

Fresh Tissue Embedding on Agarose
(A) Schematic of the protocol.
(B–E) Side and top pictures of the different steps.
Fresh Tissue Sectioning Using a Vibratome and Placement on AFM-Compatible Dishes
Fresh tissue blocks in cryomolds are cut in a vibratome. Sections are collected on AFM-compatible dishes, where they can be stained without prior fixation to evaluate orientation and identify areas of interest.
-
7.
Transport fresh tissue blocks embedded in solidified agarose (appears opaque) to a vibratome.
-
8.
Warm sample to 20°C–25°C for 10 min.
-
9.
Liberate the agarose-stabilized tissue block from the cryomold and trim the surrounding agarose using a disposable razor blade. Trim around the specimen leaving 3 mm of agarose on the top and side and 5 mm on the bottom to assure that the sample is not touching the vibratome plate (Figure 4).
-
10.
Orient the agarose-stabilized tissue block so area of interest is facing up using forceps and gently place it on a couple of drops of superglue on the vibratome base. Allow it to bind for 20 min at 20°C–25°C.
Figure 4.

Diagram of Preparation of Agarose-Stabilized Tissue Block for Vibratome Cutting
(A) Bird’s eye view of a vibratome stage showing the relative positions of the sample and blade, and approaching direction.
(B) Transverse plane of the agarose block: gray area indicates non-interesting area, for example, adjacent healthy tissue; yellow area indicates interesting area, for example, tumor vascularized stroma; brown area indicates non-usable area, for example, necrosis in liver metastasis (LM) or lumen chamber of primary tumor (pTU).
(C) Sagittal plane of the agarose block. 3 mm of agarose are left on the sides and 5 mm on the bottom of the tissue block. Dot lines along a, b, c, d indicate cutting paths, associated with height of blade and layer of tissue.
(D) Transverse plane of the agarose slides: layers are corresponding to paths in (C): b and d layers should be discarded. Coordinate axes on the bottom-left corner indicate the axis and plane.
Note: To orient the tissue pay attention to the tissue color and texture. Healthy liver is much more pink than colorectal metastasis, which has more of a white tone.
-
11.
Fill the chamber of the vibratome with cold D-PBS and ice.
-
12.
Fill as many 12-wells plate wells with 1 mL ice-cold D-PBS as needed. Place on ice bucket.
-
13.
Cut 100 μm sections using the vibratome. In short, lock the metal base with the agarose-stabilized tissue in the cold D-PBS-filled chamber. Secure a double-edge razor blade on the holder, with a sharpened edge facing upward. Carefully cut off the excess agarose from the block by advancing the vibratome (Methods Video S1).
-
14.
Spread the section evenly in the cold D-PBS. Due to surface tension, most tissue sections will spread. If that is not the case gently help using the forceps.
-
15.
Pick up the floating sections by lifting them with a 10 mm round coverslip held by the forceps (end of Methods Video S1).
-
16.
Wash and remove excess agarose by transferring each section into a 6-well well with 2.5 mL of D-PBS at 37°C for 5 min. To transfer the slices pick them up with a round coverslip as done in step 15.
-
17.
Transfer slices to pre-prepared 12-well plates on ice bucket. Upon submersion of the forceps the slice will float and thus can be deposited in the well.
-
18.
Transfer the fresh section (as done in step 15) from the 12-well into a poly-L-lysine-coated AFM-compatible glass bottom dishes with 1 mL 50% FBS in D-PBS (Methods Video S2).
-
19.
Remove most of the liquid gently by pipetting on the dish wall and let the tissue section settle down and spread evenly. Sandwich the section with a second clean, uncoated coverslip and apply very mild pressure to help the attachment to the poly-L-lysine-coated AFM-compatible glass bottom dish.
-
20.
Once the tissue section is firmly attached, carefully fill the AFM-compatible dish with 1 mL of D-PBS with proteinase inhibitors without EDTA (1 pill in 10 mL, according to the manufacturer), remove the coverslip and store till measurements within the same day at 4°C. The proteinase inhibitor together with the 4°C storage will slow down sample degradation.
Figure 5.

Comparison of Tissue Elasticity without (w/o) and with EDTA (n = 25/25; p = 0.0181; t = 2.446, df = 48)
Measurements performed on same tissue grid before and after addition of media with EDTA. Figure modified from Shen et al. (2020).
Tissue Cryosectioning Using Cryostat and Placement of Sections on AFM-Compatible Dishes and Slides for Imaging
Frozen tissue blocks in cryomolds are cut in a cryostat. Two sets of sections are collected, a first set for imaging-based assessments (quality control, evaluation of necrosis and immunochemistry) (steps 23–25) and a second set for AFS measurements (steps 26 and 27).
-
21.
Transport frozen tissue blocks in cryomolds on top of dry ice to a cryostat.
-
22.
Cut 30 μm sections using a cryostat at −20°C.
Note: A brush is useful to help guide the emerging sections from the knife blade. To prevent sections from melting some practice will be required. Try to cut each section in less than 30 s (Methods Video S3).
-
23.
For imaging transfer the sections directly to a 20°C–25°C microscope slide by touching the frozen tissue with the slide (Methods Video S3).
Note: Silanized and poly-L-lysine-coated slides improve the adherence.
-
24.
To evaluate tissue preservation and orientation, stain the first slide of each set with H&E (Mendoza and Bishop, 2020).
-
25.
For sections that will be immunostained immerse the slide immediately into a fixative (i.e., 4% PFA).
-
26.
For AFS measurements transfer the section to a RT AFM-compatible glass bottom dish with the help of a brush and immediately fill with 1 mL of D-PBS with proteinase inhibitors without EDTA till AFS measurements (Methods Video S4).
-
27.
Store at 4°C till measurement on the same day.
Note: Left-over tissue can be stored back into disposable cryomold with a layer of OCT on top to prevent freeze-drying during storage at −80°C.
Note: For long-term storage, a moistened tissue can be placed next to the sample in a paper box to prevent desiccation (particularly in a frost-free freezer).
Atomic Force Spectroscopy and Fluorescence Imaging of Tissue Sections
In order to identify the relevant areas for AFS, tissue sections can be inspected by phase contrast microscopy, or immunostained without prior fixation and visualized by fluorescence microscopy on the AFM (steps 28–32). If live immunostaining is performed, coat the cantilever with Pluronic (step 33) in parallel to save time.
-
28.
For live immunostaining, instead of filling dishes with D-PBS on steps 20 (for fresh slices) or 26 (for frozen slices), block tissue section with 5% serum (from the source of the desired secondary antibody) in D-PBS with proteinase inhibitors without EDTA for 2 h at RT.
-
29.
Incubate the tissue section with primary antibody (e.g., rabbit anti-collagen I to visualize interstitial collagen (1:100 dilution), rabbit anti-αSMA (1:100 dilution) to visualize fibroblasts) in 5% serum, 0.2% Triton in D-PBS with proteinase inhibitors without EDTA for 1 h at RT.
Note: Triton can be present in the staining buffers as we have shown that it does not have an effect on tissue mechanics (Figure 6).
Figure 6.

Comparison of Tissue Elasticity without (w/o) and with Triton X-100 (n = 25/25; p = 0.7112; t = 0.3725, df = 48)
Measurements performed on same tissue grid before and after addition of media with Triton X-100. Figure modified from Shen et al. (2020).
-
30.
Wash sample three times for 5 min each with D-PBS with proteinase inhibitors without EDTA.
-
31.
Incubate with appropriate secondary antibody conjugated to a fluorophore (e.g., Alexa Fluor 546 goat anti-rabbit secondary antibody (1:200 dilution)) in 5% serum, 0.2% Triton in D-PBS with proteinase inhibitors without EDTA for 1 h at RT.
Note: When available we recommend using conjugated antibodies as this reduces the risk of slice detachment.
-
32.
Wash tissue slices several times with D-PBS with proteinase inhibitors without EDTA.
-
33.
Coat the bead-attached cantilever with 1% Pluronic for 30 min – 1 h to avoid adhesion to the sample. For this, mount the cantilever to the glassblock and either hang it into a dish or mount it to the AFM head. Lower it into a dish with Pluronic, so that the cantilever is freely submerged in liquid without touching surfaces.
-
34.
Rinse cantilever by submerging it several times in dish with PBS and calibrate the cantilever in liquid.
-
35.
Wipe the bottom surface of the AFM-compatible dishes with the tissue slice with tissue paper and mount on the AFM sample stage.
-
36.
Align the cantilever to the center of the dish. Switch to phase contrast and/or fluorescence imaging to choose an area of interest on the tissue.
-
37.
To ensure the fastest rate of elastic measurement without viscoelastic deformation the optimal indentation speed needs to be experimentally determined for each sample type as done in (Rosenbluth et al., 2006). In short, test a whole range and identify the maximum speed (to maximize throughput) with consistent Young’s modulus. We found that for colorectal cancer a velocity of 0.4 μm/s was ideal (Figure 7).
Note: More sampling points will provide higher spatial resolution but lengthen the time required for mapping. We suggest to use a 5 × 5 sample grid to map a 20 × 20 μm2 area at an indentation velocity of 0.4 μm/s, which can be completed in ~30 min.
-
38.
For a single measurement, move the cantilever to the area of interest and perform a standard force-curve. Repeat three times on the same location.
Note: We performed our indentation curves with a force of 2 nN as it provided enough depth to fit the Hertz model to the top 200 nm upon contact but at the same time did not damage the tissue upon repeated measurements.
-
39.
For automated mapping of the region of interest, switch to Force-Map mode, where the cantilever collects individual force-displacement curves at each point within a defined sample grid.
Note: Avoid drying of the cantilever between switching samples.
Figure 7.

Approaching Speed and Sampling Frequency Optimization
(A) Pearson comparison analysis of the correlation between 0.4 μm/s and 5 μm/s (n = 100; r = 0.7001).
(B) Pearson comparison analysis of the correlation between 0.4 μm/s and 20 μm/s (n = 100; r = 0.1642).
(C) Pearson comparison analysis of the correlation between 0.4 μm/s and 2 μm/s (n = 25; r = 0.6534).
(D) Quantification of tissue stiffness with different sampling frequency on the same 20 × 20 μm2 human tissue area (n = 25/100; p = 0.8491; t = 0.1906, df = 123). Displayed dots are the average of three measurements. Figure modified from Shen et al. (2020).
Expected Outcomes
With the current protocol users will be able to section fresh or snap frozen tissues using a vibratome or cryostat, respectively. Next, they will be able to quantify tissue elasticity using an AFM in spectroscopy mode on single points or tissue grids (Figure 8).
Figure 8.

Exemplary Outcome of Our Protocol
(A) Left: Representative immunostainings of αSMA (green, to label fibroblasts and thus desmoplastic area). Scale bar, 50 μm. Right: Matching stiffness maps (20 × 20 μm) of yellow outlined area from cryosections of pTU and LM.
(B) Quantification of average tissue stiffness in pTU (n = 36) and LM (n = 40) as in (A) (p < 0.0001; t = 6.111, df = 74). Values are displayed as mean ± SEM and compared by unpaired Student’s t test. Displayed dots are the average of three measurements. Figure modified from Shen et al. (2020).
Quantification and Statistical Analysis
Force–time curves were analyzed using the JPK Data Processing Software. The Hertz model was used to determine the elastic properties of the tissue. To avoid surface artifacts only up to 10% of the sample height can be indented Yoffe, 1984. We only considered the upper 200 nm of tissue for all fits (Figure 9). Tissue samples were assumed to be incompressible and a Poisson’s ratio of 0.5 was used in the calculation of the Young’s elastic modulus.
Figure 9.

Exemplary Force Curves of Measurements with Different Elastic Modulus (E) with Hertz Model Fit in Green
All experiment statistical analyses were performed using GraphPad Prism 7.0a software (GraphPad Software). Results are presented as the mean±SEM of biological replicates. Each dot represents a biological replicate. Statistical methods for comparison of experimental group in each experiment are indicated in the figure legends. p values <0.05 are considered significant. ∗p < 0.05, ∗∗p< 0.01, ∗∗∗p < 0.001.
Limitations
The protocol presented here was used to measure the stiffness of primary colorectal tumors and liver metastases, which are rich in desmoplastic stroma areas. Other tissues, like healthy lung or mucinous adenocarcinoma of the digestive system, which have a less prominent extracellular matrix will require an optimization of the sectioning protocol. For example, by pre-inflating mouse lungs before embedding in agarose (Liu and Tschumperlin, 2011). Also, tissues that break down upon freeze-thaw without prior fixation (i.e., brain tissue) should be avoided.
Troubleshooting
Problem 1
The cryo-tissue is difficult to section into smooth thin sheets and shows horizontal strips or wrinkles (step 22).
Potential Solution
Check if the blade is dull.
Problem 2
Tissue lifts up during AFM measurement (steps 37–39).
Potential Solution
It is important to allow tissue sections to attach to the glass bottom of the AFM-compatible dishes firmly. Once tissue sections detach, there is no good method for re-attachment. In such a situation, there are two potential solutions, depending on the size of the detached area: (1) If the area is small, move the AFM tip to a nearby area that is still attached firmly. (2) If the area is large, prepare an alternative sample with another section from the same tissue block in another dish.
Problem 3
Cantilever calibration (step 1 of bead gluing to cantilevers) or tissue elasticity measurement (steps 37–39 of step-by-step protocol) is really noisy.
Potential Solutions
Make sure system is properly equilibrated and isolated: no item is touching the air table or the scope body itself and there is no air flow. If during cantilever calibration in air (step 1 of bead gluing to cantilevers) the cantilever sticks to the dish, one can also use an indium tin oxide coated glass that is grounded to avoid potential electrostatics. In liquid (steps 37–39 of step-by-step protocol) noise could also come from particles floating in the sample dish, make sure to wash enough to get rid of those.
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Alba Diz-Muñoz (diz@embl.de).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
This study did not generate/analyze any code.
Acknowledgments
We thank Martin Bergert, Sergio Lembo, Andrea Imle, and Leanne Strauss for critical reading of the manuscript. This study was supported by funds from the European Molecular Biology Laboratory and by the Deutsche Forschungsgemeinschaft (DFG) (DI 2205/2-1) to A.D.M. and by the DFG (SCHM 2560/3-1), the Ernst Jung Career Advancement Grant, and the Heidelberger Stiftung Chirurgie to T.S.
Author Contributions
A.D.M. and T.S. supervised the study. A.D.M designed and directed the AFM experiments and analysis. Y.S. optimized the protocol for tissue measurements and performed patient samples analysis. Y.S., A.D.M., and T.S. wrote and approved the manuscript.
Declaration of Interests
The authors declare no competing interests.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.xpro.2020.100167.
Contributor Information
Thomas Schmidt, Email: thomas.schmidt1@med.uni-heidelberg.de.
Alba Diz-Muñoz, Email: diz@embl.de.
References
- Boettcher H.S., Knudsen J.C., Andersen P.H., Danscher A.M. Technical note: Effects of frozen storage on the mechanical properties of the suspensory tissue in the bovine claw. J. Dairy Sci. 2014;97:2969–2973. doi: 10.3168/jds.2013-7208. [DOI] [PubMed] [Google Scholar]
- Harris A.R., Charras G.T. Experimental validation of atomic force microscopy-based cell elasticity measurements. Nanotechnology. 2011;22:345102. doi: 10.1088/0957-4484/22/34/345102. [DOI] [PubMed] [Google Scholar]
- Hertz H. Ueber die Berührung fester elastischer Körper. Journal für die reine und angewandte Mathematik. 1882;1882:156–171. [Google Scholar]
- Instruments - Bruker J.P.K. Attaching microspheres to cantilevers using the NanoWizard® Life Science stage and AFM head. 2014. https://www.jpk.com/app-technotes-img/AFM/pdf/jpk-tech-spheres-on-tip.14-1.pdf
- Liu F., Tschumperlin D.J. Micro-mechanical characterization of lung tissue using atomic force microscopy. JoVE. 2011 doi: 10.3791/2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez J.I., Kang I., You W.-K., McDonald D.M., Weaver V.M. In situ force mapping of mammary gland transformation. Integr. Biol. 2011;3:910–921. doi: 10.1039/c1ib00043h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendoza A., Bishop J. Staining methods in frozen section: best lab practices. 2020. https://blog.ucdmc.ucdavis.edu/labbestpractice/index.php/2020/03/15/staining-methods-in-frozen-section-best-lab-practices/
- Rosenbluth M.J., Lam W.A., Fletcher D.A. Force microscopy of nonadherent cells: a comparison of leukemia cell deformability. Biophys. J. 2006;90:2994–3003. doi: 10.1529/biophysj.105.067496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y., Wang X., Lu J., Salfenmoser M., Wirsik N.M., Schleussner N., Imle A., Freire Valls A., Radhakrishnan P., Liang J. Reduction of liver metastasis stiffness improves response to bevacizumab in metastatic colorectal cancer. Cancer Cell. 2020;37:800–817.e7. doi: 10.1016/j.ccell.2020.05.005. [DOI] [PubMed] [Google Scholar]
- Wang X., Freire Valls A., Schermann G., Shen Y., Moya I.M., Castro L., Urban S., Solecki G.M., Winkler F., Riedemann L. YAP/TAZ orchestrate VEGF signaling during developmental angiogenesis. Dev Cell. 2017;42:462–478.e7. doi: 10.1016/j.devcel.2017.08.002. [DOI] [PubMed] [Google Scholar]
- Yoffe E.H. Modified Hertz theory for spherical indentation. Philos. Mag. A. 1984;50:813–828. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate/analyze any code.