

Summary
Positive-strand RNA viruses replicate in distinct membranous structures called replication organelles (ROs). Mechanistic studies of RO formation have been difficult because perturbations affecting viral replication have an impact on viral protein amounts, thus affecting RO biogenesis. Here, we present a detailed guide on how to use a replication-independent expression system, designated pIRO (plasmid-induced replication organelle formation), inducing bona fide flavivirus ROs in transfected cells. This will be useful for mechanistic studies of viral and cellular factors driving flavivirus RO biogenesis.
For complete details on the use and execution of this protocol, please refer to Cerikan et al. (2020).
Subject Areas: Cell Biology, Microscopy, Molecular Biology
Graphical Abstract
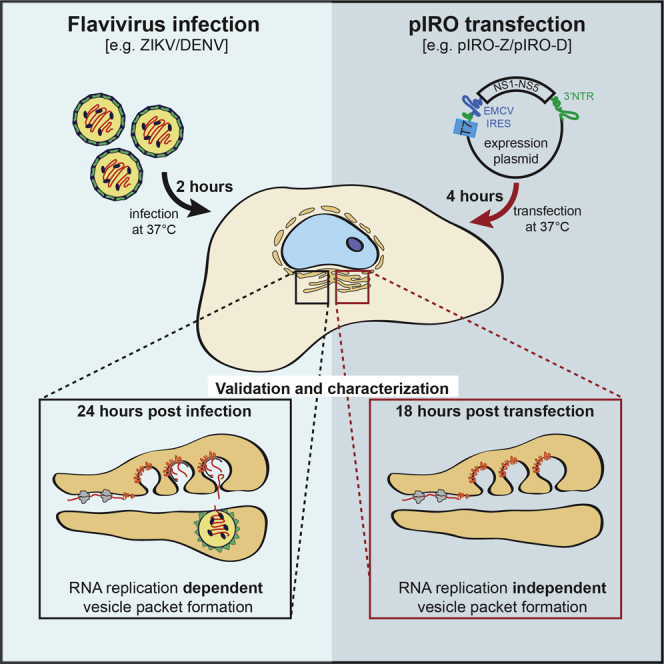
Highlights
-
•
A replication-independent system to form flaviviral replication organelles is described
-
•
The system is referred to as pIRO, an acronym for plasmid-induced replication organelle
-
•
The pIRO system has been established for dengue (pIRO-D) and Zika virus (pIRO-Z)
-
•
The pIRO system can be used under low biosafety conditions
Positive-strand RNA viruses replicate in distinct membranous structures called replication organelles (ROs). Mechanistic studies of RO formation have been difficult because perturbations affecting viral replication have an impact on viral protein amounts, thus affecting RO biogenesis. Here, we present a detailed guide on how to use a replication-independent expression system, designated pIRO (plasmid-induced replication organelle formation), inducing bona fide flavivirus ROs in transfected cells. This will be useful for mechanistic studies of viral and cellular factors driving flavivirus RO biogenesis.
Before You Begin
Solutions are prepared following the recipes in the Materials and Equipment section. Solutions which are prepared in advance and can be stored are indicated. A complete list of Materials and Equipment required is given in the Key Resources Table.
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| α-Beta Actin | Sigma-Aldrich | A5441 |
| α-GAPDH | Santa Cruz Biotechnology | Sc-47724 |
| α-RTN3 | Santa Cruz Biotechnology | Sc-374599 |
| α-PDI | Sigma-Aldrich | P7496 |
| α-DENV NS3 | GeneTex | GTX629477 |
| α-DENV NS4B | GeneTex | GTX124250 |
| α-DENV NS1 | Welsch et al., 2009 | N/A |
| α-DENV NS3 | Miller et al., 2006 | N/A |
| α-DENV NS4B | Miller et al., 2006 | N/A |
| α-DENV NS5 | Miller et al., 2006 | N/A |
| α-ZIKV NS1 | GeneTex | GTX634158 |
| α-ZIKV NS2B | GeneTex | GTX133318 |
| α-ZIKV NS3 | GeneTex | GTX133320 |
| α-ZIKV NS4A | GeneTex | GTX133704 |
| α-ZIKV NS4B | GeneTex | GTX133321 |
| α-ZIKV NS5 | GeneTex | GTX133327 |
| Alexa Fluor 488 donkey anti-mouse IgG | Thermo Fisher Scientific | A-21202 |
| Alexa Fluor 568 donkey anti-rabbit IgG | Thermo Fisher Scientific | A10042 |
| Goat anti-mouse IgG-HRP | Sigma-Aldrich | A4416 |
| Goat anti-rabbit IgG-HRP | Sigma-Aldrich | A6154 |
| Bacterial and Virus Strains | ||
| DENV isolate 16681; synthetic genome | Fischl and Bartenschlager, 2013 | N/A |
| ZIKV strain H/PF/2013; synthetic genome | Münster et al., 2018 | N/A |
| ZIKV strain H/PF/2013 | European Virus Archive | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Benzonase | Merck Millipore | 3440287 |
| Bromophenol Blue | Bio-Rad | 1610404 |
| CaCl2 | Sigma-Aldrich | C5670-100G |
| Cacodylate acid sodium trihydrate | Serva | 15540.02 |
| Chloroform | Sigma-Aldrich | 32211-1L |
| DAPI Fluoromount-G | Southern Biotech | 0100-20 |
| DBA hardener | Carl Roth | 8623.1 |
| DMEM | Gibco | 2124909 |
| DMP 30 | Carl Roth | 8621.1 |
| DTT | Roche | 20871725 |
| Ethanol absolute | Sigma-Aldrich | 1024282500 |
| Fetal bovine serum | Gibco | 26140079 |
| Formvar | Electron Microscopy Sciences | #15800 |
| Glutaraldehyde EM grade | Electron Microscopy Sciences | E16210 |
| Glycerol | Honeywell | 15523-1L |
| Glycidyl ether | Carl Roth | 8619.1 |
| HPLC water | Sigma-Aldrich | 34877-2.5L |
| HCl | Honeywell | 35328-1L |
| KCl | Sigma-Aldrich | P5833-500G |
| Lead citrate | Electron Microscopy Sciences | #17800 |
| MgCl2 | Sigma Life Science | M8266-1KG |
| Milk powder | Carl Roth | T145.2 |
| MEM non-essential amino acids | Gibco | 11140-035 |
| MirusIT-LT1 transfection reagent | Mirus | 91094359 |
| MNA | Carl Roth | 8639.1 |
| NaCl | Bernd Kraft | 04160.5600 |
| NaOH | Fisher Scientific | 1310-73-2 |
| OptiMEM reduced medium | Gibco | 31985-047 |
| Paraformaldehyde 16% EM grade | Science Services | E15700 |
| Penicillin/Streptomycin | Gibco | 15140-122 |
| Sodium dodecyl sulfate | Serva | 151-21-3 |
| Sucrose | Carl Roth | 4621.1 |
| Triton X-100 | Merck Millipore | 9036-19-5 |
| Tween 20 | Carl Roth | 9127.1 |
| Uranyl acetate dihydrate | Serva | 77870.10 |
| Western Lightning Plus-ECL | Perkin Elmer | 203-19251 |
| Zeocin | Thermo Fisher | R25001 |
| Experimental Models: Cell Lines | ||
| Huh7/Lunet T7 | Appel et al., 2005 | N/A |
| Software and Algorithms | ||
| ImageJ (FIJI) | Schindelin et al., 2012 | https://imagej.nih.gov/ij/ |
| GraphPad Prism 5.0 | LaJolla, CA, USA | https://www.graphpad.com/scientific-software/prism/ |
| NIS – Elements Advanced Research | Nikon | https://www.microscope.healthcare.nikon.com/products/software/nis-elements/nis-elements-advanced-research |
| Adobe Illustrator 5.1 | San Jose, CA, USA | https://www.adobe.com/products/illustrator.html |
| Other | ||
| Automated Cell Counter TC20 | Bio-Rad | 1450102 |
| EM grids (100 mesh) | Plano | G2410D |
| PVDF membrane 0.45 μm | Merck Millipore | IPVH00010 |
Materials and Equipment
CRITICAL: All solutions and materials used for cell culture have to be sterilized either by filtration or autoclaving.
Mammalian Cell Culture Medium
Timing: <1 h
-
1.
Complete cell culture medium is prepared by adding the supplements specified in the table below to Dulbecco’s Modified Eagle Medium (DMEM):
| Component | Amount |
|---|---|
| DMEM basal medium | 500 mL |
| Fetal bovine serum | 10% |
| Penicillin/Streptomycin | 100 U/mL |
| Streptomycin | 100 μg/mL |
| MEM Non-essential amino acids | 1% |
Zeocin Stock and Working Solutions
Note: For maintaining a stable expression of the T7 RNA polymerase in Huh7/Lunet-T7 cells, zeocin is added freshly to the cell culture medium. The zeocin stock solution of 100 mg/mL can be stored at −20°C for up to 18 months.
-
2.
To avoid repetitive freeze-thaw cycles of the zeocin stock solution, a ready-to-use solution of 1 mg/mL can be prepared by diluting zeocin (100 mg/mL) in DMEM complete medium. Aliquots of 1 mL are stored at −20°C.
Note: Zeocin is added into the cell culture medium at a final concentration of 5 μg/mL.
Solutions for Electron Microscopy
-
3.Preparation of the 0.2 M cacodylate buffer.
-
a.Dissolve 21.4 g of cacodylate acid sodium trihydrate in 400 mL of distilled water.
-
b.Adjust the pH with HCl and/or NaOH to 7.2–7.4.
-
c.Adjust the volume to 500 mL with distilled water.
-
a.
Note: Cacodylate buffer can be stored at 4°C.
-
4.
Fixative buffer for electron microscopy samples is prepared by adding all supplements specified in the table below to cacodylate buffer:
| Reagent | Stock Concentration | Final Concentration | Amount |
|---|---|---|---|
| Cacodylate buffer | 0.2 M | 50 mM | 1.25 mL |
| KCl | 1 M | 50 mM | 0.25 mL |
| MgCl2 | 0.1 M | 2.6 mM | 0.13 mL |
| CaCl2 | 0.1 M | 2.6 mM | 0.13 mL |
| Paraformaldehyde | 16% | 1% | 0.3125 mL |
| Glutaraldehyde | 25% | 2.5% | 0.5 mL |
| Sucrose | N/A | 2% | 0.1 g |
| HPLC H2O | N/A | N/A | to 5 mL |
| Total | 5 mL |
Note: Fixative buffer should be prepared fresh on the day of usage.
Note: Stock solutions of KCl, MgCl2, and CaCl2 can be prepared in advance and stored at ∼22°C. We recommend preparing all solutions with HPLC-grade water.
-
5.Contrast enhancement of samples with uranyl acetate (UA) and lead citrate.
-
a.Prepare a 3% UA stock solution by dissolving 3 g of UA in 100 mL EM graded ddH2O.
-
b.Aqueous lead citrate is prepared according to Reynolds (1963).
-
c.Solutions are stored wrapped in aluminum foil at 4°C.
-
a.
Note: We recommend filtering the solutions before use. In particular lead citrate forms insoluble complexes with CO2. To avoid CO2, we recommend using degassed ddH2O.
-
6.
To prepare graded ethanol series (from 50% to 100%) dilute absolute ethanol (99.9%) to the final % (v/v) in ddH2O. Graded ethanol solutions are stored in flammable storage cupboards at ∼22°C.
-
7.
Epoxy resin for flat embedding is prepared by mixing the following chemicals while stirring:
| Reagent | Amount |
|---|---|
| Glycidyl ether | 48 g |
| DBA hardener | 19 g |
| MNA hardener | 33 g |
| → stir for 30 min until phase-free | |
| DMP 30 | 2 g |
| → stir for 30 min until phase-free | |
| Total | 100 mL |
Preparation of Formvar Film-Coated EM Grids for Electron Microscopy
-
8.Formvar Film-coated EM mesh grids are prepared according to Sherman (2014) with slight modifications.
-
a.Prepare a 1.2% formvar solution in chloroform. Impurities can be removed by filtering the solution using a 0.2 μm Whatman filter.
-
b.Clean glass slides using distilled water and lens paper.Note: Do not “over-clean” the glass slides as this may affect the release of the formvar films.
-
c.Fill a slide chamber (needs to have a thistle tube) 3–4 cm deep with formvar. To minimize evaporation of chloroform try to keep it covered.
-
d.Place the clean glass slide in the chamber and let it settle for 2–3 min.
-
e.Drain the formvar from the chamber in a slow but steady stream.
-
f.Let the slide dry for 2–3 min.
-
g.Remove the slide from the chamber and remove any residual liquid by touching the bottom with a filter paper.
-
h.Scrape edges of slide and ∼0.5 cm from the bottom of the slide using a razor blade to loosen the film.
-
i.Fill a container with clean water and sweep the surface with a lens paper to remove dust.
-
j.Slowly lower the slide into the water at a ∼45° angle to float off the film.Note: Films should be silver in color. If films appear gold or gray they are too thick or too thin, respectively.
-
k.Place the mesh grids onto the film with the shiny or dull side down. We recommend to stay consistent and to note which side of the mesh grid has the film.
-
l.Collect the formvar-coated grids by lowering a piece of parafilm at a ∼45° angle and remove it with a steady but quick down and turn motion.
-
m.Excess of water is removed using a Kimwipe or filter paper.
-
n.Formvar-coated grids are placed in a Petri dish for drying.
-
a.
Alternatives: Instead of formvar one can also use Pioloform FN 65 (Plano).
Solutions for Immunofluorescence Microscopy
Note: Cells grown on glass coverslips are fixed with 4% paraformaldehyde (PFA) in PBS.
-
9.
Dissolve 80 g of paraformaldehyde in 1.4 L H2O.
-
10.
Add 1 mL of 1 M NaOH and stir gently on a heating block at 60°C.
-
11.
Once solution appears clear, add 200 mL of 10× PBS. Cool solution down to ∼22°C.
-
12.
Adjust the pH with HCl to 7.4.
-
13.
Adjust the volume to 2 L with H2O.
-
14.
Solution can be filtered through a 0.45 μm membrane filter.
-
15.
The final 4% PFA solution is stored as 13 mL aliquots at −20°C.
Solutions for Western Blot Analysis
Note: For western blot analysis, cells are directly lysed in 2× sample buffer. A 6× sample buffer stock can be prepared ahead of time and stored at −20°C in 2 mL aliquots.
-
16.
Sample buffer for western blot analysis is prepared by combining all reagents as specified in the table below:
| Reagent | Stock Concentration | Final Concentration | Amount |
|---|---|---|---|
| ddH2O | N/A | N/A | 5.375 mL |
| Tris-HCl [pH 6.8] | 1 M | 360 mM | 18 mL |
| SDS | 1 M | 180 mM | 9 mL |
| DTT | 1 M | 300 mM | 15 mL |
| Glycerol | 100% | 5.25% | 2.625 mL |
| Bromophenol blue | N/A | 0.3% | 0.15 g |
| Total | 50 mL |
Note: For degrading contaminating nucleic acids, we add 1 μL of the benzonase (stock: 10KU) to 100 μL sample buffer before heating at 95°C for 5 min.
Step-By-Step Method Details
General Description of the Construct Design
To enable investigation of the flaviviral replication organelle biogenesis independent of viral replication, an expression plasmid is required which can continuously be transcribed and translated by the host cell machinery. Since flaviviruses replicate in the cytoplasm and therefore, the viral RNA genome is not spliced, expression of flaviviral constructs by conventional expression plasmids bears the risk of inadvertent splicing, thus affecting the expression of viral proteins. Therefore, we employ a cytoplasmic transcription/translation system originally developed by Fuerst et al. (1986). For this, parts of the viral genome (ETm-3′ UTR; Figure 1) are inserted into the pTM expression plasmid, which allows cytoplasmic transcription via the promoter of the T7 RNA polymerase. By using the Huh7/Lunet-T7 cell line, which stably expresses the T7 RNA polymerase, viral RNA is continuously produced upon DNA transfection. To promote RNA translation, an EMCV IRES element is added to the 5′ end of the viral open reading frame encoding the polyprotein. To ensure the generation of viral RNAs with an authentic 3′UTR, the hepatitis D virus ribozyme was inserted downstream of the ZIKV or DENV 3′ UTR (Figure 1). The present protocol describes the procedure for inducing and analyzing replication organelles of Zika virus (the pIRO-Z system). However, the same method can be applied to dengue virus replication organelles, simply by switching the backbone plasmid from pIRO-Z to pIRO-D.
Figure 1.

Design of the pIRO Construct
Schematic representation of the pIRO (pTM/Δ 5′SLAB–3′WT-Ribozyme) expression plasmid. The pIRO construct is composed of the T7 RNA polymerase promoter (blue box), a short part of the capsid coding region containing the 5′ cyclization sequence (CS), the internal ribosome entry site (IRES) of the encephalomyocarditis virus (EMCV) to facilitate high-level RNA translation, the NS1 to NS5 coding region, the 3′ non-translated region (NTR), and the HDV ribozyme for generating authentic 3′ ends. The nucleotide positions of the ZIKV H/PF/2013 (GenBank accession number KJ776791.2) and DENV-2 genome (GenBank accession number NC_001474.2) used to generate the constructs are given below each element in brackets. Sequences of ZIKV are given in green; sequences corresponding to DENV are given in purple.
Plasmids Transfection and Validation of Experiments
Timing: ∼2 weeks in total
Huh7/Lunet-T7 cells are seeded for transfection experiments which will be analyzed 18 h.p.t. (pIRO-Z) or 20 h.p.t. (pIRO-D) by western blot, immunofluorescence, and electron microscopy.
Seeding of Cells
-
1.
One day prior to transfection, Huh7/Lunet-T7 cells are seeded either on glass coverslips in 24-well plates or 6-well plates at a cell density of 3 × 104 cells/well and 2 × 105 cells/well, respectively. Cells grown on glass coverslips in 24-well plates are used to estimate the transfection efficiency via immunofluorescence microscopy and to analyze the ultrastructural membranous rearrangements via electron microscopy. Cells seeded in a 6-well plate are subjected to western blot analysis for verifying polyprotein expression and processing.
Transfection of Cells
-
2.
On the day of transfection, change medium of seeded cells at least 30 min prior to transfection.
Note: We recommend replacing the medium by 500 μL/well fresh and pre-warmed DMEM complete medium for 24-well plates and 1 mL/well in case of a 6-well plate.
-
3.Prepare the transfection mixture in sterile reaction tubes:
-
a.Place OptiMEM reduced serum medium (24-well plate: 100 μL; 6-well plate: 400 μL) into a sterile reaction tube.
-
b.Add plasmid DNA (24-well plate: 500 ng; 6-well plate: 2 μg) and gently vortex to mix completely.
-
c.Add Mirus TransIT-LT1 transfection reagent (24-well plate: 1.5 μL; 6-well plate: 6 μL) to the DNA mixture. Make sure while adding the transfection reagent to not touch the sides of the reaction tube.
-
d.Mix completely by vortexing.
-
e.To allow complex formation, incubate mixture at ∼22°C for 20 min.
-
a.
Note: As recommended by the manufacturer, warm the transfection reagent to ∼22°C and gently vortex before usage.
Note: If more than one well is being transfected, scale up accordingly and prepare a transfection master mix.
-
4.
Transfection mixtures are added to the cells in a drop-wise manner. To ensure even distribution of the TransIT-LT1 reagent:DNA complexes, gently rock the plates back-and-forth and side-to-side.
-
5.
Incubate plates for 4 h at 37°C and 5% CO2 before changing the transfection medium to fresh DMEM complete. Once the medium is exchanged, expression of the polyprotein is allowed for a maximum of 18 h (pIRO-Z) or 20 h (pIRO-D).
Collecting Samples
-
6.Immunofluorescence microscopy samples.
-
a.Cells are washed three times with sterile PBS prior to fixation with 4% paraformaldehyde in PBS (step 10). Fixation is allowed for 10 min at ∼22°C.
-
b.Fixed cells are washed three times with sterile PBS. Samples can either be directly further processed or stored at 4°C.
-
a.
Pause Point: PFA fixed cells can be stored for up to 1 week at 4°C. If samples are stored at 4°C we recommend to cover the coverslips with 1 mL of PBS.
-
7.Electron microscopy samples.
-
c.Cells are washed three times with sterile PBS prior to fixation with EM fixative buffer (step 4). Fixation is allowed for at least 30 min at ∼22°C.
-
d.Samples can either be directly further processed for EPON flat embedding or stored at 4°C in fixation buffer for up to 3 days.
-
c.
-
8.Western blot samples.
-
e.Cells are lysed by adding 100 μL/well of 2× sample buffer (step 16) supplemented with 1 μL of benzonase (stock: 10KU). Degradation of nucleic acids is allowed for 5 min at ∼22°C.
-
f.Transfer samples to 1.5 mL reaction tubes and heat samples at 95°C for 5 min.
-
g.Samples can either immediately subjected to SDS-PAGE or stored at −20°C.
-
e.
Characterization of the pIRO-Z Construct
-
9.Estimation of the transfection efficiency.
-
a.Cells are permeabilized with 0.5 mL 0.2% Triton X-100 in PBS for up to 10 min at ∼22°C.
-
b.Permeabilized cells are washed three times with 1 mL PBS before being incubated for 1 h in 0.5 mL blocking buffer (10% FCS in PBS) at ∼22°C.
-
c.Primary antibodies against ZIKV NS4B are diluted 1:250 in 3% bovine serum albumin (BSA) in PBS.
-
d.Incubation with primary antibodies can either be done at 4°C for up to 16 h or 2 h at ∼22°C.
-
e.Coverslips are washed three times with PBS-T (0.01% Tween20) 10 min each.
-
f.Alexa Fluorophore 568-conjugated secondary antibodies are diluted 1:1,000 in 3% BSA in PBS.
-
g.Cells are incubated with secondary antibodies for 1 h at ∼22°C in the dark.
-
h.Samples are washed three times with PBS-T (0.01% Tween20) 10 min each.
-
i.Before mounting, coverslips are rinsed once with ddH2O.
-
j.Coverslips are mounted on microscopy slides using 5 μL of Fluoromount-G supplemented with DAPI to stain nuclear DNA.
-
k.Samples are air dried for at least 30 min at ∼22°C before being analyzed on a Nikon Eclipse Ti microscope.
-
l.To estimate the transfection efficiency, large overview pictures (5 × 5) are recorded using the 20× magnification objective. Transfection efficiency is calculated by determining the number of NS4B-positive cells over the total number of cells (Figure 2).
-
a.
Figure 2.

Transfection Efficiency of Huh7/Lunet-T7 Cells with pIRO-Z
(A) Representative IF pictures of Huh7/Lunet-T7 cells transfected with pIRO-Z at 18 h post transfection. Cells were stained using a NS4B-specific antibody. Scale bar, 200 μm.
(B) Transfection efficiency was determined by counting the number of NS4B positive cells.
-
10.Processing of the polyprotein
-
a.15 μL of protein sample is subjected to a 15% SDS-PAGE.
-
b.Electrophoresis is performed for 2 h at constant 100 V.
-
c.Proteins are transferred onto a 0.45 μm PVDF membrane at 350 mA for 1 h at 4°C using the wet blot transfer system of Bio-Rad.
-
d.Membranes are blocked in 5% milk in PBS.
-
e.Primary antibodies (ZIKV NS1, NS2B, NS3, and NS4A) are diluted 1:1,000 in 1% milk in PBS.
-
f.Membranes are incubated at 4°C for up to 16 h.
-
g.On the next day, membranes are washed three times with PBS-T (0.1% Tween20), 10 min each.
-
h.HRP-conjugated secondary antibodies (anti-mouse and anti-rabbit) are diluted in 1% milk in PBS.
-
i.Membranes are incubated for 1 h at ∼22°C.
-
j.Membranes are washed three times with PBS-T (0.1% Tween20), 10 min each.
-
k.Western blots are developed using the Western Lightning Plus-ECL reagent.
-
l.Images are taken using the ChemoStar imager software package.
-
m.Western blot images are analyzed using the FIJI software package (Figure 3A).
-
a.
Figure 3.

Functional Characterization of the pIRO-Z Construct
(A) Huh7/Lunet-T7 cells were either transfected (left column) or infected (right column) with pIRO-Z or the H/PF/2013 strain (MOI 10), respectively. Whole cell lysates were subjected to western blot analysis, and polyprotein processing was determined by probing for NS1, NS2B, NS4A, and NS3. GAPDH was used as loading control.
(B) Representative transmission electron microscopy images showing replication organelles (yellow arrowheads) induced upon pIRO-Z transfection or ZIKV infection. Mock transfected cells served as control, and only regular ER was observed (yellow arrow). Scale bars, 150 nm.
-
11.Formation of the replication organelles.CRITICAL: While embedding the samples it is important to never let them dry out. Every step should be performed in a fume-hood and one should wear appropriate protection clothes.
-
a.Flat embedding for EM.
-
i.Rinse samples five times with 50 mM cacodylate buffer, 5 min each.
-
ii.Incubate with 2% osmium tetroxide in 50 mM cacodylate buffer for at least 40 min on ice.
-
iii.Wash samples three times with EM-grade water.
-
iv.Samples are incubated for up to 16 h with 0.5% aqueous uranyl acetate at 4°C.
-
v.On the next day, samples are rinsed three times with EM-grade water.
-
vi.Dehydration is performed using a graded ethanol series (from 50% to 100%).
-
vii.Flip coverslips on epoxy resin filled capsules.
-
viii.Epon is polymerized at 60°C for at least 48 h.
-
i.
-
b.Thin sectioning for transmission electron microscopy.
-
i.Trim polymerized sample to a 0.5 × 0.5 mm sized section using a razor blade.
-
ii.Mount trimmed block into an ultramicrotome holder.
-
iii.Align the specimens’ surface with the diamond knife in all axis.CRITICAL: Before sectioning clean the diamond knife with methanol.
-
iv.Fill the boat of the diamond knife with H2O up to the knifes’ edge.
-
v.70 nm thin sections are cut automatically by the microtome.
-
i.
-
c.Section retrieval.
-
i.Floating sections are picked up with a loop and transferred onto a formvar-coated TEM grid.
-
ii.Dry the grid by air at ∼22°C.
-
i.
-
d.Contrast enhancement.
-
i.Fix a parafilm on a paper or on a table.
-
ii.Transfer grids with sections to aqueous uranyl acetate (3%) droplets and incubate for 5 min.
-
iii.Wash grids with ddH2O by transferring the grid onto droplets and leaving them for 30 s on. Repeat two times.
-
iv.Grids are transferred to aqueous lead citrate droplets and are incubated for 5 min.
-
v.Wash grids by following instructions in step iii.
-
vi.Grids are dried at ∼22°C before being analyzed on a JEOL JEM-1400 transmission electron microscope (Figure 3B).
-
i.
-
a.
Expected Outcomes
Routinely we obtain a transfection efficiency of 30%–50%, which we determine by standard immunofluorescence microscopy. Transfected cells expressing the ZIKV polyprotein are identified by NS4B staining, while the total number of cells is determined by DAPI staining (see Figure 3H+I in Cerikan et al. (2020)).
Note: For quantification, we recommend to always include and analyze a mock sample for each independent IF experiment. Mock samples are used to determine the antibody background signal for quantifying the number of positive cells.
Note: We recommend analyzing large overview pictures (5 × 5) instead of single pictures as transfection might occur in clusters of cells.
We seed 2 × 105 cells/well for western blot analysis to investigate the expression and processing of the viral polyprotein. Removal of the 5′ SLAB element has no effect on that. Furthermore, amounts of ZIKV proteins are comparable to a normal ZIKV infection (Figure 3) (see Figure 3C in Cerikan et al. (2020)).
Electron microscopy results demonstrate that 6–10 out of 20 cells will contain replication organelles, reflecting the previously determined transfection efficiency (30%–50%) by immunofluorescence microscopy. However, variation in numbers of vesicle packets per cell frequently occur as expression levels of the polyprotein may vary between cells. For quantification of vesicle packets, at least 20 cells per experiment are analyzed (see Figure 3E–G in Cerikan et al. (2020)).
Limitations
Since EM does not allow the screening of large number of cells, transfection efficiency must be high in order to identify cells containing replication organelles. Transfection efficiency of Huh7/Lunet-T7 cells depends on 1) the confluency and condition of the cell culture and 2) the passage number of the cell culture. The optimal Huh7/Lunet-T7 culture used for experiments should be at 80%–90% confluent and have passage number <P30. If cells are too confluent on the day of the experiment, one must split the cells.
Troubleshooting
Problem
Cell death upon transfection (Step-By-Step Method Details, steps 1–5).
Potential Solution
Cell death can occur upon transfection using Mirus-LT1 when cells are seeded at a too low density. Make sure that cells are seeded as a monolayer with 70% confluency. Huh7/Lunet-T7 cells should be seeded at a density of 3–4 × 104 cells/well and 2 × 105 cells/well of 24- and 6-well plates, respectively.
Problem
Transfection efficiency is low (Step-By-Step Method Details, step 9).
Potential Solution
Determine confluency of cells. If they are too confluent, they can no longer be transfected. If cell density is too low, cells are prone to die upon transfection. In addition, make sure that while preparing the transfection mixture DNA is first mixed with OptiMEM by vortexing before transfection reagent is added. Complex formation should be allowed for 20 min and not longer. In case transfection efficiency is repeatedly low, we recommend preparing a fresh plasmid DNA stock and using a fresh vial of Mirus TransIT LT1 transfection reagent.
Problem
Cell death due to protein overexpression (Step-By-Step Method Details, step 10).
Potential Solution
We recommend performing an initial kinetic experiment to determine the best time point for harvesting samples. Based on our experiences, expression of pIRO-Z or pIRO-D starts to become toxic to the cells at around 18 h or 20 h post transfection, respectively.
Problem
Cells are showing ultrastructural rearrangements which are not known to occur in infected cells (Step-By-Step Method Details, step 11).
Potential Solution
It is of utmost importance to never let the coverslip dry out completely. While changing the liquid in the 24-well plate it is best to leave residual liquid in each well. Drying of the cells will cause structural damage, which can only be seen when analyzing the samples with an electron microscope.
Problem
Cells are breaking out of the 70 nm section (Step-By-Step Method Details, step 11b).
Potential Solution
Cells can break out of the 70 nm section when epoxy resin infiltration is not complete. Make sure, while embedding your samples, to allow the epoxy resin solution to infiltrate the cells. You can enhance infiltration by leaving the coverslips on epoxy resin filled capsules for 1 h at ∼22°C before polymerizing the samples at 60°C.
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed and will be fulfilled by the Lead Contact, Ralf Bartenschlager (Ralf.Bartenschlager@med.uni-heidelberg.de).
Materials Availability
All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and Code Availability
The published protocol includes all [datasets/code] generated or analyzed during this study.
Acknowledgments
We thank Marie Bartenschlager and Micha Fauth for excellent technical assistance and all members of the Molecular Virology Department for their support. We thank the Electron Microscopy Core Facility at Heidelberg University for their support and access to their equipment. This work was supported by the Deutsche Forschungsgemeinschaft (DFG), grants Ba1505/8-1 and 240245660-SFB 1129, and the German Center for Infection Research (DZIF), project numbers 8029801806 and 8029705705, all to R.B. C.J.N. was funded in part by a European Molecular Biology Organization (EMBO) long-term fellowship (ALTF466-2016).
Author Contributions
S.G. performed the experiments and analyzed the data for pIRO-Z. Conceptualization and methodology of this system was done by B.C., M.C., C.J.N., and R.B. S.G. drafted the manuscript. All others contributed to the protocol by editing the manuscript. Funding acquisition was done by R.B.
Declaration of Interests
The authors declare no competing interest.
Contributor Information
Sarah Goellner, Email: sarah.goellner@med.uni-heidelberg.de.
Ralf Bartenschlager, Email: ralf.bartenschlager@med.uni-heidelberg.de.
References
- Appel N., Pietschmann T., Bartenschlager R. Mutational analysis of hepatitis C virus nonstructural protein 5A: potential role of differential phosphorylation in RNA replication and identification of a genetically flexible domain. J. Virol. 2005;79:3187–3194. doi: 10.1128/JVI.79.5.3187-3194.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerikan B., Goellner S., Neufeldt C.J., Haselmann U., Mulder K., Chatel-Chaix L., Cortese M., Bartenschlager R. A non-replicative role of the 3′ terminal sequence of the dengue virus genome in membranous replication organelle formation. Cell Rep. 2020;32:107859. doi: 10.1016/j.celrep.2020.107859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischl W., Bartenschlager R. High-throughput screening using dengue virus reporter genomes. Methods Mol. Biol. 2013;1030:205–219. doi: 10.1007/978-1-62703-484-5_17. [DOI] [PubMed] [Google Scholar]
- Fuerst T.R., Niles E.G., Studier F.W., Moss B. Eukaryotic transient-expression system based on recombinant vaccinia virus that synthesizes bacteriophage T7 RNA polymerase. Proc. Natl. Acad. Sci. U S A. 1986;83:8122–8126. doi: 10.1073/pnas.83.21.8122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller S., Sparacio S., Bartenschlager R. Subcellular localization and membrane topology of the dengue virus type 2 non-structural protein 4B. J. Biol. Chem. 2006;281:8854–8863. doi: 10.1074/jbc.M512697200. [DOI] [PubMed] [Google Scholar]
- Münster M., Płaszczyca A., Cortese M., Neufeldt C.J., Goellner S., Long G., Bartenschlager R. A reverse genetics system for Zika virus based on a simple molecular cloning strategy. Viruses. 2018;10:1–17. doi: 10.3390/v10070368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds E.S. The use of lead citrate at high pH as an electron-opaque stain in electron microscopy. J. Cell Biol. 1963;17:208–212. doi: 10.1083/jcb.17.1.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B. Fiji: An open-source platform for biological-image analysis. Nat. Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman D. 2014. https://www.emsdiasum.com/microscopy/technical/techtips/formvar_film-coated_grids.aspx
- Welsch S., Miller S., Romero-Brey I., Merz A., Bleck C.K.E., Walther P., Fuller S.D., Antony C., Krijnse-Locker J., Bartenschlager R. Composition and three-dimensional architecture of the Dengue virus replication and assembly sites. Cell Host Microbe. 2009;5:365–375. doi: 10.1016/j.chom.2009.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The published protocol includes all [datasets/code] generated or analyzed during this study.