

Summary
Mosaic analysis by dual recombinase-mediated cassette exchange (MADR) is a technology that allows stable and locus-specific integration of transgenic elements into recipient cells carrying loxP and FRT sites. Nevertheless, most cell lines lack these recombination-specific sites. This protocol describes a method to introduce the minimum requirements into cells, leading to the generation of de novo primary MADR recipient cells or MADR “Proxy” cells. These cell lines allow the combinatorial use of a wide range of transgenic elements through MADR.
For complete details on the use and execution of this protocol, please refer to Kim et al. (2019).
Subject Areas: Flow Cytometry/Mass Cytometry, Model Organisms, Molecular Biology, Gene Expression, CRISPR
Graphical Abstract
Highlights
-
•
De novo generation of Proxy-MADR cells through homology-directed repair
-
•
MADR reaction validation
-
•
Generation of multiple Proxy-MADR cells through MADR reaction
-
•
Versatility of colorless Proxy-MADR cells
Mosaic analysis by dual recombinase-mediated cassette exchange (MADR) is a technology that allows stable and locus-specific integration of transgenic elements into recipient cells carrying loxP and FRT sites. Nevertheless, most cell lines lack these recombination-specific sites. This protocol describes a method to introduce the minimum requirements into cells, leading to the generation of de novo primary MADR recipient cells or MADR “Proxy” cells. These cell lines allow the combinatorial use of a wide range of transgenic elements through MADR.
Before You Begin
Experimental Design Considerations
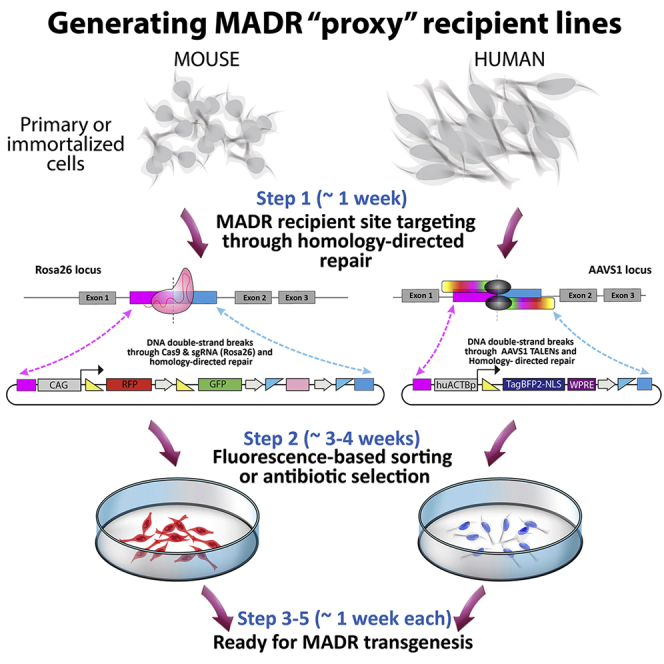
Three main aspects should be considered when generating Proxy-MADR cell lines. These include the locus to be targeted, the strategy to edit the locus, and the design of the DNA sequence or “cassette” to be inserted into the locus.
- 1.
Note: These loci are chosen because they are known as safe harbor sites. They have been shown to remain transcriptionally active and do not cause any negative side effects on cells when transgenes are inserted (Lombardo et al., 2011; Soriano, 1999).
-
2.Choose a strategy to edit the desired loci. For this protocol CRISPR-Cas9 or transcription activator-like effector nucleases (TALENs) are used to edit the loci through double-strand breaks (DSBs) and homology-directed repair (HDR).
-
a)For the Neuro-2a cell line we used a sgRNA to target the Rosa26 locus to induce DSBs through spCas9 (Figure 1A). The sgRNA used for this protocol is listed on the key resources table (Chu et al., 2015). We recommend combining the sgRNA and spCas9 in a single plasmid, here we used pX330-U6-sg.ROSA26-Chimeric_BB-CBh-eSpCas9(1.1), called sgRosa26-Cas9.
-
b)For the HEK293T cell line we used a pair of TALENs (TALEN-left and TALEN-right) to induce DSBs in the AAVS1 locus (Figure 1C) (Mandegar et al., 2016). These are referred to as AAVS1-TALENs plasmids.
-
a)
Note: CRISPR/Cas9 and TALENs were used in mouse and human cells, respectively, but any of these options can be used interchangeably depending on the availability and the experimental design.
-
3.Design the DNA cassette to be inserted at the desired locus. The cassette should include one or more reporter transgenes flanked by loxP and FRT sites embedded in a plasmid donor repair template for HDR.
-
a)For the mouse Rosa26 locus, we used the Rosa26 mT/mG plasmid which is the same that was used for engineering the Rosa26 mT/mG mouse (Muzumdar et al., 2007). The plasmid carries a cassette under the CAG promoter with tdTomato and EGFP transgenes flanked by loxP and FRT sites. The whole cassette is flanked by homology arms for the Rosa26 locus. Referred to here as Rosa26 mT/mG donor template (Figure 1A).
-
b)For the human AAVS1 locus, we designed the AAVS1-Actp-loxP-TagBFP2-V5-NLS-WPRE-FRT plasmid which contains a cassette with a TagBFP2 reporter transgene fused to a V5 epitope tag, a nuclear localization sequence (NLS) followed by a woodchuck hepatitis virus post-transcriptional regulatory element (WPRE) and a poly (A) element, all flanked by loxP and FRT sites under the human B-actin promoter. The whole cassette is flanked by homology arms for the AAVS1 locus. This plasmid is referred to as AAVS1 TagBFP2-NLS donor template (Figure 1C).
-
a)
Note: Any reporter transgene can be used to visualize and select the targeted cell population. Nuclear reporters can more unambiguously delineate cells—especially with small cytoplasmic content.C ytoplasmic/membrane reporters are better for morphological discrimination, but any reporter can be used.
CRITICAL: The minimum requirements of the plasmid donor repair template for HDR are two homology arms for the specific locus, a promoter, and a reporter transgene flanked by one loxP site and one FRT site.
Figure 1.

De Novo Generation of Proxy-MADR Cells
(A) Schematic showing Rosa26 locus editing to Rosa26 “mT/mG” locus through CRISPR-Cas9 and HDR.
(B) Neuro-2a Rosa26 “mT/mG” Proxy-MADR cell line expressing tdTomato.
(C) Schematic showing AAVS1 locus editing to AAVS1 “TagBFP2-NLS” locus through TALENs and HDR.
(D) HEK293T AAVS1 “TagBFP2-NLS” Proxy-MADR cell line expressing nuclear TagBFP2-NLS. Scale bar, 50 μm.
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Experimental Models: Cell lines | ||
| Mouse: Neuro-2a | ATCC | Cat# CCL-131 |
| Human: HEK293T | ATCC | Cat# CRL-3216 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| DMEM, High Glucose | Thermo Fisher Scientific | Cat# 11965118 |
| Penicillin-Streptomycin-Amphotericin (PSA) | Thermo Fisher Scientific | Cat# 15240096 |
| 1× PBS | Thermo Fisher Scientific | Cat# 10010023 |
| Poly-L-lysine 0.01% | Sigma-Aldrich | Cat# P4707 |
| Fetal Bovine Serum (FBS) heat inactivated | Thermo Fisher Scientific | Cat# 16140071 |
| Lipofectamine 3000 Transfection Reagent | Thermo Fisher Scientific | Cat# L3000015 |
| Opti-MEM Reduced Serum Medium | Thermo Fisher Scientific | Cat# 31-985-062 |
| Oligonucleotides | ||
| sgRNA targeting sequence: ROSA26 locus: gACTCCAGTCTTTCTAGAAGA (“g” added for efficient hU6 transcription) | (Chu et al., 2015) | Addgene Plasmid #64219 |
| Recombinant DNA | ||
| plasmid: pX330-U6-sg.Rosa26-Chimeric_BB-CBh-eSpCas9(1.1) | (Kim et al., 2019) | N/A |
| plasmid: AAVS1-TALENs | Gift: Conklin and Mandegar (Mandegar et al., 2016) | N/A |
| Template plasmid: Rosa26 mT/mG | (Muzumdar et al., 2007) | Addgene Plasmid #17787 |
| Template plasmid: AAVS1-Actp-loxP-TagBFP2-V5-NLS-WPRE-FRT | This paper | N/A |
| plasmid: pCAG-FlpO-2A-Cre EV | (Kim et al., 2019) | Addgene Plasmid #129419 |
| plasmid: MADR pDonor TagBFP2-3xFlag (cyto) WPRE | (Kim et al., 2019) | Addgene Plasmid #129421 |
| plasmid: MADR pDonor-smFP-myc (bright) WPRE | (Kim et al., 2019) | N/A |
| plasmid: MADR pDonor-mScarlet-3XSpot WPRE | This paper | N/A |
| plasmid: MADR pDonor-rtTA-V10-AU1-P2A-puro-WPRE | (Kim et al., 2019) | N/A |
| plasmid: MADR pDonor-PIP-NLS-Venus-P2A-mCherry-hGEM1/110 | (Kim et al., 2019) | N/A |
| Software and Algorithms | ||
| Incucyte S3 Software | Essen BioScience | https://www.essenbioscience.com/es/products/software/incucyte-s3-software-v2018b/ |
| ImageJ software | NIH | https://imagej.nih.gov/ij/ |
| Other | ||
| Incucyte S3 system | Essen BioScience | https://www.essenbioscience.com/en/products/incucyte/incucyte-s3/ |
Step-By-Step Method Details
Delivery of Nucleases and Template Plasmids for HDR
Timing: 1 week
The purpose of this step is to deliver all the molecular requirements to edit the desired locus with a specific cassette for further MADR applications.
-
1.Coat the plate with Poly-L-lysine.
-
a)Add enough 0.01% Poly-L-lysine to coat a 6-well plate (0.5 mL per well).
-
b)Incubate the coated plate in an incubator at 37°C for 30 min.
-
c)Remove the Poly-L-lysine and wash the plate once with PBS. Remove all PBS.
-
d)Allow the plate to dry completely either in the hood or in an incubator for faster drying.
-
a)
Note: Poly-L-lysine is not necessary, but it is useful to maintain attachment of the cells to the plate after the transfection step. Poly-D-lysine should be used in cell lines that are able to digest Poly-L-lysine.
Note: All cell culture is performed in a biosafety level II cabinet and a humidified incubator at 37°C and 5% CO2.
-
2.Plate the cells.
-
a)Plate the Neuro-2a and HEK293T cells around 30% confluency using high glucose DMEM supplemented with 10% HI-FBS, 1% PSA and 1% glutamax.
-
b)Change media every 3 days.
-
c)Cells are ready to be transfected when they reach 60%–70% confluency.Note: The time needed to reach 60%–70% confluency depends on the type and number of cells plated previously.
-
d)Aspirate old media and add 1 mL of fresh media 1 h before transfection.
-
a)
Note: For transfection of cells we used Lipofectamine 3000 but any in vitro transfection method can be used.
-
3.Transfection of plasmid donor repair template and nuclease plasmids.
-
a)Mix the plasmids in a 1:1 ratio w/w.
-
i.For the Neuro-2a cells mix 1,250 ng of the sgRosa26-Cas9 plasmid and 1,250 ng of the Rosa26 mT/mG donor template for a total of 2.5 μg DNA in sterile water or TE buffer.
-
ii.For the HEK293T cells mix 850 ng of each of the AAVS1-TALENs plasmids and 850 ng of the AAVS1 TagBFP2-NLS donor template for a total of 2.55 μg DNA in sterile water or TE buffer.
-
i.
-
b)In a 1.5 mL tube, dilute each plasmid suspension mix in 250 μL of Opti-MEM medium and add 5 μL of P3000 reagent.
-
c)In a separate 1.5 mL tube, dilute 5 μL of Lipofectamine 3000 in 250 μL of Opti-MEM medium.
-
d)Add diluted DNA to diluted Lipofectamine 3000 and mix well. The final volume should be approximately 500 μL for each plasmid mix. Incubate the reaction at 20°C–25°C for 15 min.
-
e)Add the final suspension dropwise into to their respective cell culture, directly into the media.
-
i.Add the mix containing the sgRosa26-Cas9 plasmid and Rosa26 mT/mG donor template to the Neuro-2a cells.
-
ii.Add the mix containing the AAVS1-TALENs plasmids and AAVS1 TagBFP2-NLS donor template to the HEK293T cells.
-
i.
-
f)Incubate the reaction at 37°C in a sterile incubator for 24 h.
-
g)Add fresh medium and replace it every three days.
-
a)
Note: The quantity of plasmid and media that is described in this step is for cell cultures plated in 1 well of a 6-well plate with a surface area of 9 cm2. For smaller or bigger plates, adapt the final quantity based on the manufacturer’s protocol.
Clone Selection
The purpose of this step is to enrich the cell population that successfully integrated the DNA cassette from the donor template into their respective loci.
-
4.Identify the cells that express the reporter transgenes with a fluorescence microscope.
-
a)For this protocol identify the tdTomato positive Neuro-2a cells.
-
b)For this protocol identify the nuclear TagBFP2-NLS positive HEK293T cells.
-
a)
-
5.Select the positive cells by fluorescence-activated cell sorting (FACS).
-
a)Maintain and expand the cells for around 2 weeks.Note: Varying intensities of fluorescent signals can be observed due to the episomal copy number expression.
- b)
-
a)
Note: We recommend freezing the cells at least 1 month post selection to allow the cells to dilute out the episomal DNA. Allow the cells to be in culture 2–3 days after thawing for further experiments. For improved viability, cells should be split at least once after thawing before use in experiments.
Note: Loss of fluorescent reporter intensity should not be observed because the cassette has been inserted in a safe harbor under strong promoters.
dRMCE Validation in Proxy-MADR Cells
Two requirements are needed to validate dual recombinase-mediated cassette exchange (dRMCE) (Osterwalder et al., 2010) in the Proxy-MADR cells. First, a promoter-less donor plasmid which can have any transgene flanked by loxP and FRT sites in the same orientation as the targeted locus. In addition, the open reading frame (ORF) should be preceded by phosphoglycerate kinase (PGK) and trimerized SV40 polyadenylation signals (i.e., 4× polyA) or equivalent elements to avoid transcription from unintegrated episomes and randomly integrated whole plasmids. The ORF should be followed by poly(A) elements but may be interceded by a WPRE element to promote RNA stability and facilitate transgene expression. Here, this plasmid is referred to as MADR pDonor (Figures 2A and 2C). Second, a plasmid that allows for the expression of the two recombinases which recognize the loxP and FRT sites, Cre and Flp, respectively. In this protocol, one plasmid expressing both recombinases is used, called pCAG-FlpO-2A-Cre EV (Figures 2A and 2C). See Kim et al., 2019 and Rincon Fernandez Pacheco et al., 2020 accompanying STAR protocol for a detailed description and construction of these plasmids.
-
6.Transfection of MADR pDonor and pCAG-FlpO-2A-Cre EV.Note: Any in vitro transfection methodology can be used. In this protocol, Lipofectamine 3000 is used.
-
a)Coat plates with Poly-L-lysine as described in step 1.
-
b)Plate cells and let them grow until they reach ∼60% confluency. See step 2.
-
c)Prepare a mix of plasmids containing the pCAG-FlpO-2A-Cre EV and the MADR pDonor. The suggested ratio is 1:2 w/w, pCAG-FlpO-2A-Cre EV: MADR pDonor.
-
i.Mix 800 ng of pCAG-FlpO-2A-Cre EV and 1,600 ng of MADR pDonor TagBFP2-3xFlag (cyto) WPRE for a total of 2.4 μg DNA in sterile water or TE buffer.
-
ii.Mix 800 ng of pCAG-FlpO-2A-Cre EV and 1,600 ng of MADR pDonor-mScarlet-3XSpot WPRE for a total of 2.4 μg DNA in sterile water or TE buffer.CRITICAL: It is highly recommended to have Flp and Cre in the same plasmid to enhance the dRMCE (Anderson et al., 2012).Note: The MADR pDonor TagBFP2-3xFlag (cyto) WPRE plasmid contains a TagBFP2 transgene with three flag epitope tags flanked upstream by one loxP site and downstream by one FRT site (Figure 2A). The MADR pDonor-mScarlet-3XSpot WPRE contains an mScarlet (Bindels et al., 2016) transgene with three spot tags flanked upstream by one loxP site and downstream by one FRT site (Figure 2C).
-
i.
-
d)Proceed with the Lipofectamine 3000 transfection as described in steps 3b–3d.
-
e)Add the final suspension dropwise into to their respective cell culture, directly into the media.
-
i.Add the mix containing the pCAG-FlpO-2A-Cre EV and MADR pDonor-TagBFP2-3xFlag (cyto) WPRE to the Neuro-2a Rosa26 “mT/mG” Proxy-MADR cells.
-
ii.Add the mix containing the pCAG-FlpO-2A-Cre EV and MADR pDonor-mScarlet-3XSpot WPRE to the HEK293T AAVS1 “TagBFP2-NLS” Proxy-MADR cells.
-
i.
-
f)Incubate the reaction at 37°C in a sterile incubator for 24 h.
-
g)Add fresh medium and replace it every three days.
-
a)
-
7.Visualization of transgene expression.Note: If recombination has occurred through dRMCE, cells should express the transgenes present in the promoter-less MADR pDonor.
-
a) Confirm the expression of the MADR pDonor transgene under a fluorescence microscope.Note: In our experience, the expression of transgenes can be visualized 48 h post transfection. This is longer than is required for transfection or nucleofection, presumably because the transgene must integrate before expression—which requires FlpO and Cre expression and the subsequent multistep dRMCE reaction.
-
i.For this protocol, look for a blue fluorescent signal in the Neuro-2a Rosa26 “mT/mG” Proxy-MADR cells under a fluorescence microscope (Figure 2B).Note: For the Neuro-2a Rosa26 “mT/mG” Proxy-MADR cells it is highly probable to see EGFP-positive cells as the tdTomato transgene got excised due to the CRE-loxP recombination (Figure 2B). For a more accurate discussion, see Kim et al, 2019 and Rincon Fernandez Pacheco et al., 2020 accompanying STAR protocol.
-
ii.For this protocol, look for a red fluorescent signal in the HEK293T AAVS1 “TagBFP2-NLS” Proxy-MADR cells under a fluorescence microscope (Figure 2D).
-
i.
-
a
Figure 2.

Dual Recombinase-Mediated Cassette Exchange Validation (dRMCE) in Proxy-MADR Cells
(A) Schematic showing dRMCE or Cre-mediated excision in the Neuro-2a Rosa26 “mTmG” locus.
(B) Neuro-2a Rosa26 “mT/mG” Proxy-MADR cells expressing tdTomato, EGFP, or TagBFP2 after dRMCE or Cre-mediated excision.
(C) Schematic showing dRMCE in the HEK293T AAVS1 “TagBFP2-NLS” locus.
(D) HEK293T AAVS1 “TagBFP2-NLS” Proxy-MADR cells expressing mScarlet after dRMCE. Scale bar, 50 μm.
Swapping of MADR Elements
After validating the functionality of the Proxy-MADR cells, it is possible to swap any cassette using MADR and purify the cells based on the newly inserted cassette. Thus, no more locus editing through HDR or similar processes is needed. For this protocol, we performed experiments with the Neuro-2a Rosa26 “mT/mG” Proxy-MADR cells, but it can be applied to any cell that has all the requirements to apply MADR.
Note: One of the disadvantages of the Neuro-2a Rosa26 “mT/mG” Proxy-MADR cells, in terms of visualization, is that any further cassette integration through MADR should avoid red or green fluorescent reporter transgenes. That is why a blue fluorescent reporter transgene was used in the previous experiment. To have more options available to select desired cells, e.g., to use a MADR pDonor containing a red fluorescent reporter, it is suggested to work with the EGFP-positive cells that went through Cre and Flp-mediated excision (Figure 3A).
Figure 3.

Generation of Multiple Neuro-2a Rosa26 Proxy-MADR Cell Lines
(A) Schematic showing Cre- and Flp-mediated excision and subsequent swapping of cassettes through dRMCE in the Rosa26 locus.
(B) Different Neuro-2a Rosa26 Proxy-MADR cell lines after Cre- and Flp-mediated excision and serial swapping of cassettes through dRMCE. Proxy-MADR cell lines: Neuro-2a Rosa26 “mT/mG” expressing tdTomato, Neuro-2a Rosa26 “mG” expressing EGFP, Neuro-2a Rosa26 “mScarlet” expressing mScarlet, and Neuro-2a Rosa26 “dark-PuroR” expressing puromycin N-acetyl-transferase (PAC). Scale bar, 50 μm. (B) Adapted with permission from Kim et al. (2019); copyright 2019 Elsevier.
-
8.Generation of EGFP-positive cells through Cre and Flp-mediated excision.Note: Cre-mediated excision is enough to get EGFP-positive cells. For this protocol Flp expression is suggested as well to get one FRT site in the locus to facilitate subsequent dRMCE (Figure 3A).
-
a)Plate the Neuro-2a Rosa26 “mT/mG” Proxy-MADR cells in a Poly-L-lysine coated plate and let them grow until they reach ∼60% confluency. See steps 1 and 2.
-
b)Use Lipofectamine 3000 to transfect the pCAG-FlpO-2A-Cre EV plasmid.
-
i.Mix 2,500 ng of the pCAG-FlpO-2A-Cre EV plasmid.
-
i.
-
c)Proceed with the Lipofectamine 3000 transfection described in steps 3b–3d.
-
d)Add the final suspension dropwise, directly into the media and incubate at 37°C for 24 h.
-
e)Add fresh medium and replace it every three days.
-
f)EGFP-positive cells should be visualized under a fluorescent microscope 24 h post transfection (Figure 3B).
-
g)Expand the cells and sort the EGFP-positive cells by FACS. Perform sorting as many times as necessary to have 100% EGFP-positive cells. See step 5. This cell line is now called Neuro-2a Rosa26 “mG” Proxy-MADR (Figure 3B).
-
a)
Note: In our experience 2 to 3 rounds of sorting by FACS is enough to achieve 100% purity.
Note: Removing tdTomato from the mTmG-Neuro-2a Proxy-MADR cell line allows for the use of red fluorescent proteins as reporter transgenes.
-
9.Swap the EGFP cassette of the Neuro-2a Rosa26 “mG” Proxy-MADR cells with a new one containing a different transgene. In this protocol, a MADR pDonor containing a red fluorescent reporter, mScarlet, is used (Figure 3A).
-
a)Plate the Neuro-2a Rosa26 “mG” Proxy-MADR cells in a Poly-L-lysine coated plate and let them grow until they reach ∼60% confluency. See steps 1 and 2.
-
b)Use Lipofectamine 3000 to transfect the pCAG-FlpO-2A-Cre EV and the MADR pDonor-mScarlet-3XSpot WPRE plasmids. The suggested ratio is 1:2 w/w, pCAG-FlpO-2A-Cre EV: MADR pDonor-mScarlet-3XSpot WPRE.
-
i.Mix 800 ng of pCAG-FlpO-2A-Cre EV and 1,600 ng of MADR pDonor-mScarlet-3XSpot WPRE.
-
i.
-
c)Proceed with the Lipofectamine 3000 transfection described in steps 3b–3d.
-
d)Add the final suspension dropwise, directly into the media and incubate at 37°C for 24 h.
-
e)Add fresh medium and replace it every three days.
-
f)mScarlet-positive cells should be visualized under a fluorescent microscope 48 h post transfection (Figure 3B).
-
g)Expand the cells and sort the mScarlet-positive cells by FACS. See step 5. This cell line is now called Neuro-2a Rosa26 “mScarlet” Proxy-MADR (Figure 3B).
-
a)
-
10.Swap the mScarlet cassette of the Neuro-2a Rosa26 “mScarlet” Proxy-MADR cells with a new one containing a colorless transgene.Note: Cassettes can be swapped an unlimited number of times from any cell that contains the minimum requirements to perform MADR. As proof of principle in this protocol, swap the mScarlet cassette of the Neuro-2a Rosa26 “mScarlet” Proxy-MADR cells using the MADR pDonor-rtTA-V10-AU1-P2a-puro-WPRE which contains a colorless puromycin resistance transgene, puromycin N-acetyltransferase (PAC) (Figure 3A).
-
a)Plate the Neuro-2a Rosa26 “mScarlet” Proxy-MADR cells in a Poly-L-lysine coated plate and let them grow until they reach ∼60% confluency. See steps 1 and 2.
-
b)Use Lipofectamine 3000 to transfect the pCAG-FlpO-2A-Cre EV and the MADR pDonor-rtTA-V10-AU1-P2a-puro-WPRE plasmids. The suggested ratio is 1:2 w/w, pCAG-FlpO-2A-Cre EV: MADR pDonor-rtTA-V10-AU1-P2a-puro-WPRE.
-
i.Mix 800 ng of pCAG-FlpO-2A-Cre EV and 1,600 ng of MADR pDonor-rtTA-V10-AU1-P2a-puro-WPRE.
-
i.
-
c)Proceed with the Lipofectamine 3000 transfection described in steps 3b–3d.
-
d)Add the final suspension dropwise, directly into the media and incubate at 37°C for 24 h.
-
e)Add fresh medium and replace it every three days.
-
f)Colorless cells should be visualized 48 h post transfection.Note: Due to the lack of a fluorescent reporter transgene, the colorless cells can be hard to visualize among the fluorescent-positive cells. Expansion and puromycin selection of the cells will help to visualize them.
-
g)Expand the cells for around 1 week and select them by adding puromycin to the culture media at a final concentration of 1 μg/mL.
-
h)Change media every three days and add fresh puromycin for 2–3 weeks. This cell line is now called Neuro-2a Rosa26 “dark-PuroR” Proxy-MADR (Figure 3B).
-
a)
Note: The final concentration of puromycin depends on the cell type used, thus, a puromycin dose titration is highly suggested. It is also recommended to select the cells through FACS in case there are remaining fluorescent cells that developed resistance to puromycin.
Note: The “dark-PuroR” Proxy-MADR cells are extremely helpful when selection through any fluorescent transgene is desired.
-
11.Swap the colorless cassette of the Neuro-2a Rosa26 “dark-PuroR” Proxy-MADR cells for any cassette containing a visible reporter transgene.Note: One of the main advantages of the colorless Proxy-MADR cells is that there is a broad spectrum of reporter transgenes that can be used to select the desired cells. In this protocol, we show the utility of the Neuro-2a Rosa26 “dark-PuroR” Proxy-MADR cells through the transfection of three MADR pDonors each containing a different fluorescent reporter transgene and the transfection of one MADR pDonor containing two fluorescent reporter transgenes in the same cassette (Figures 4A and 4C).
-
a)Plate the Neuro-2a Rosa26 “dark-PuroR” Proxy-MADR cells in a Poly-L-lysine coated plate and let them grow until they reach ∼60% confluency. See steps 1 and 2.
-
b)Use Lipofectamine 3000 to transfect the pCAG-FlpO-2A-Cre-EV and the respective MADR pDonors. The suggested ratio is 1:2 w/w, pCAG-FlpO-2A-Cre-EV: MADR pDonor.
-
i.Mix 350 ng of pCAG-FlpO-2A-Cre EV and 700 ng of each of the following plasmids: MADR pDonor-TagBFP2-3xFlag (cyto) WPRE, MADR pDonor smFP-myc (bright) WPRE, and MADR pDonor-mScarlet-3XSpot WPRE.Note: The MADR pDonor smFP-myc (bright) WPRE contains a cassette that codes for a “spaghetti monster” fluorescent protein (smFP) with multiple Myc tags that also emits GFP fluorescence (Viswanathan et al., 2015).
-
ii.Mix 800 ng of pCAG-FlpO-2A-Cre EV and 1,600 ng of MADR pDonor-PIP-NLS-mVenus-P2A-mCherry-hGEM1/110.Note: The MADR pDonor-PIP-NLS-mVenus-P2A-mCherry-hGEM1/110 plasmid contains a cassette with two reporter transgenes, mVenus and mCherry. Expression is dependent on the cell cycle phase. See Fluorescent Ubiquitination-based Cell Cycle Indicator (FUCCI) (Grant et al., 2018; Sakaue-Sawano et al., 2008).
-
i.
-
c)Proceed with the Lipofectamine 3000 transfection as described in steps 3b–3d.
-
d)Add the final suspension dropwise into to their respective cell culture, directly into the media and incubate at 37°C for 24 h.
-
e)Add fresh medium and replace it every three days.
-
f)Confirm the expression of the MADR pDonors under a fluorescent microscope.
-
i.Look for blue, red, and green signals in the Neuro-2a Rosa26 “dark-PuroR” Proxy-MADR cells that were transfected with the three MADR pDonors (Figure 4B).Note: We have realized that SM-Myc-Bright is not as bright as EGFP, thus, performing immunocytochemistry (ICC) is helpful to identify if cells integrated the cassette.
-
ii.Look for green and red signals in the Neuro-2a Rosa26 “dark-PuroR” Proxy-MADR cells transfected with the MADR pDonor Green-Red-FUCCI. Expand the cells and sort the mVenus and mCherry-positive cells by FACS. See step 5. This cell line is now called Neuro-2a Rosa26 “Green-Red-FUCCI” Proxy-MADR (Figure 4D).
-
i.
-
a)
Figure 4.

Versatility of “Dark” Proxy-MADR Cells
(A) Schematic showing competence among pDonors to integrate their cassettes through dRMCE into the Rosa26 “PuroR” locus.
(B) Neuro-2a Rosa26 “dark-PuroR” Proxy-MADR cells expressing tdTomato, smFP-Bright, or TagBFP2 after dRMCE.
(C) Schematic showing integration of the Green-Red-FUCCI cassette through dRMCE into the Rosa26 “PuroR” locus.
(D) Neuro-2a Rosa26 “Green-Red FUCCI” Proxy-MADR cells expressing mVenus and mCherry after dRMCE and subsequent purification. Scale bar, 50 μm.
Validation of Green-Red-FUCCI System on Proxy-MADR Cells
The purpose of this step is to validate the FUCCI system in Proxy-MADR cells. Live-cell time-lapse imaging of proliferating cells is a great method for studying cell cycle dynamics and it can be used to validate FUCCI which is a good system to identify cell cycle phases.
-
12.Live-cell time-lapse imaging of Neuro-2a Rosa26 “Green-Red-FUCCI” Proxy-MADR cells.
-
a)Add enough Poly-L-lysine to coat a 96-well plate (100 μL per well). See step 1.CRITICAL: It is highly recommended to coat any plate to maintain cell attachment for easier visualization.
-
b)Plate ∼10,000 Neuro-2a Rosa26 “Green-Red-FUCCI” Proxy-MADR cells and let them grow at least 24 h before imaging.
-
c)In this protocol, the Incucyte S3 System and 20× objective were used to take images under phase, green, and red fluorescence. Images were taken every 30 min for 40 h.Note: The image collection time depends on the cell type, fluorescence channels, number of plates, etc. We recommend standardizing the proper time collection.
-
d)For this protocol, Incucyte S3 Software and ImageJ were used to collect and process live-cell time-lapse imaging of the Neuro-2a Rosa26 “Green-Red-FUCCI” Proxy-MADR cells (Methods Video S1).Methods Video S1. Time-Lapse Imaging of Neuro-2a Rosa26 “Green-Red FUCCI” Proxy-MADRCells Phase, green, and red fluorescence channels on the left panel. Green and red fluorescence channels on the right panel (Related to point 12 of the protocol).Download video file (7.5MB, mp4)
-
a)
Expected Outcomes
MADR reaction in Proxy-MADR cells is easier to perform than in primary cells that come from a transgenic mouse that meet the requirements to perform MADR, e.g., Gt(ROSA)26Sortm4(ACTB-tdTomato,-EGFP)Luo/J, usually called mTmG mouse (Muzumdar et al., 2007). An important reason is that commonly used cell lines (e.g., HEK293T) are in general easier to transfect than primary cells, thus allowing the use of standard methods like Lipofectamine. The efficiency of the recombination through MADR varies depending on multiple factors like the type of the cell, method of transfection, available recipient loci, size of the MADR pDonor, among others. In our experience, the efficiency of the recombination in Proxy-MADR cells is between 0.5% and 15% 1 week post transfection after which further selection is required to enrich the desired cell population. One of the main important factors to increase the recombination is the transfection efficiency.
In principle, any line amenable to HDR can be targeted with these methods, and we recently generated multiple clones of human induced pluripotent stem cells with an efficiency of roughly 5%–10% using a MaxCyte scalable transfection system. Subsequent swapping of MADR cassettes with Lipofectamine was successful in about 5% of recipient cells. Taken together, generating MADR cells is a technique that is easily accomplished by most labs experienced in cell culture and enables a variety of unique opportunities for downstream generation of stable transgenic cell lines.
Limitations
The main limitation of the Proxy-MADR cells is that there is no certainty about the copy number of MADR recipient sites per cell. There are two main reasons, first is that it is difficult to control the number of the MADR cassettes integrated into cells through HDR via either CRSPR/Cas9 or TALENs. The second is that some commercial cell lines, especially tumor cells such as Neuro-2a, have extra chromosomes thus allowing the insertion of more cassettes into desired loci through HDR. If there are more than on locus edited per cell, then it is possible to have more than one copy of MADR cassettes through dRMCE. Nevertheless, in our experience it is difficult to see cells with more than one reporter expression when two or more different pDonors are co-transfected (Figure 4B).
Troubleshooting
Problem
After HDR induction the positive cells that were sorted are no longer positive.
Potential Solution
Culture the induced cells for at least two extra weeks before proceeding with extra FACS or antibiotic selection. Plasmids that have not been integrated and are causing transient expression should be washed out after this time. In our experience, after 1 month of HDR induction and post selection, all MADR “proxy” cells maintain a constant transgene expression because the cassette has been inserted in a safe harbor under a strong promoter, e.g., CAG or Actin promoters.
Adding an extra antibiotic resistance transgene into the cassette facilitates and reduces the costs of the FACS selection.
Problem
Low MADR recombination efficiency
Potential Solution
Proxy-MADR cells can be transfected a second, and even a third time with the same recombinases and MADR pDonors 96 h after the first MADR reaction. It is also possible to remove the Lipofectamine 48 h post transfection instead of 24 h.
Also, empirically determining the ideal MADR pDonor to recombinase ratio is a key element for maximizing the dRMCE reaction.
Problem
Lipofection has an extremely low efficiency in primary cell lines.
Potential Solution
An electrofection method, e.g., nucleofection, is suggested if lipofection has a low efficiency when transfecting primary cells. Use the same plasmids to perform HDR and maintain the same ratios. Remember to adapt the final concentration of DNA based on the electrofection method.
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Josh Breunig (joshua.breunig@cshs.org).
Materials Availability
Selected plasmids are available from Addgene: http://www.addgene.org/browse/article/28203812/
Data and Code Availability
This study did not generate datasets and codes.
Acknowledgments
We acknowledge support from TJ’s Dream Team HEADing for a Cure, a Samuel Oschin Comprehensive Cancer Institute (SOCCI) Cancer Research Forum Award, the Board of Governors RMI of Cedars-Sinai (to J.J.B.). J.J.B. was supported by NIH grants R33CA202900, R33CA236687, and R03NS101529, American Cancer Society grant RSG-16-217-01-TBG, and SOCCI Jack Mishkin Discovery, P&G, and Cancer Biology Awards.
Author Contributions
J.J.B. conceived the in vitro recombination strategy. A.E.A. and N.K. wrote the manuscript and prepared the figures under the supervision of J.J.B. All authors read and edited the final version of this manuscript.
Declaration of Interests
Cedars-Sinai has filed for patent protection for in vivo dual recombinase-mediated cassette exchange and disease models thereof.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.xpro.2020.100184.
References
- Anderson R.P., Voziyanova E., Voziyanov Y. Flp and Cre expressed from Flp-2A-Cre and Flp-IRES-Cre transcription units mediate the highest level of dual recombinase-mediated cassette exchange. Nucleic Acids Res. 2012;40:e62. doi: 10.1093/nar/gks027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bindels D.S., Haarbosch L., van Weeren L., Postma M., Wiese K.E., Mastop M., Aumonier S., Gotthard G., Royant A., Hink M.A. MScarlet: A bright monomeric red fluorescent protein for cellular imaging. Nat. Methods. 2016;14:53–56. doi: 10.1038/nmeth.4074. [DOI] [PubMed] [Google Scholar]
- Chu V.T., Weber T., Wefers B., Wurst W., Sander S., Rajewsky K., Kühn R. Increasing the efficiency of homology-directed repair for CRISPR-Cas9-induced precise gene editing in mammalian cells. Nat. Biotechnol. 2015;33:543–548. doi: 10.1038/nbt.3198. [DOI] [PubMed] [Google Scholar]
- Cooper M.J., Lippa M., Payne J.M., Hatzivassiliou G., Reifenberg E., Fayazi B., Perales J.C., Morrisn L.J., Templeton D., Piekarz R.L. Safety-modified episomal vectors for human gene therapy. Proc. Natl. Acad. Sci. U S A. 1997;94:6450–6455. doi: 10.1073/pnas.94.12.6450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devkota S. The road less traveled: Strategies to enhance the frequency of homology-directed repair (HDR) for increased efficiency of CRISPR/Cas-mediated transgenesis. BMB Reports. 2018;51:437–443. doi: 10.5483/BMBRep.2018.51.9.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaj T., Gersbach C.A., Barbas C.F. ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2013;31:397–405. doi: 10.1016/j.tibtech.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant G.D., Kedziora K.M., Limas J.C., Cook J.G., Purvis J.E. Accurate delineation of cell cycle phase transitions in living cells with PIP-FUCCI. Cell Cycle. 2018;17:2496–2516. doi: 10.1080/15384101.2018.1547001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homann S., Hofmann C., Gorin A.M., Nguyen H.C.X., Huynh D., Hamid P., Maithel N., Yacoubian V., Mu W., Kossyvakis A. A novel rapid and reproducible flow cytometric method for optimization of transfection efficiency in cells. PLoS One. 2017;12 doi: 10.1371/journal.pone.0182941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim G.B., Rincon Fernandez Pacheco D., Saxon D., Yang A., Sabet S., Dutra-Clarke M., Levy R., Watkins A., Park H., Abbasi Akhtar A. Rapid generation of somatic mouse mosaics with locus-specific, stably integrated transgenic elements. Cell. 2019;179:251–267.e24. doi: 10.1016/j.cell.2019.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombardo A., Cesana D., Genovese P., di Stefano B., Provasi E., Colombo D.F., Neri M., Magnani Z., Cantore A., lo Riso P. Site-specific integration and tailoring of cassette design for sustainable gene transfer. Nat. Methods. 2011;8:861–869. doi: 10.1038/nmeth.1674. [DOI] [PubMed] [Google Scholar]
- Mandegar M.A., Huebsch N., Frolov E.B., Shin E., Truong A., Olvera M.P., Chan A.H., Miyaoka Y., Holmes K., Spencer C.I. CRISPR interference efficiently induces specific and reversible gene silencing in human iPSCs. Cell Stem Cell. 2016;18:541–553. doi: 10.1016/j.stem.2016.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muzumdar M.D., Tasic B., Miyamichi K., Li N., Luo L. A global double-fluorescent cre reporter mouse. Genesis. 2007;45:593–605. doi: 10.1002/dvg.20335. [DOI] [PubMed] [Google Scholar]
- Osterwalder M., Galli A., Rosen B., Skarnes W.C., Zeller R., Lopez-Rios J. Dual RMCE for efficient re-engineering of mouse mutant alleles. Nat. Methods. 2010;7:893–895. doi: 10.1038/nmeth.1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rincon Fernandez Pacheco D., Sabet S., Breunig J.J. Preparation, Assembly, and Transduction of Transgenic Elements Using Mosaic Analysis with Dual Recombinases (MADR) Star Protocols. 2020;1 doi: 10.1016/j.xpro.2020.100199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaue-Sawano A., Kurokawa H., Morimura T., Hanyu A., Hama H., Osawa H., Kashiwagi S., Fukami K., Miyata T., Miyoshi H. Visualizing spatiotemporal dynamics of multicellular cell-cycle progression. Cell. 2008;132:487–498. doi: 10.1016/j.cell.2007.12.033. [DOI] [PubMed] [Google Scholar]
- Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain [1] Nat. Genet. 1999;21:70–71. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- Viswanathan S., Williams M.E., Bloss E.B., Stasevich T.J., Speer C.M., Nern A., Pfeiffer B.D., Hooks B.M., Li W.P., English B.P. High-performance probes for light and electron microscopy. Nat. Methods. 2015;12:568–576. doi: 10.1038/nmeth.3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Cells Phase, green, and red fluorescence channels on the left panel. Green and red fluorescence channels on the right panel (Related to point 12 of the protocol).
Data Availability Statement
This study did not generate datasets and codes.