

Summary
Mapping the genomic locations of chromatin-associated proteins, such as transcription factors and histone modifications, is key to understanding the mechanisms of transcriptional regulation. ChIPmentation offers a simple and robust way of investigating the genomic binding sites of a protein using relatively low-input material. Here, we present a detailed protocol for the key steps that lead to a successful ChIPmentation experiment, as well as a quick analysis pipeline to examine the data.
For complete details on the use and execution of this protocol, please refer to Schmidl et al. (2015). For example data produced by this protocol, please refer to Henriksson et al. (2019) and Zhang et al. (2019).
Subject Areas: Genomics, Sequencing, ChIPseq, Chromatin immunoprecipitation (ChIP)
Graphical Abstract
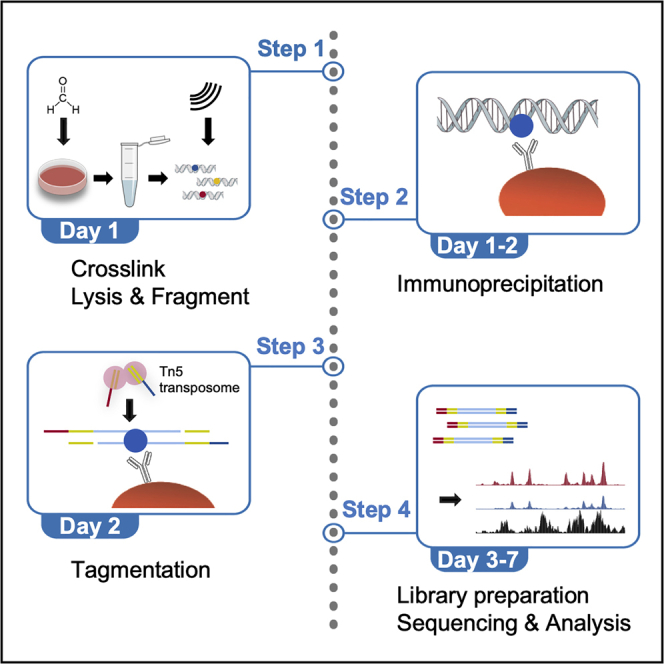
Highlights
-
•
Preparation of a transposase-assisted ChIP-seq library
-
•
A simpler and cheaper approach than traditional ChIP-seq
-
•
A sensitive and reliable method for transcription factors and histone modifications
Mapping the genomic locations of chromatin-associated proteins, such as transcription factors and histone modifications, is key to understanding the mechanisms of transcriptional regulation. ChIPmentation offers a simple and robust way of investigating the genomic binding sites of a protein using relatively low-input material. Here, we present a detailed protocol for the key steps that lead to a successful ChIPmentation experiment, as well as a quick analysis pipeline to examine the data.
Before You Begin
There are many existing ChIP-seq protocols with different modifications. Many of them are lengthy and difficult to carry out. We have found ChIPmentation is the simplest and the most robust to implement in a molecular biology lab, especially for people who have already had the experience with the ChIP technique. The ChIPmentation method is modular, where it contains a ChIP module and a library preparation module. Experienced researchers can just stick to their own ChIP protocol and start following the procedures described here after washing the IP (i.e., step 13). People with no previous ChIP experience are recommended to follow the exact procedures described in this protocol. It is also recommended to read the full protocol before starting in order to get a feeling about the timing and work load in each step.
Compared to other ChIP-seq methods, one advantage of ChIPmentation is its sensitivity. We routinely use 5 × 105 cells to profile histone modifications and 5 × 106 cells to study transcription factors. The minimum cell number required for a successful ChIPmentation experiment in our hands is 104 cells for histone modifications and 105 cells for transcription factors. However, it is worth noting that the number of cells required for a successful ChIPmentation experiment depends on many factors, such as the abundance of the protein/modification of interest and the efficiency of the antibody. The other major advantage of ChIPmentation is simplicity. Sequencing adapters are added by the transposase Tn5, and library PCR is performed immediately after reverse crosslinking and DNA purification. The third advantage is the cost. Only a small amount of Tn5 transposase is needed per library. Nowadays, we always use ChIPmentation even when the cell number is not a constraint, such as cell lines.
Prepare Reagents and Buffers
Timing: 3 h
-
1.Prepare the following buffers, sterilize by using .22 μm filter units. See Materials and Equipment for buffer recipes.
-
a.100× protease inhibitor cocktail stock
-
b.11% formaldehyde
-
c.1.25 M Glycine
-
d.Blocking Solution
-
e.Sonication/IP Buffer
-
f.RIPA Wash Buffer
-
g.Low Salt Wash Buffer
-
h.High Salt Wash Buffer
-
i.LiCl Wash Buffer
-
j.10 mM Tris-HCl, pH 8.0
-
k.1× TE 50 mM NaCl
-
l.ChIP Elution Buffer
-
m.2× Tagmentation DNA (TD) Buffer
-
a.
-
2.
Prepare oligo stocks by resuspend primers in ddH2O to reach 100 μM stock concentration. See Table 1 below for sequences. We order them from Sangon Biotech (Shanghai) with standard desalt purification.
Table 1.
ChIPmentation Oligos Used for Library PCR (Illumina Nextera Index Primers)
| Oligo Name | Oligo Sequence (5′ to 3′) |
|---|---|
| N701 | CAAGCAGAAGACGGCATACGAGATTCGCCTTAGTCTCGTGGGCTCGG |
| N702 | CAAGCAGAAGACGGCATACGAGATCTAGTACGGTCTCGTGGGCTCGG |
| N703 | CAAGCAGAAGACGGCATACGAGATTTCTGCCTGTCTCGTGGGCTCGG |
| N704 | CAAGCAGAAGACGGCATACGAGATGCTCAGGAGTCTCGTGGGCTCGG |
| N705 | CAAGCAGAAGACGGCATACGAGATAGGAGTCCGTCTCGTGGGCTCGG |
| N706 | CAAGCAGAAGACGGCATACGAGATCATGCCTAGTCTCGTGGGCTCGG |
| N707 | CAAGCAGAAGACGGCATACGAGATGTAGAGAGGTCTCGTGGGCTCGG |
| N710 | CAAGCAGAAGACGGCATACGAGATCAGCCTCGGTCTCGTGGGCTCGG |
| N711 | CAAGCAGAAGACGGCATACGAGATTGCCTCTTGTCTCGTGGGCTCGG |
| N712 | CAAGCAGAAGACGGCATACGAGATTCCTCTACGTCTCGTGGGCTCGG |
| N714 | CAAGCAGAAGACGGCATACGAGATTCATGAGCGTCTCGTGGGCTCGG |
| S502 | AATGATACGGCGACCACCGAGATCTACACCTCTCTATTCGTCGGCAGCGTC |
| S503 | AATGATACGGCGACCACCGAGATCTACACTATCCTCTTCGTCGGCAGCGTC |
| S505 | AATGATACGGCGACCACCGAGATCTACACGTAAGGAGTCGTCGGCAGCGTC |
| S506 | AATGATACGGCGACCACCGAGATCTACACACTGCATATCGTCGGCAGCGTC |
| S507 | AATGATACGGCGACCACCGAGATCTACACAAGGAGTATCGTCGGCAGCGTC |
| S508 | AATGATACGGCGACCACCGAGATCTACACCTAAGCCTTCGTCGGCAGCGTC |
| S510 | AATGATACGGCGACCACCGAGATCTACACCGTCTAATTCGTCGGCAGCGTC |
| S511 | AATGATACGGCGACCACCGAGATCTACACTCTCTCCGTCGTCGGCAGCGTC |
Sonication Test and Antibody Test I
Note: It is very important to find the right condition for sonication and a good antibody for the immunoprecipitation of the protein of your interest. In general, one can refer to the ENCODE and modENCODE guidelines (Landt et al., 2012). This section describes some extra details on how to check sonication and perform an initial test on antibodies. Sonication serves two purposes: to solubilize chromatin and to fragment DNA to a size range that is suitable for next generation sequencing. However, during the sonication process, we found the epitopes of some proteins can be destroyed as well. The main purpose here is to find a balance between getting the DNA to the right range and maintain the protein integrity at the same time. This is the most variable process in the entire protocol, because sonication is highly dependent on the machine in use. We have successful experience using both probe sonicators and water bath sonicators.
-
3.
Follow steps 1–3 and 5–6 from the Step-by-Step Method Details. We prepare cells that are enough for 4–5 aliquots (5 × 106 per aliquot) for the sonication test.
-
4.After resuspending cell pellet at a concentration of 5 × 106 cells per 300 μL Sonication/IP Buffer (i.e., step 6):Note: both the cell concentration and the volume can influence sonication results. We routinely used 300 μL Sonication/IP Buffer to resuspend cells from 105 to 5 × 106. When more than 5 × 106 cells are used, we scale up the volume to maintain a concentration of 5 × 106 per 300 μL volume. If the final volume exceeds the recommendation of the sonicator, make aliquots to perform sonication.
-
a.Take out 25 μL lysate and mix with 75 μL ChIP Elution Buffer and 1 μL Proteinase K (20 mg/mL). Leave the reaction on a thermomixer at 65°C, with shaking at 1,400 rpm for at least 6 h (or overnight, 12–16 h) for the reverse crosslink. This is the no sonication DNA input.
-
b.Take out 32 μL lysate and mix with 8 μL 5× SDS Loading Buffer. Boil at 99°C for 10 min. This is the no sonication protein input.
-
a.
-
5.If using a water bath sonicator, aliquot 300 μL lysate into 1.5 mL Eppendorf tubes (or other tubes required by the sonicator manufacturer). Perform a sonication time course with a recommended ON/OFF cycle setting. For example, if using a Bioruptor Pico, a time course of 2, 4, 6, 8 min with 30 s ON/30 s OFF can be used for an initial trial. At the end of each time course:
-
a.Take out 25 μL lysate from an aliquot and mix with 75 μL ChIP Elution Buffer and 1 μL Proteinase K (20 mg/mL). Leave the reaction on a thermomixer at 65°C, with shaking at 1,400 rpm for at least 6 h (or overnight, 12–20 h) for the reverse crosslink. This is for the DNA size check.
-
b.Take out 32 μL lysate and mix with 8 μL 5× SDS Loading Buffer. Boil at 99°C for 10 min. This is for the protein check.
-
a.
Note: When using the Bioruptor water bath sonicator, the maximum recommended volume in a 1.5 mL Eppendorf tube is 300 μL. Therefore, it is not possible to just use one tube for the entire time course due the volume needed for the DNA and protein analysis. We normally prepare multiple tubes, one for each time point.
-
6.
Analyze all the protein samples using western blot with the antibody of your choice. We include in the Key Resources Table the antibodies used in this particular protocol, but the antibodies will depend on the factors of interest. Four examples are shown in Figure 1.
CRITICAL: This is the initial test of the antibody and protein integrity. There are two things to check here. First, in the no sonication input (0 min), a single (or major) clear band around the predicted size of the protein of interest is present. In Figure 1, all four antibodies satisfy this standard. Second, the protein of interest remains detectable during sonication. In Figure 1, different proteins perform differently. STAT6 and Pol II start to become less visible after 4 min, while IRF4 and CTCF remain relatively stable until 8 min. See the next step to decide which condition to use. For some lowly expressed proteins, more concentrated cell lysate needs to be used to visualize them on the western blot.
-
7.
Purify all the DNA samples after the reverse crosslink using the Qiagen minElute PCR Purification Kit. Determine the DNA concentration using a Nanodrop. Run equal amount of DNA (we routinely use 500–1,000 ng) on a 1.5% Agarose gel. An example is shown in Figure 2.
Figure 1.

Checking the Antibody and Protein Integrity before and during the Sonication Time Course
The sonication time and antibodies in use are indicated in the figure.
Figure 2.

DNA Size Distribution during the Sonication Time Course
Lanes 1 and 7 are NEB 2-log DNA ladder. Lane 2 is the input DNA before sonication. Lanes 3–6 are purified input DNA sonicated with 2, 4, 6, 8 min.
Antibody Test II
In the previous section, the sonication condition is determined and whether the antibody is able to detect the protein of interest is also tested. In this section, procedures are described to test if the antibody can immunoprecipitate the protein of interest after formaldehyde crosslinking. The success of antibody in this test does not necessarily guarantee a successful ChIPmentation experiment, but it provides some useful information about the antibody and the immunoprecipitation condition.
-
8.Follow steps 1–16 from the Step-by-Step Method Details.
-
a.At the step 6, take 32 μL lysate and mix with 8 μL 5× SDS Loading Buffer. Boil at 99°C for 10 min. This is the input sample and can be stored in −20°C and will be used in the next day.
-
b.At the step 8, before washing, put the immunoprecipitation on the magnetic stand, and take out 32 μL lysate and mix with 8 μL 5× SDS Loading Buffer. Boil at 99°C for 10 min. This is the supernatant.
-
c.After the step 16, instead of adding tagmentation mix, add 40 μL 1× SDS Loading Buffer (diluted in Sonication/IP Buffer from 5× SDS Loading Buffer) to the beads. Boil at 99°C for 10 min. This is the IP sample.
-
a.
-
9.
Analyze all the samples (input, supernatant and IP) using western blot with the same antibody. An example using an anti-FOXM1 antibody is shown in Figure 3.
Figure 3.

ChIP-Western Results Demonstrating the FOXM1 Antibody Is Able to Immunoprecipitate the Protein of Interest (i.e., FOXM1) after the Whole ChIP Procedure
The identities of the samples and bands are indicated in the figure.
Figure 4.

ChIP-Western Results with a Mouse Monoclonal V5 Antibody in a Cell Line Stably Expressing V5-Tagged FOXM1
Note the presence (indicated by arrows) of FOXM1-V5, the heavy and light chains of IgG in the supernatant when using Protein G Dynabeads without crosslinking the antibody to the beads. All the aforementioned bands disappear when the antibody is crosslinked to the Protein G Dynabeads or Pan Mouse IgG Dynabeads are used.
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Stat6 Antibody (M-20) | Santa Cruz Biotechnology | sc-981 |
| FOXM1 Antibody (C-20) | Santa Cruz Biotechnology | sc-502 |
| IRF-4 Antibody (M-17) | Santa Cruz Biotechnology | sc-6059 |
| Anti-RNA polymerase II CTD antibody (8WG16) | Abcam | ab817 |
| Anti-CTCF Antibody | Millipore | Cat# 07-729 |
| Anti-V5 antibody, Mouse monoclonal | Sigma | V8012 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 10× PBS, pH 7.4 | Thermo Fisher | AM9624 |
| BSA | Sigma | V900933 |
| 1 M HEPES, pH 7.5 | Thermo Fisher | Cat# 15630106 |
| 5 M NaCl | Sigma | S6546 |
| 0.5 M EDTA, pH 8.0 | Thermo Fisher | AM9260G |
| 0.5 M EGTA, pH 8.0 | Sigma | E3889 |
| Triton X-100 | Sigma | Cat# 93443 |
| Sodium Deoxycholate (DOC) | Sigma | Cat# 30970 |
| 10% Sodium Dodecyl Sulfate (SDS) | Thermo Fisher | Cat# 15553027 |
| 1 M Tris-HCl, pH 8.0 | Thermo Fisher | Cat# 15568025 |
| 1 M MgCl2 | Thermo Fisher | AM9530G |
| IGEPAL CA-630 | Sigma | I8896 |
| LiCl | Sigma | Cat# 62476 |
| Proteinase K | Thermo Fisher | AM2546 |
| Protease Inhibitor Cocktail | Roche | Cat# 1697498 |
| N,N-Dimethylformamide | Sigma | D4551 |
| 37% Formaldehyde solution | Sigma | Cat# 252549 |
| Glycine | Sigma | G8790 |
| 20× EvaGreen | Biotium | Cat# 31000-T |
| NEBNext High-Fidelity 2× PCR Master Mix | NEB | M0541 |
| Dynabeads Protein A for Immunoprecipitation | Thermo Fisher | 10001D |
| Dynabeads Protein G for Immunoprecipitation | Thermo Fisher | 10003D |
| Dynabeads Pan Mouse IgG | Thermo Fisher | 11041 |
| AmpureXP for PCR Purification | Beckman Coulter | A63881 |
| VAHTS DNA Clean Beads | Vazyme Biotech | N411 |
| Critical Commercial Assays | ||
| Illumina Tagment DNA TDE1 Enzyme and Buffer kit | Illumina | Cat# 20034197 |
| Fapon Tnp Library Prep Kit for Illumina | Fapon Biotech | NK001 |
| MinElute PCR Purification Kit | Qiagen | Cat# 28004 |
| Agilent High Sensitivity DNA Kit | Agilent | Cat# 5067-4626 |
| Deposited Data | ||
| ChIP-seq data | (Zhang et al., 2019) | ArrayExpress: E-MTAB-6165 |
| Software and Algorithms | ||
| Fastp | (Chen et al., 2018) | https://github.com/OpenGene/fastp |
| HISAT2 | (Kim et al., 2019) | https://github.com/DaehwanKimLab/hisat2 |
| Picard | https://github.com/broadinstitute/picard | http://broadinstitute.github.io/picard/ |
| MACS2 | (Zhang et al., 2008) | https://github.com/macs3-project/MACS |
| bdg2bw | https://gist.github.com/taoliu/2469050 | https://gist.github.com/taoliu/2469050 |
| fetchChromSizes | (Kent et al., 2002) | http://hgdownload.soe.ucsc.edu/admin/exe/ |
| bedClip | (Kent et al., 2002) | http://hgdownload.soe.ucsc.edu/admin/exe/ |
| bedGraphToBigWig | (Kent et al., 2002) | http://hgdownload.soe.ucsc.edu/admin/exe/ |
| Samtools | (Li et al., 2009) | http://www.htslib.org/ |
Materials and Equipment
Buffers
Alternatives: all the chemicals and solutions listed in the Key Resources Table can be purchased from different suppliers for your own convenience, provided they are all molecular biology grade.
100× Protease Inhibitor Cocktail stock (aliquot and store at −20°C)
| Reagent | Final Concentration | Amount |
|---|---|---|
| Protease Inhibitor Cocktail | 100× | 1 tablet∗ |
| ddH2O | n/a | 1 mL |
| Total | n/a | 1 mL |
∗If Protease Inhibitor cocktail solution is purchased, ignore this table and use the solution according the supplier’s recommendation.
11% formaldehyde solution (freshly prepared each time, and prepare just enough for the experiment to reduce the waste)
| Reagent | Final Concentration | Amount |
|---|---|---|
| 1 M HEPES, pH 7.5 | 50 mM | 0.5 mL |
| 5 M NaCl | 100 mM | 0.2 mL |
| 0.5 M EDTA, pH 8.0 | 1 mM | 20 μL |
| 0.5 M EGTA, pH 8.0 | 0.5 mM | 10 μL |
| 37% formaldehyde | 11% | 2.97 mL |
| ddH2O | n/a | 6.3 mL |
| Total | n/a | 10 mL |
1.25 M Glycine (store at room temperature: 20°C–25°C)
| Reagent | Final Concentration | Amount |
|---|---|---|
| Glycine | 1.25 M | 9.38 g |
| ddH2O | n/a | Add to 100 mL |
| Total | n/a | 100 mL |
Blocking Solution (store at 4°C)
| Reagent | Final Concentration | Amount |
|---|---|---|
| 10× PBS (pH 7.4) | 1× | 10 mL |
| BSA | 0.5% | 0.5 g |
| ddH2O | n/a | Add to 100 mL |
| Total | n/a | 100 mL |
Sonication/IP Buffer (store at 4°C)
| Reagent | Final Concentration | Amount |
|---|---|---|
| 1 M HEPES, pH 7.5 | 50 mM | 5 mL |
| 5 M NaCl | 140 mM | 2.8 mL |
| 0.5 M EDTA, pH 8.0 | 1 mM | 0.2 mL |
| 10% Triton X-100 | 1% | 10 mL |
| 10% DOC | 0.1% | 1 mL |
| 10% SDS | 0.1% | 1 mL |
| ddH2O | n/a | 80 mL |
| Total | n/a | 100 mL |
RIPA Wash Buffer (store at 4°C)
| Reagent | Final Concentration | Amount |
|---|---|---|
| 1 M Tris-HCl, pH 8.0 | 50 mM | 5 mL |
| 5 M NaCl | 150 mM | 3 mL |
| 0.5 M EDTA, pH 8.0 | 2 mM | 0.4 mL |
| 10% IGEPAL-CA630 | 1% | 10 mL |
| 10% DOC | 0.1% | 1 mL |
| 10% SDS | 0.1% | 1 mL |
| ddH2O | n/a | 79.6 mL |
| Total | n/a | 100 mL |
Low Salt Wash Buffer (store at 4°C)
| Reagent | Final Concentration | Amount |
|---|---|---|
| 1 M Tris-HCl, pH 8.0 | 20 mM | 2 mL |
| 5 M NaCl | 150 mM | 3 mL |
| 0.5 M EDTA, pH 8.0 | 2 mM | 0.4 mL |
| 10% Triton X-100 | 1% | 10 mL |
| 10% SDS | 0.1% | 1 mL |
| ddH2O | n/a | 83.6 mL |
| Total | n/a | 100 mL |
High Salt Wash Buffer (store at 4°C)
| Reagent | Final Concentration | Amount |
|---|---|---|
| 1 M Tris-HCl, pH 8.0 | 20 mM | 2 mL |
| 5 M NaCl | 500 mM | 10 mL |
| 0.5 M EDTA, pH 8.0 | 2 mM | 0.4 mL |
| 10% Triton X-100 | 1% | 10 mL |
| 10% SDS | 0.1% | 1 mL |
| ddH2O | n/a | 76.6 mL |
| Total | n/a | 100 mL |
LiCl Wash Buffer (store at 4°C)
| Reagent | Final Concentration | Amount |
|---|---|---|
| 1 M Tris-HCl, pH 8.0 | 10 mM | 1 mL |
| 5 M LiCl | 250 mM | 5 mL |
| 0.5 M EDTA, pH 8.0 | 1 mM | 0.2 mL |
| 10% IGEPAL CA-630 | 1% | 10 mL |
| 10% DOC | 0.5% | 5 mL |
| ddH2O | n/a | 78.8 mL |
| Total | n/a | 100 mL |
10 mM Tris-HCl, pH 8.0 (store at 4°C)
| Reagent | Final Concentration | Amount |
|---|---|---|
| 1 M Tris-HCl, pH 8.0 | 10 mM | 1 mL |
| ddH2O | n/a | 99 mL |
| Total | n/a | 100 mL |
1× TE + 50 mM NaCl (store at 4°C)
| Reagent | Final Concentration | Amount |
|---|---|---|
| 1 M Tris-HCl, pH 8.0 | 10 mM | 1 mL |
| 5 M NaCl | 50 mM | 1 mL |
| 0.5 M EDTA, pH 8.0 | 1 mM | 0.2 mL |
| ddH2O | n/a | 97.8 mL |
| Total | n/a | 100 mL |
2× Tagmentation DNA (TD) Buffer (store at −20°C and discard after one or two months)
| Reagent | Final Concentration | Amount |
|---|---|---|
| 1 M Tris-HCl, pH 8.0 | 20 mM | 20 μL |
| 1 M MgCl2 | 10 mM | 10 μL |
| N,N-Dimethylformamide | 20% | 200 μL |
| ddH2O | n/a | 770 μL |
| Total | n/a | 1 mL |
ChIP Elution Buffer (store at room temperature: 20°C–25°C)
| Reagent | Final Concentration | Amount |
|---|---|---|
| 1 M Tris-HCl, pH 8.0 | 50 mM | 50 μL |
| 0.5 M EDTA, pH 8.0 | 10 mM | 20 μL |
| 10% SDS | 1% | 100 μL |
| ddH2O | n/a | 830 μL |
| Total | n/a | 1 mL |
Equipment
Sonicator: Diagenode Bioruptor Pico
Alternatives: Other systems such as Bioruptor Plus, Covaris, and probe sonicators.
Magnet: DynaMag-2
Alternatives: Any magnet with tube racks.
qPCR machine: QuantStudio1 Real-Time PCR Machine.
Alternatives: Any qPCR machine.
Library QC: Agilent Bioanalyzer 2100
Alternatives: Other systems such as Agilent TapeStation, Fragment Analyzer, Caliper LabChip GX.
Step-By-Step Method Details
Fixation of Cells and Binding Antibodies to Dynabeads
This section fixes cultured cells with formaldehyde, which preserves protein-DNA and protein-protein interactions. In addition, antibody-beads complex is prepared for immunoprecipitation. When working with tissues, use an appropriate method to dissociate the tissue into single-cell suspension and start the protocol from step 2b. We normally use 5 × 105 cells for profiling histone modifications and 5 × 106 cells for transcription factors.
-
1.
Add 1/10 volume of 11% formaldehyde solution directly to the culture media in plates. Swirl briefly and incubate at room temperature (20°C–25°C) for 10 min.
-
2.Add 1/10 volume of 1.25 M Glycine, swirl briefly and incubate at room temperature (20°C–25°C) for 5 min to stop formaldehyde crosslinking.
-
a.For adherent cells, remove all liquid in the plate, and rinse with ice-cold 1× PBS (pH 7.4) twice. Collect cells into 1 mL ice-cold 1× PBS (pH 7.4) supplied with 1% Fetal Bovine Serum (FBS) using a cell scraper, and transfer to 1.5 mL Eppendorf tubes. Spin at 4°C for 5 min at 1,000 × g, and discard supernatant.
-
b.For suspension cells:
-
i.Transfer enough cells into either Eppendorf tubes or conical tubes. If no serum is in the culture media, add 1/100 volume of FBS.
-
ii.Spin at 4°C for 5 min at 1,000 × g, and discard supernatant.
-
iii.Resuspend cell pellet with the same amount of ice-cold 1× PBS (pH 7.4), spin at 4°C for 5 min at 1,000 × g, and discard supernatant. Repeat once.
-
i.
-
a.
-
3.
Cell pellets can be snap freezed in liquid nitrogen and store in −80°C for at least 6 months. Or cell pellets can be used immediately. See the next section.
-
4.Bind antibodies to Dynabeads. We use 10 μL Dynabeads and 1 μg antibody per ChIP. Protein A or G Dynabeads, or Pan Mouse IgG Dynabeads are chosen based on the primary antibody being used and the results from the previous section. Prepare each ChIP individually in different Eppendorf tubes.
-
a.Mix 10 μL Dynabeads with 500 μL Blocking Solution in the tube, and collect beads using DynaMag-2. Allow beads to set at the side of the tube. Invert twice or three times to collect beads at the tube cap. No need to centrifuge. Remove the supernatant with an aspirator.
-
b.Add 500 μL Blocking Solution to wash the beads. This can be done by removing the rack from the magnet and inverting the rack with tubes still in place for 20 times or until the beads are evenly distributed in the Blocking Solution.
-
c.Repeat the above wash twice, to reach a total of three washes.
-
d.Resuspend the washed beads in 250 μL Blocking Solution, add 1 μg antibody, and put on a rotator at 4°C for at least 6 h or overnight (12–20 h).
-
a.
Sonication and Immunoprecipitation of Chromatin
This section describes the procedures to solubilize and break chromatin into appropriate size range, and use the specific antibody to immunoprecipitate the DNA bound by the protein of interest.
-
5.
If using the frozen pellet from the steps described above, take the pellet from −80°C and thaw on ice.
-
6.
Resuspend the pellet of appropriate cell numbers in 300 μL Sonication/IP Buffer with freshly added 1× protease inhibitor cocktails, and sonicate on a Bioruptor Pico (or the alternatives) for an appropriate number of cycles based on the results from Before You Begin section.
-
7.Incubate the chromatin with antibody-beads complex:
-
a.Centrifuge the sonicated chromatin at 16,000 × g at 4°C for 10 min.
-
b.During the 10-min centrifugation time, wash the antibody-beads complex from step 4d three times with 500 μL Blocking Solution in the same way as described in steps 4a and 4b.
-
c.Save 2 μL supernatant from step 7a, and store in −20°C as the input sample, and transfer the rest supernatant to the washed antibody-beads complex. Incubate overnight (12–20 h) at 4°C on a rotator.
-
a.
Note: There should be very tiny or no visible pellet after the centrifugation at step 7a.
Wash Beads, Tagmentation on Beads, and Reverse Crosslinking
This section describes the procedures in to wash the immunoprecipitation and add sequencing adapters via tagmentation by Tn5. Then the crosslink is reversed by heating at 65°C. All wash steps are done at the bench with wash buffers kept on ice.
-
8.
Put the immunoprecipitation on DynaMag-2 to collect the beads at the side of the tube. Invert twice or three times to collect beads at the tube cap. No need to centrifuge.
-
9.
Remove the supernatant, and the buffer at the cap using an aspirator or a pipette.
-
10.
Wash once with 500 μL RIPA Wash Buffer. This can be done by removing the tube rack from the magnet, add the buffer and invert by hand with the tube still on the rack for 15–20 times or until the beads are evenly distributed in the buffer.
-
11.
Wash once with 500 μL Low Salt Wash Buffer.
-
12.
Wash once with 500 μL High Salt Wash Buffer.
-
13.
Wash once with 500 μL LiCl Wash buffer.
-
14.
Wash twice with 500 μL 10 mM Tris-HCl, pH 8.0.
Note: The washes from step 11 to 14 are performed in the same way as described in step 10.
-
15.
Collect the beads to the bottom of the tube by a brief centrifugation at 100 × g for 30 s.
-
16.
Put the tube on DynaMag-2 and remove trace of Tris-HCl.
-
17.
Resuspend the beads thoroughly with 30 μL tagmentation mix, which consists of 15 μL 2× TD Buffer + 14 μL ddH2O + 1 μL Tn5. The Tn5 can be from either the Illumina Tagment DNA TDE1 Enzyme and Buffer kit or the Fapon Tnp Library Prep Kit for Illumina. You only need one kit, not both.
-
18.
Take the 2 μL input sample from −20°C, and mix with 30 μL tagmentation mix (the same as above).
-
19.
Put both the IP and input samples on the thermomixer to incubate at 37°C for 5 min with 800 rpm shaking.
Figure 5.

Schematic View of the Tagmentation Process on the Beads
-
20.Stop the tagmentation reaction:
-
a.For input samples, directly add 70 μL ChIP Elution Buffer, add 1 μL of Proteinase K (20 mg/mL) and leave at 65°C on a thermomixer with 1,400 rpm shaking for at least 6 h or overnight (12–20 h).
-
b.For IP samples, wash beads with 500 μL Low Salt Wash Buffer twice, then with 500 μL 1× TE 50 mM NaCl once. Perform the wash in the same way as described in step 10.
-
c.Briefly centrifuge the IP samples to collect beads at the bottom of the tube. Put the tubes to DynaMag-2 and remove trace of 1× TE 50 mM NaCl.
-
d.Add 100 μL ChIP Elution Buffer to the beads, add 1 μL Proteinase K (20 mg/mL) and briefly vortex until the beads become homogeneous.
-
e.Leave all samples at 65°C on a thermomixer with 1,400 rpm shaking for at least 6 h or overnight (12–20 h).
-
a.
DNA Purification and Library Preparation
This section describes the procedures of DNA purification and library preparation. See Figure 6 below for the schematic view of this section with PCR details.
-
21.
Purify DNA from both input and IP samples using the Qiagen minElute PCR Purification Kit according to manufacturer’s instructions. Elute the DNA in 11 μL Elution Buffer from the kit twice, which generally yields 20 μL DNA.
Note: There is no need to quantify the DNA concentration at this stage. Use all for the next step.
-
22.
Setup the library PCR reaction per sample as follows:
| Reagent | Volume |
|---|---|
| Purified DNA from step 21 | 20 μL |
| 10 μM S5xx Primer | 2.5 μL |
| 10 μM N7xx Primer | 2.5 μL |
| NEBNext High-Fidelity 2× PCR Master Mix | 25 μL |
| Total | 50 μL |
-
23.
Run a pre-amplification PCR using the following condition:
| Pre-Amplification PCR Cycling Conditions | |||
|---|---|---|---|
| Steps | Temperature | Time | Cycles |
| Gap fill-in | 72°C | 5 min | 1 |
| Initial Denaturation | 98°C | 1 min | 1 |
| Denaturation | 98°C | 10 s | 4 cycles |
| Annealing | 63°C | 30 s | |
| Extension | 72°C | 20 s | |
| Hold | 10°C | Forever | |
Note: The combination of S5xx and N7xx primers identifies a sample. Therefore, different samples should use different combinations of S5xx and N7xx primers. If you do not have many samples, it is recommended to use different N7xx primers, because the index in the N7xx primer is sequenced first on an Illumina machine.
-
24.
After the pre-amplification, take out 9 μL of the reaction, and mix with 1 μL 10× EvaGreen and perform a qPCR analysis to decide the optimal cycle number. Leave the rest 41 μL reaction on ice.
-
25.
Use the following cycling condition to perform a qPCR analysis, and monitor the amplification curve in linear scale.
| qPCR Cycling Conditions | |||
|---|---|---|---|
| Steps | Temperature | Time | Cycles |
| Initial Denaturation | 98°C | 1 min | 1 |
| Denaturation | 98°C | 10 s | 35 cycles |
| Annealing | 63°C | 30 s | |
| Extension | 72°C | 20 s (acquire data) | |
-
26.
Determine the cycle number N, where the amplification curve reach half way of saturation. In the examples shown in the Figure 7 below, N = 11, 14, and 14 for the three different samples.
Note: The cycle number should be chosen at the exponential phase, before reaching saturation.
-
27.
Once the optimal cycle number is decided, amplify the rest 41 μL reaction for a further of N cycles, using the following condition:
| Post-Amplification PCR Cycling Conditions | |||
|---|---|---|---|
| Steps | Temperature | Time | Cycles |
| Initial Denaturation | 98°C | 1 min | 1 |
| Denaturation | 98°C | 10 s | N cycles |
| Annealing | 63°C | 30 s | |
| Extension | 72°C | 20 s | |
| Hold | 10°C | forever | |
-
28.
Purify the library PCR product using 1.2× beads ratio using AmpureXP for PCR Purification beads or VAHTS DNA Clean Beads, according to manufacturer’s instructions. Elute the library using 30 μL Elution Buffer from the Qiagen minElute PCR Purification kit or 10 mM Tris-HCl, pH 8.0.
Figure 6.

Schematic View of Steps of DNA Purification and Library Purification
Figure 7.

Amplification Plot of qPCR
Expected Outcomes
The quality and quantity of the purified library should be checked by an Agilent Bioanalyzer 2100 machine or the like. We use the Agilent High Sensitivity DNA Kit and follow exactly the steps described in the manufacturer’s manual. Figure 8 shows a few examples of successful libraries and a failed one in different machines. One should expect at least 4 nM at the region between 200 bp and 1,000 bp.
Note: The shape of the size distribution of the library depends on many factors, such as the sonication and the protein of being analyzed. The majority of the DNA should fall between 200 and 1,000 bp. We found the large fragments (>1,000 bp) do not affect quantification or sequencing at all. Therefore, we just leave them as they are. Asterisks indicate primer leftover, which can be removed by a further beads purification if needed.
Figure 8.

Examples of Successful and Failed Libraries on an Agilent Bioanalyzer 2100 and Caliper LabChip GX
Asterisks indicate primer leftover.
If the libraries look good, send for sequencing. We normally perform 50 bp pair end sequencing, but single end sequencing can also be used. We followed the ENCODE ChIP-seq guide (Landt et al., 2012) to sequence at least 20 million reads for each experiment, which is usually enough for point source factors such as many transcription factors. For other factors that have broad binding patterns, such as H3K27me3 and H3K9me3, deeper sequencing is needed.
Quantification and Statistical Analysis
Unfortunately, a successful library preparation does not mean a successful ChIPmentation experiment. Some preliminary computational analysis on the data need to be performed to see if the ChIPmentation experiment is working or not.
Sequence Read Alignment
Use the following command for the read alignment and format conversion:
hisat2 -p {threads} --no-temp-splicesite --no-spliced-alignment -x {genome} -1 {read1.fq.gz} -2 {read2.fq.gz} --summary-file mapping_stats.txt | samtools view -@ {threads} -ShuF 4 -q 30 - | samtools sort - -T {factor}_tmp -o {factor}_q30_sorted.bam
Note: The above command is in one single line. Change {threads} to the number of cores you want to use, {genome} to the hisat2 genome index, {read1.fq.gz} and {read2.fq.gz} to your sequencing read file names, {factor} to the name of the factor that is being investigated. The commands align the reads to the genome, remove un-aligned reads, sort the reads by coordinates and only keep reads with mapping quality higher than 30.
Peak Calling
Take the output bam file from the Sequence Read Alignment step, and use the following command to identify the genomic binding sites of the factor of interest:
macs2 -t {factor}_q30_sorted.bam -c input_q30_sorted.bam -g {genome_size} -f BAMPE -q 0.01 -B --SPMR -n {factor}
Note: The above command is in one single line. Change {factor} to the name of the factor that is being investigated, and {genome_size} to the appropriate genome size or species code. Check the MACS2 manual for more information.
There will be a file in bedGraph format called {factor}_treat_pileup.bdg generated after the MACS2 peak calling. It is recommended to convert it to the bigWig format for the visualization. First, get the chromosome sizes for the genome, using the human genome hg38 as an example:
fetchChromSizes hg38 > hg38.chrom.sizes
Then, use the following command for the conversion:
bdg2bw {factor}_treat_pileup.bdg hg38.chrom.sizes
Note: For more information about different file formats, check the UCSC genome browser documentation: https://www.genome.ucsc.edu/FAQ/FAQformat.html
Assessment of Results
It is difficult to define universal rules to check whether a ChIPmentation experiment works or not. In our experience, the peak file generated during the peak calling process by MACS2 should contain thousands or even tens of thousands of peaks when an experiment is successful. However, some factors may have very few binding peaks. We have found visual inspection of the binding signal in the bigWig file using UCSC genome browser can be very helpful. See Troubleshooting 3.
First, look at the chromosome-wide view of the experiment. For successful experiments, clear “spikes” should be apparently visible to eyes, and there should be many comparing to the input sample. For failed experiments, it is relatively flat. When zooming in into specific target genes, you should see smooth bell-curve shaped peaks. The peak and the background can be easily discriminated by eyes. See examples in Figure 9.
Figure 9.

UCSC Genome Browser Screenshot of ChIPmentation Examples
Both successful (S) and possibly failed (F) experiments are shown. On the left, the entire mouse chromosome 19 is shown. On the right, the locus of the Tbx3 gene is displayed.
Limitations
The nature of ChIPmentation is essentially ChIP-seq. Many limitations that restrict the use of ChIP-seq also apply to ChIPmentation. The technique still requires a ChIP-grade antibody that can recognize and pull down its target protein after formaldehyde crosslinking. In general, finding a ChIP-grade antibody is difficult and time consuming. Alternatively, stable cell line expressing an epitope tagged version of the protein of interest can be generated, and an antibody against the tag can be used for ChIPmentation. For example, the ENCODE project has used the 3xFLAG tag to investigate genomic locations of hundreds of chromatin-associated proteins (Partridge et al., 2020), but this cannot be achieved in primary cells and tissues.
In addition, ChIPmentation only simplifies the procedure and increase the sensitivity of the library preparation steps. It does not change the chromatin immunoprecipitation part of the protocol. Therefore, the cell number required for a ChIPmentation experiment is relatively low comparing to the traditional ChIP-seq methods. In our hands, the minimum cell number for a successful ChIPmentation experiment is 104 for profiling histone modifications and 105 for investigating transcription factors. However, these numbers are still high and prohibit the profiling of rare material. If cell number is very limited, other methods such as uliChIP-seq (Brind’Amour et al., 2015), CUT&RUN (Skene and Henikoff, 2017), STAR ChIP (Zhang et al., 2016), CUT&TAG (Kaya-Okur et al., 2019), ACT-seq (Carter et al., 2019), itChIP-seq (Ai et al., 2019) can be used.
Troubleshooting
Problem 1
Poor sonication results: the majority of fragments are too large (>1 kb), or too small (100–200 bp) or heterogeneous (the presence of both large and small fragments at the same time), or not enough input DNA to visualize on agarose gels due to low number of cells.
Potential Solution
When either large fragments or small fragments are present, adjust the sonication condition accordingly. The most straightforward approach is to change the number of cycles of the sonication, but sometimes, one needs to change the ON/OFF time. For histone modification, small fragments may not be a big problem.
We have found heterogeneous sonication often results from heterogeneous crosslinking. This often happens for cells that grow in colonies (i.e., not monolayer) or for primary cells not properly dissociated from tissues. Optimize your system to get good single-cell suspension first (i.e., use trypsin, collagenase etc.), and start from step 1 of the Step-By-Step Method Details section to crosslink the cells in solution.
If cells number is limited, and not enough DNA is recovered, one can reverse crosslink and purify DNA from the entire sample (instead of taking a fraction out).
Problem 2
Low yield of immunoprecipitated DNA: this can be reflected at the qPCR stage where N (is step 26) is high (>14) or a flat profile on Bioanalyzer or the like.
Potential Solution
It should be noted that high number of cycles does not necessarily mean a failed experiment, but a flat profile indicates the experiment probably failed. This could be due to low abundance of the protein of interest, or low antibody affinity to the protein. Increase the starting number of cells to see if it helps. In addition, trying different antibodies or adding an epitope tag to the protein of interest often help. Commonly-used tags with good antibodies include 3xFLAG, V5, and 3xHA. See the ENCODE ChIP-seq guide (Landt et al., 2012) for more details.
Problem 3
Library preparation is successful, but the sequencing results suggest low signal-to-noise ratio. This is the most frequently encountered problem according to our experience.
Potential Solution
Like suggested in the previous section, a successful library does not necessarily mean a successful ChIPmentation. Include a positive control antibody, such as a transcription factor antibody that has been tested successfully in ChIP-seq/ChIPmentation by neighboring labs to make sure the protocol is working as intended. If the positive control works, but the actual experiment fails, try to change to a different antibody or considering adding tags to the protein of interest.
Another reason could be the protein of interest does not interact with DNA tightly or directly, a dual crosslinking step can be used. Formaldehyde can crosslink both protein-DNA and protein-protein interactions, but it has poor efficiency of crosslinking protein-protein interactions due to its short spacer arm. A protein-protein crosslinker with a longer spacer arm (such as EGS) can be used to secure protein-protein interaction first, then formaldehyde is used to crosslink protein-DNA interaction. Check (Zeng et al., 2006) for details.
Finally, the right crosslinking condition also needs to be tested. If under-crosslinking happens, the protein will not be efficiently crosslinked to DNA and the bound DNA may be lost during the washes. If over-crosslinking happens, the protein epitope will be destroyed and the antibody will not be able to recognize the protein of interest. Both cases result in low signal-to-noise ratio. We suggest choosing a time course of crosslinking and perform the ChIPmentation experiment in parallel to find the condition that gives the best signal-to-noise ratio.
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Xi Chen (chenx9@sustech.edu.cn).
Materials Availability
This study did not generate new unique reagents. All materials used in this study are commercially available, and the detailed information can be found in the Key Resources Table.
Data and Code Availability
This study did not generate new data or code.
Acknowledgments
We thank all members from the Chen lab and the Zhang lab for the help with the experiments. Work in the Zhang lab is supported by grants from National Natural Science Foundation of China (31471389 and 31970812).
Author Contributions
X.C. and W.Z. conceived the project. A.D.S. provided advice and supervised the optimization of the sonication and antibody test. W.X. and Y.Y. carried out the experiments extensively in many different human and mouse cell lines. All authors discussed the potential improvement on optimization and participated in the writing and editing of the manuscript.
Declaration of Interests
The authors declare no competing interests.
Contributor Information
Wensheng Zhang, Email: zhangwensheng@suda.edu.cn.
Xi Chen, Email: chenx9@sustech.edu.cn.
References
- Ai S., Xiong H., Li C.C., Luo Y., Shi Q., Liu Y., Yu X., Li C., He A. Profiling chromatin states using single-cell itChIP-seq. Nat. Cell Biol. 2019;21:1164–1172. doi: 10.1038/s41556-019-0383-5. [DOI] [PubMed] [Google Scholar]
- Brind’Amour J., Liu S., Hudson M., Chen C., Karimi M.M., Lorincz M.C. An ultra-low-input native ChIP-seq protocol for genome-wide profiling of rare cell populations. Nat. Commun. 2015;6:6033. doi: 10.1038/ncomms7033. [DOI] [PubMed] [Google Scholar]
- Carter B., Ku W.L., Kang J.Y., Hu G., Perrie J., Tang Q., Zhao K. Mapping histone modifications in low cell number and single cells using antibody-guided chromatin tagmentation (ACT-seq) Nat. Commun. 2019;10:3747. doi: 10.1038/s41467-019-11559-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S., Zhou Y., Chen Y., Gu J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018;34:i884–i890. doi: 10.1093/bioinformatics/bty560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henriksson J., Chen X., Gomes T., Ullah U., Meyer K.B., Miragaia R., Duddy G., Pramanik J., Yusa K., Lahesmaa R. Genome-wide CRISPR Screens in T Helper Cells Reveal Pervasive Crosstalk between Activation and Differentiation. Cell. 2019;176:882–896.e18. doi: 10.1016/j.cell.2018.11.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaya-Okur H.S., Wu S.J., Codomo C.A., Pledger E.S., Bryson T.D., Henikoff J.G., Ahmad K., Henikoff S. CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat. Commun. 2019;10:1930. doi: 10.1038/s41467-019-09982-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent W.J., Sugnet C.W., Furey T.S., Roskin K.M., Pringle T.H., Zahler A.M., Haussler D. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D., Paggi J.M., Park C., Bennett C., Salzberg S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019;37:907–915. doi: 10.1038/s41587-019-0201-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landt S.G., Marinov G.K., Kundaje A., Kheradpour P., Pauli F., Batzoglou S., Bernstein B.E., Bickel P., Brown J.B., Cayting P. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res. 2012;22:1813–1831. doi: 10.1101/gr.136184.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., Subgroup, 1000 Genome Project Data Processing The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partridge E.C., Chhetri S.B., Prokop J.W., Ramaker R.C., Jansen C.S., Goh S.-T., Mackiewicz M., Newberry K.M., Brandsmeier L.A., Meadows S.K. Occupancy maps of 208 chromatin-associated proteins in one human cell type. Nature. 2020;583:720–728. doi: 10.1038/s41586-020-2023-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidl C., Rendeiro A.F., Sheffield N.C., Bock C. ChIPmentation: fast, robust, low-input ChIP-seq for histones and transcription factors. Nat. Methods. 2015;12:963–965. doi: 10.1038/nmeth.3542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skene P.J., Henikoff S. An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. eLife. 2017;6:e21856. doi: 10.7554/eLife.21856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng P.-Y., Vakoc C.R., Chen Z.-C., Blobel G.A., Berger S.L. In vivo dual cross-linking for identification of indirect DNA-associated proteins by chromatin immunoprecipitation. Biotechniques. 2006;41:694–698. doi: 10.2144/000112297. [DOI] [PubMed] [Google Scholar]
- Zhang B., Zheng H., Huang B., Li W., Xiang Y., Peng X., Ming J., Wu X., Zhang Y., Xu Q. Allelic reprogramming of the histone modification H3K4me3 in early mammalian development. Nature. 2016;537:553–557. doi: 10.1038/nature19361. [DOI] [PubMed] [Google Scholar]
- Zhang W., Chronis C., Chen X., Zhang H., Spalinskas R., Pardo M., Chen L., Wu G., Zhu Z., Yu Y. The BAF and PRC2 complex subunits Dpf2 and Eed antagonistically converge on Tbx3 to control ESC differentiation. Cell Stem Cell. 2019;24:138–152.e8. doi: 10.1016/j.stem.2018.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W. Model-based analysis of ChIP-seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This study did not generate new data or code.