

Abstract
Juvenile polyposis syndrome (JPS) and hereditary haemorrhagic telangiectasia (HHT) are rare autosomal dominant diseases, where symptoms manifest at childhood. A 32-year-old man with no family history of JPS or HHT with SMAD4 gene mutation who developed signs and symptoms only at the age of 32, when he was an adult. In this article, we highlight the steps taken to diagnose this rare pathology, explain its pathophysiology and management.
Keywords: endoscopy, genetics, gastrointestinal surgery
Background
SMAD4 juvenile polyposis syndrome (JPS) with hereditary haemorrhagic telangiectasia (HHT) mutation is a rare disease with a high rate of morbidity. Our article may bring more awareness to this rare pathology and assist clinicians in establishing a diagnosis earlier.
Case presentation
A 32-year-old man presented to a regional general surgical unit for severe symptomatic anaemia. Within a span of 4 months his haemoglobin dropped by 62 points from 137 gL (N: 135–180 gL). His medical history included epilepsy on Valproate, heart arrhythmia on Flecainide, carpal tunnel syndrome and C5 nerve root compression. He is a non-smoker, rare alcohol user, has two dogs at home, and two children. His family history included his grandfather who had bowel cancer diagnosed at 78 years old, and a grandmother with breast cancer.
The first imaging modality was a CT scan showing a large fungating mass in the body of the stomach extending into the pylorus and first part of the duodenum (figure 1). He underwent upper gastrointestinal endoscopy (UGE) to investigate the pathology. Histopathology frequently returned as benign hyperplastic tissue. The first histopathology results reported gastric mucosa with minor foveolar hyperplasia, stromal oedema with minor increase in chronic inflammatory cells. There was no evidence of helicobacter organisms, active gastritis or atypia. Initial scope findings showed carpet-like polyps lining the gastric antrum and body.
Figure 1.

CT abdomen + pelvis + contrast showing a large mass extending from distal stomach to the duodenum.
On a second UGE, tunnel biopsies were performed, showing the carpet-like polyps have progressed into the first part of the duodenum, in comparison to previous UGE (figure 2). The biopsy returned as Helicobacter heilmannii-associated chronic gastritis with hyperplastic changes of fundic gland polyp (figure 3).
Figure 2.

Paris classification 0-Isp carpet-like granular patterned polyp with blood ooze contributing to the patient’s anaemia.
Figure 3.

Histopathology of gastric body showing Helicobacter heilmanii (black arrow) gram-negative spirochete organisms within the gastric pits.
Serum ANA, ANCA, antiparietal cell antibodies, Cytomegalovirus infection serology, hepatitis panel, serum protein electrophoresis, Helicobacter serology and CA 72–4 tumour marker were ordered. Results came back negative except for a positive Helicobacter IgG serology. H. heilmannii was treated with two courses of Helicobacter eradication regime. Repeat UGE, 2 months later, showed no evidence of Helicobacter organism. Histopathology results were hyperplastic gastric polyp with background of mild chronic inflammation. Repeat UGE 3 months after the initial UGE showed Paris classification 0-Isp in the duodenum (figure 4) and the polyps have extended further into the oesophagus (figure 5). An endoscopic mucosal resection was performed on the oesophageal polyp with histopathology returning as juvenile polyp. Narrow-band imaging of a polyp in the gastric body showed irregular microvascular and irregular microsurface pattern (figure 6).1
Figure 4.

Paris classification: 0-Isp in the duodenum (black arrow).
Figure 5.

Paris classification 0-Isp (arrow) found in the upper third of the oesophagus. Endoscopic mucosal resection was performed.
Figure 6.

Narrow-band imaging of a polyp in the gastric body showing irregular microvascular and irregular microsurface pattern, using the vascular plus surface pattern (vs) classification system. Black arrow points to irregular microvascular vessels (tortuous and branched). Yellow arrow is the marginal crypt epithelium (villous structure). Wide open pit is shown by the red arrow.
A colonoscopy revealed two 2–3 mm polyps in the ascending colon and caecum, and one 5 mm semipedunculated polyp in the sigmoid colon. The colonic lesions resembled Kudo’s Pit pattern IIIL (figure 7). Histopathology of caecum and descending colon polyps returned as inflammatory juvenile polyps. Capsule endoscopy revealed scattered 0–1 p small bowel polyps (figures 8 and 9) and areas of telangiectasia (figure 10).
Figure 7.
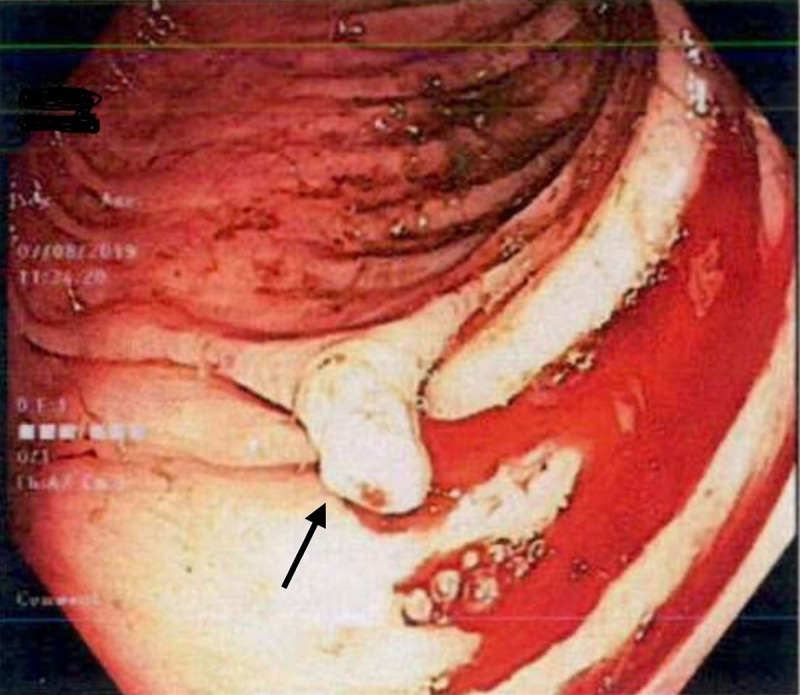
Colonoscopy showing a pedunculated polyp Paris classification 0-Isp in the descending colon. Histopathology reported as inflammatory juvenile polyp.
Figure 8.

Pillcam showing a small bowel polyp, Paris classification 0-Ip.
Figure 9.

Pillcam image showing a duodenal polyp (black arrow).
Figure 10.

Pillcam showing an angioectasia (black arrow) in the small bowel.
Peutz-Jegher’s syndrome and HHT were suspected in the differential diagnoses thus he was referred for genetic screening; where a SMAD4 gene mutation was confirmed. MR bowel (figure 11), MR liver and MR brain were performed which all excluded arteriovenous malformations (AVMs) in the brain and liver. We are currently awaiting report from the pulmonologist to exclude lung AVMs.
Figure 11.

TGFβ/SMAD4 signalling pathway. created with Biorender.com. TGFβ, transforming growth factor-β.
The patient had blood and iron infusion-dependent bleeding resulting in symptomatic anaemia. His polyp burden was massive and could not be managed endoscopically. He underwent a total gastrectomy. Intraoperatively, a Roux-En-Y reconstruction, an oesophageal-jejunostomy, and an access loop was created to assess the duodenal polyps. The histopathology report for the stomach returned as as hyperplastic epithelium with cystically dilated glands, consistent with juvenile polyps with no evidence dysplasia or malignancy. The biopsied duodenal polyp returned as Brunner’s gland hyperplasia. Total gastrectomy itself is a major surgery and has complications associated with it. For our patient, he developed a small bowel obstruction secondary to adhesion 4 months post gastrectomy and underwent a laparotomy with adhesiolysis.
Differential diagnosis
Differential diagnoses included gastric polyps due to valproic acid use, Cronkhite-Canada syndrome and Menetrier’s disease. There has been a case report associating recurrent gastric polyposis in a 56-year-old woman using valproic acid and carbamazepine.2 Cronkhite-Canada syndrome is a rare, non-inherited disorder characterised by numerous hamartomatous polyposis throughout the gastrointestinal (GI) tract including characteristic carpet-like polyps in the stomach and the small intestine.3 Menetrier’s disease is characterised by hypertrophic growth of the gastric fundus mucosa. It was not until we referred our patient to genetic screening that we were able to confirm the SMAD4 genetic mutation, leading us towards the diagnosis of JPS with HHT.
Outcome and follow-up
Our patient’s anaemia was treated with periodic iron transfusions. He underwent a total gastrectomy, complicated by adhesion and had to undergo a laparotomy with adhesiolysis. He has been referred to a pulmonologist to exclude and for surveillance for vascular malformations in the lung. He is scheduled for annual surveillance UGE and colonoscopy.
Discussion
JPS and HHT are two rare and distinct autosomal dominant conditions. However, those with SMAD4 mutations may develop combination of both JPS and HHT.
JPS is a rare, usually autosomal dominant condition that affects about 1 in 100 000–160 000 individuals. It carries a high malignant potential predisposing those affected to colorectal, GI and pancreatic cancers.4 First described in 1964 by Dr McColl,5 signs of anaemia such as pallor, lethargy and shortness of breath with symptoms usually manifesting in young children rather than adults. It is inherited in an autosomal dominant pattern. The risk of those with JPS developing GI cancers is 50%.
Diagnostic criteria of JPS:
At least five juvenile colorectal polyps in the absence of family history of JPS, or
Any juvenile polyposis in other parts of the GI tract, or
Any number of juvenile polyps with a family history of JPS.
HHT or Osler-Weber-Rendu syndrome is an autosomal dominant vascular condition affecting 1 in 5000–10 000 in Europe.6 The condition is characterised by vascular lesions throughout the body including the oral mucosa, liver, lungs and brain. Complications include pulmonary AVMs that may lead to haemothorax. Cerebral manifestations include strokes, cerebral abscess and catastrophic haemorrhages.
The Curaçao diagnostic criteria of HHT, needs to meet three or more :
Recurrent spontaneous epistaxis
Telangiectasias in lips, oral cavity, fingers, nose
Visceral lesions such as GI telangiectasia, pulmonary, cerebral and hepatic AVMs.
Family history of first-degree relative with HHT.
Genetics
Two genes have been implicated in JPS: SMAD4 and Bone Morphogenic Protein 1A (BMPR1A). These two genes are involved in the signalling pathway of transforming growth factor-β (TGFβ), a serine/threonine kinase receptor. In normal epithelium, TGFβ is involved in tumour suppression by regulating the cell cycle, inducing apoptosis and inhibiting angiogenesis of tumour. A significant source of TGFβ secretion comes from fibroblasts. However, in tumour cells, TGFβ can inherit pro-oncogenic properties including aiding cancer cells with metastases and immune system evasion. The mechanism is not clearly elucidated, but research has shown that tumour cells are able to develop mutations resulting in non-responsive TGFβ receptors or mutations in the SMAD signalling pathway.7 8
The SMAD4 gene, previously known as the deleted in pancreatic cancer 4gene as it was found to be mutated in 50% of pancreatic adenocarcinomas. The SMAD family of genes are important in TGFβ signalling pathways. They directly affect transcription TGFβ target genes. SMAD4 is the common mediator of both TGFβ and bone morphogenic protein (BMP) signalling pathway.
TGFβ has two receptors: type I and type II. When TGFβ binds, the two receptors dimerise, and phosphorylate SMAD2 and SMAD3 causing these proteins to bind together. When phosphorylated, the SMAD2/3 complex results in a conformational change allowing it to bind SMAD4. Together, SMAD2/3/4 move into the nucleus to transcribe important downstream cell regulatory proteins (figure 11).
SMAD4 specifically is the common regulator in both TGFβ and BMP signalling. It consists of 12 exons and 10 introns. SMAD4 regulates the G1/S check point by causing the cell cycle to arrest in the G1 phase. The SMAD2/3/4 complex induces apoptosis by encouraging transcription of proapoptotic genes. Mutations in SMAD4 have been found in intrahepatic cholangiocarcinoma, pancreatic adenocarcinoma and colorectal cancer.9–12
A prospective study found patients with JPS with SMAD4 mutations were associated with more polyps in the gastric region, with significantly increased risk of upper GI malignancy, and worse prognosis than those with BMPR1A mutation.13 A small group of those with SMAD4 mutations also develop HHT. Those with SMAD4 mutations can develop HHT, JPS or a combination of both.
This is a unique case of a SMAD4 mutation JPS with HHT presenting in an adult at age 32 with no family history. The decision-making process was difficult because of his lack of a family history, and inconclusive histological results. Differential diagnoses considered included side-effect of valproic acid, Peutz-Jegher’s syndrome, Cronkite Canada syndrome, Osler-Weber Rendu syndrome and Menetrier’s disease.
H. heilmannii is a zoonotic microbe found in dogs. In our case, H. heilmannii was a distractor in the decision-making process.
Given that the majority of JPS is diagnosed in the paediatric population, it is prudent to be cognizant that the SMAD4 JPS–HHT condition may manifest in the adult population, may present in those with no family history and may present at a later age. Wider and earlier recognition of this rare condition and awareness of its sequelae will not only avoid the risks associated with the condition but will also benefit the offspring and future generations. The offspring of the affected parent will have a 50% chance of inheriting the mutated gene. Referral to genetic screening would allow at-risk family members to plan and initiate the process of endoscopic screening. The benefit of early endoscopic screening is allowing for early endoscopic polypectomy which reduces the risk of bleeding, cancer and/or intestinal obstruction.14 It has been shown those with SMAD4 JPS–HHT variants are at an increased risk of early onset colorectal cancer as early as 28 years of age.15
There is a paucity of data regarding surveillance recommendations for JPS with SMAD4 mutation. A few studies have recommended baseline endoscopic screening of small intestine and colon at age 15 or when symptoms first start.16 Another study has recommended initial screening with only colonoscopy at 15–18 years then repeat every 1–2 years after. However, this guideline recommends commencement of UGE at age 25 and repeat every 1–2 years.17 A more updated guideline recommends commencing surveillance with both UGE and colonoscopy at 15 years of age or when symptoms start, if the endoscopy results are normal then repeat surveillance every 2–3 years. If a polypectomy was performed or if polyps were found in initial endoscopies then annual endoscopies should be performed until patient is polyp free. This guideline also recommend MR abdomen every 2–3 years for surveillance of the small bowel.18
In families with JPS/HHT with SMAD4 gene mutation, the affected parent’s offspring should be offered genetic testing before 15 years old as HHT findings begin earlier than polyps. Early surveillance for HHT will be beneficial as complications of HHT include pulmonary, brain and liver AVMs. Surveillance and early detection with CT or MRIs may prevent life-threatening haemorrhage, haemothorax and strokes.
For our patient, a multidisciplinary team involving surgeons, gastroenterologists and geneticists have made the clinical decision that he should have annual gastroscopies and colonoscopies to monitor his polyp burden and to perform polypectomies as needed. Capsule endoscopy will also be performed for him every 2 years. His offspring will have surveillance starting at the age of 15 years old, and earlier if any signs or symptoms present earlier.
Patient’s perspective.
I’ve been feeling very up and down, just always in pain, and different. Eating now I have some pain, very uncomfortable, now I can eat a little bit more now. I constantly have this sickness type/butterfly/quinging type feeling these days. I recently got married, this has past year has been pretty stressful. I worry about whether I will be able to make it back home every time I’m admitted to the hospital. I’m back at work now. I’m relived I finally know what I have, but no one knows what will happen in the future. I hope the kids don’t get it. I feel hopeful in a way, and take each day as it comes. This condition is painful, but I will push through it and try to get on with it.
Learning points.
Juvenile polyposis syndrome (JPS) and hereditary haemorrhagic telangiectasia (HHT) with SMAD4 mutation can present in those with no family history and can be difficult to diagnose.
Given that the majority of JPS is diagnosed in the paediatric population, it is prudent to be cognizant that JPS may also manifest in the adult population and usually present with symptomatic anaemia.
Genetic screening was a crucial stepping stone in helping us confirm our patient’s diagnosis.
Patients with JPS and HHT due to SMAD4 mutation should undergo various imaging modalities such as MRI or CT with contrast to exclude vascular lesions especially in the brain, liver and lung.
Wider and earlier recognition of this rare condition and awareness of its sequelae will not only avoid the risks associated with the condition but will also benefit the offspring and future generations. Earlier recognition allows the commencement of endoscopic surveillance for the affected parent’s offspring. Thus, allowing for earlier endoscopic polypectomies, reducing the polyp burden and potentially reducing the risk of requiring major surgeries including a total gastrectomy or a colectomy.
Footnotes
Contributors: WC wrote the article. CP provided interpretation of endoscopic findings, and insights on pathophysiology. PR provided explanation and description of pathology.
Funding: The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests: None declared.
Patient consent for publication: Obtained.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Muto M, Yao K, Sano Y. Atlas of endoscopy with narrow band imaging. Springer, 2015. [Google Scholar]
- 2.Ozturk O, Koklu S, Koklu H. Recurrent gastric polyposis in a patient using antiepileptic drugs for a long time. Gastroenterol Nurs 2017;40:152–3. 10.1097/SGA.0000000000000281 [DOI] [PubMed] [Google Scholar]
- 3.Faria MAG, Basaglia B, Nogueira VQM, et al. A case of adolescent Cronkhite-Canada syndrome. Gastroenterology Res 2018;11:64–7. 10.14740/gr912w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Coburn MC, Pricolo VE, DeLuca FG, et al. Malignant potential in intestinal juvenile polyposis syndromes. Ann Surg Oncol 1995;2:386–91. 10.1007/BF02306370 [DOI] [PubMed] [Google Scholar]
- 5.McColl I, BUSXEY HJ, VEALE AM, et al. Juvenile polyposis coli. Proc R Soc Med 1964;57:896–7. [PubMed] [Google Scholar]
- 6.Guttmacher AE, Marchuk DA, White RI. Hereditary hemorrhagic telangiectasia. N Engl J Med 1995;333:918–24. 10.1056/NEJM199510053331407 [DOI] [PubMed] [Google Scholar]
- 7.Kubiczkova L, Sedlarikova L, Hajek R, et al. TGF-β - an excellent servant but a bad master. J Transl Med 2012;10:183. 10.1186/1479-5876-10-183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jelsig A, Qvist N, Brusgaard K, et al. Hamartomatous polyposis syndromes: a review. Orphanet J Rare Dis 2014;9:101 10.1186/1750-1172-9-101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stemper TJ, Kent TH, Summers RW. Juvenile polyposis and gastrointestinal carcinoma. A study of a kindred. Ann Intern Med 1975;83:639–46. 10.7326/0003-4819-83-5-639 [DOI] [PubMed] [Google Scholar]
- 10.Howe JR, Ringold JC, Summers RW, et al. A gene for familial juvenile polyposis maps to chromosome 18q21.1. Am J Hum Genet 1998;62:1129–36. 10.1086/301840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Howe JR, Roth S, Ringold JC, et al. Mutations in the Smad4/DPC4 gene in juvenile polyposis. Science 1998;280:1086–8. 10.1126/science.280.5366.1086 [DOI] [PubMed] [Google Scholar]
- 12.Xu X, Ehdaie B, Ohara N, et al. Synergistic action of Smad4 and PTEN in suppressing pancreatic ductal adenocarcinoma formation in mice. Oncogene 2010;29:674–86. 10.1038/onc.2009.375 [DOI] [PubMed] [Google Scholar]
- 13.Aytac E, Sulu B, Heald B, et al. Genotype-defined cancer risk in juvenile polyposis syndrome. Br J Surg 2015;102:114–8. 10.1002/bjs.9693 [DOI] [PubMed] [Google Scholar]
- 14.Larsen Haidle J, Howe JR. Juvenile Polyposis Syndrome : Adam MP, Ardinger HH, Pagon RA, et al., GeneReviews®. Seattle (WA): University of Washington, Seattle, 2003. [Google Scholar]
- 15.Schwenter F, Faughnan ME, Gradinger AB, et al. Juvenile polyposis, hereditary hemorrhagic telangiectasia, and early onset colorectal cancer in patients with Smad4 mutation. J Gastroenterol 2012;47:795–804. 10.1007/s00535-012-0545-8 [DOI] [PubMed] [Google Scholar]
- 16.Howe JR, Ringold JC, Hughes JH, et al. Direct genetic testing for Smad4 mutations in patients at risk for juvenile polyposis. Surgery 1999;126:162–70. 10.1016/S0039-6060(99)70150-9 [DOI] [PubMed] [Google Scholar]
- 17.British Society for Gastroenterology, Association of Coloproctology for Great Britain and Ireland . Guidance on large bowel surveillance for people with two first degree relatives with colorectal cancer or one first degree relative diagnosed with colorectal cancer under 45 years. Gut 2002;51:v17–20. 10.1136/gut.51.suppl_5.v17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guillem JG, Friedman G. Management of hereditary colorectal cancer: a multidisciplinary approach. Cham: Springer International Publishing, 2020. [Google Scholar]