

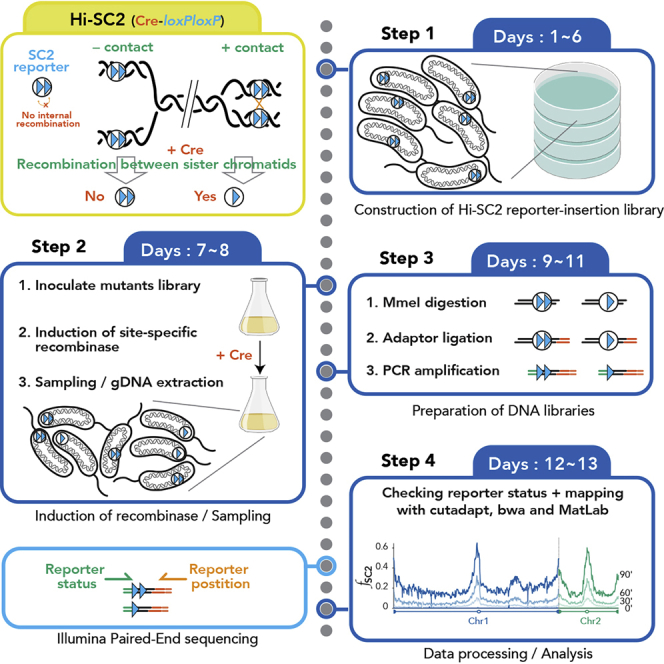
Summary
Sister chromatid interactions are a key step to ensure the successful segregation of sister chromatids after replication. Our knowledge about this phenomenon is mostly based on microscopy approaches, which have some constraints such as resolution limit and the impossibility of studying several genomic positions at the same time. Here, we present a protocol for Hi-SC2, a high-throughput sequencing-based method, to monitor sister chromatid contacts after replication at high resolution throughout the genome, which we applied to study cohesion in Vibrio cholerae.
For complete details on the use and execution of this protocol, please refer to Espinosa et al. (2020).
Subject Areas: Bioinformatics, Genomics, High Throughput Screening, Molecular Biology, Sequence analysis, Sequencing
Graphical Abstract
Highlights
-
•
Method to analyze sister chromatid contacts at high resolution
-
•
Step-by-step protocol from cell culture to bioinformatics analysis
-
•
Protocol easy to adapt to different organisms
Sister chromatid interactions are a key step to ensure the successful segregation of sister chromatids after replication. Our knowledge about this phenomenon is mostly based on microscopy approaches, which have some constraints such as resolution limit and the impossibility of studying several genomic positions at the same time. Here, we present a protocol for Hi-SC2, a high-throughput sequencing-based method, to monitor sister chromatid contacts after replication at high resolution throughout the genome, which we applied to study cohesion in Vibrio cholerae.
Before You Begin
Hi-SC2 requires expression of a site-specific recombinase (e.g., Cre or XerCD) which must be tightly regulated. The recombinase gene is highly recommended to be integrated to the host genome rather than being provided from a plasmid. Here we use as a model an engineered Vibrio cholerae strain containing a tightly regulated Cre expression cassette (PBAD-cre-invPlac; OFF by supplement of 0.1 mM Isopropyl β-D-thiogalactoside (IPTG), ON by supplement of 0.02% L-arabinose (L-ara)). Adequate engineering must be made for your organism of interest. We use a SC2 reporter containing Cre-recombination sites (loxP) in tandem, which is associated with a kanamycin (Km) resistance selection marker on a mini-transposon (Mariner transposon). The transposon is harbored on a conditional replication plasmid carrying a chloramphenicol (Cm) resistance gene. The plasmid is delivered by conjugation using as donor an Escherichia coli strain that cannot grow in the absence of 2,6-Diaminopimelic acid (DAP). Adequate engineering must be made for the delivery of the SC2 reporter in your organism of interest.
CRITICAL: It is not essential to have specific selection marker(s) for the organism of interest which will be recipient of the SC2 reporters. In the example we provide, recipient cells can be differentiated from the donor cells using a medium lacking DAP. However, the selection marker associated to the SC2 reporter should be compatible with the organism of interest. In the example we present, the recipient strain must be Km-sensitive and Cm-sensitive.
Sample Multiplexing
Illumina allows pooling different samples and sequencing multiple libraries at the same time. We recommend such sample multiplexing for two reasons: (1) We usually use ∼20 M reads to proceed informatics analysis for each sample (see below), which does not fit in a MiSeq run but is way below the capacity of NextSeq format. (2) Hi-SC2 libraries have a low initial sequence diversity, which can impede cluster calling process as is the case for Tn-seq (Mitra et al., 2015). To (partially) avoid risk of low-quality data, we use adapters containing different length and “spacer” nucleotide sequences (Table 1). It is important to use different adapters for different samples as much as possible (here are presented eight different adapters). Furthermore, it is required to use sufficient number of P7 NGS primers with different “index” sequences (See Key Resources Table, Oligonucleotides and https://support.illumina.com for detail).
Table 1.
List of Adapter Combinations
| Adapter Number | Oligo Aa | Oligo Ba | P5 NGS Oligob | Adapter Coupled-Spacer Sequencec |
|---|---|---|---|---|
| 1 | 3455 | 3456 | 3457 | CAGT |
| 2 | 3811 | 3812 | 3825 | CGTA |
| 3 | 3813 | 3814 | 3826 | ACAGT |
| 4 | 3815 | 3816 | 3827 | TACTC |
| 5 | 3817 | 3818 | 3828 | CTAGT |
| 6 | 3819 | 3820 | 3829 | TGACTC |
| 7 | 3821 | 3822 | 3830 | ATGCTA |
| 8 | 3823 | 3824 | 3831 | AGCATA |
See above Annealing Adapters.
See steps 24–26.
See step 30a.
Annealing Adapters
Timing: 6 h
-
1.
Prepare 10× Hybridization buffer.
-
2.
Turn ON the heat block and set it to 70°C.
-
3.
Prepare the following mixture:
| Reagent | Amount |
|---|---|
| 10× Hybridization buffer | 1 μL |
| 100 μM Adapter Oligo A | 1 μL |
| 100 μM Adapter Oligo B | 1 μL |
| ddH2O | 7 μL |
| Total | 10 μL |
-
4.
Incubate at 70°C for 5 min.
-
5.
Turn OFF the heat block and let cool down slowly until it reaches 21°C.
-
6.
Make aliquots of 5 μL and store at −20°C.
Note: After adapter annealing, the resulting double-stranded DNA contains two base pairs (NN) overhanging at the 3′end. We recommend preparing a small quantity of annealed adapters to prevent degradation due to several rounds of freezing and thawing and long-term storage at −20°C.
Preparation of Media
Media preparation for the pilot experiment (should be done on day 0)
-
7.
Prepare 4 LB agar plates supplemented with DAP.
-
8.
Prepare 12 LB agar plates supplemented with Km.
-
9.
Prepare 2 LB agar plates supplemented with Cm.
Media preparation for the transposon insertion (should be done on day 4)
-
10.
Prepare 1 LB agar plate supplemented with DAP and IPTG.
-
11.
Prepare 1 LB agar plate supplemented with Km and IPTG.
-
12.
Prepare 4 “large” LB agar plates supplemented with Km and IPTG. Leave them at 21°C until the use next day (dry with the lid closed).
Note: “Large” plates have a diameter of 20 cm and hold 250 mL of LB agar (instead of typical Ø 85 mm with 25 mL). It is advised to prepare the media the day before starting the conjugation, especially for the large plates, so that the plates are completely dry before the experiment.
Alternatives: We spread the SC2 reporter insertion library in four large plates to isolate ∼400,000 colonies without being too confluent. Any other kind of plates can be used (e.g., large square plates or typical Ø 85 mm plates). However, the number of plates and the volume of conjugated cells that has to be spread to each plate must be adjusted accordingly.
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, Peptides, and Recombinant Proteins | ||
| Isopropyl β-D-thiogalactoside | Sigma-Aldrich | Cat# I6758 |
| L-Arabinose | Sigma-Aldrich | Cat# A3256 |
| 2,6-Diaminopimelic acid | Sigma-Aldrich | Cat# D1337 |
| Kanamycin | Sigma-Aldrich | Cat# K4000 |
| Chloramphenicol | Sigma-Aldrich | Cat# C0378 |
| Gentamycin | Sigma-Aldrich | Cat# G1264 |
| Liquid nitrogen | N/A | N/A |
| Dream Taq Polymerase | Thermo Scientific | Cat# EP0713 |
| MmeI | New England Biolabs | Cat# R0637 |
| T4 DNA ligase (2,000,000 units/mL) | New England Biolabs | Cat# M0202M |
| Phusion polymerase | Thermo Scientific | Cat# F532 |
| Critical Commercial Assays | ||
| GenElute Bacterial Genomics DNA kit | Sigma-Aldrich | Cat# NA2110 |
| MinElute PCR purification kit | QIAGEN | Cat# 28004 |
| Pippin Prep Gel Cassette, 1.5% Agarose, dye free, w/ internal standards | SAGE Science | Cat# CDF1510 |
| Pippin Prep Gel Cassette, 2% Agarose, dye free, w/ internal standards | SAGE Science | Cat# CDF2010 |
| Deposited Data | ||
| SC2 reporter Transposon insertion in E. coli | ArrayExpress | E-MTAB-9713 |
| Hi-SC2 codes | Mendeley | https://data.mendeley.com/datasets/2p4w6h56nz/1 |
| Experimental Models | ||
|
Vibrio cholerae N16961 ChapR ΔlacZ PBAD-cre-invPlac Zeo-R, Gm-R |
(Espinosa et al., 2020) | EEV29 |
| Escherichia coli | Laboratory stock | MG1655 |
| Escherichia coli MG1655 RP4-2-Tc::[ΔMu1::aac(3)IV-ΔaphA-Δnic35-ΔMu2::zeo ΔdapA::(erm-pir) ΔrecA | (Ferrières et al., 2010) | MFDpir |
| Escherichia coli (F-) RP4-2-Tc::Mu Δdap Kan | (Demarre et al., 2005) | β2163 |
| Escherichia coli β2163 / pEE22 | (Espinosa et al., 2020) | EE14 |
| Oligonucleotides | ||
| 3459: 5′-TTGGATGATAAGTCCCCGGTC-3′ | This paper | N/A |
| 1514: 5′-TGACGAGTTCTTCTGAGC GGGACTCTGG-3′ |
This paper | N/A |
| Adapter 3455: 5′-TTCCCTACACGACGCTCTTCC GATCTCAGTNN-3′ |
(Espinosa et al., 2020) | N/A |
| Adapter 3456: 5′-ACTGAGATCGGAAGAG CGTCGTGTAGGG-3′ |
(Espinosa et al., 2020) | N/A |
| Adapter 3811: 5′-TTCCCTACACGACGCTCT TCCGATCTCGTANN-3′ |
(Espinosa et al., 2020) | N/A |
| Adapter 3812: 5′-TACGAGATCGGAAGA GCGTCGTGTAGGG-3′ |
(Espinosa et al., 2020) | N/A |
| Adapter 3813: 5′-TTCCCTACACGACGCTCT TCCGATCTACAGTNN-3′ |
(Espinosa et al., 2020) | N/A |
| Adapter 3814: 5′-ACTGTAGATCGGAAGA GCGTCGTGTAGGG-3′ |
(Espinosa et al., 2020) | N/A |
| Adapter 3815: 5′-TTCCCTACACGACGCTC TTCCGATCTTACTCNN-3′ |
(Espinosa et al., 2020) | N/A |
| Adapter 3816: 5′-GAGTAAGATCGGAAG AGCGTCGTGTAGGG-3′ |
(Espinosa et al., 2020) | N/A |
| Adapter 3817: 5′-TTCCCTACACGACGCTCT TCCGATCTCTAGTNN-3′ |
(Espinosa et al., 2020) | N/A |
| Adapter 3818: 5′-ACTAGAGATCGGAAGAG CGTCGTGTAGGG-3′ |
(Espinosa et al., 2020) | N/A |
| Adapter 3819: 5′-TTCCCTACACGACGCTCTTCC GATCTTGACTCNN-3′ |
(Espinosa et al., 2020) | N/A |
| Adapter 3820: 5′-GAGTCAAGATCGGAAGAG CGTCGTGTAGGG-3′ |
(Espinosa et al., 2020) | N/A |
| Adapter 3821: 5′-TTCCCTACACGACGCTC TTCCGATCTATGCTANN-3′ |
(Espinosa et al., 2020) | N/A |
| Adapter 3822: 5′-TAGCATAGATCGGAAG AGCGTCGTGTAGGG-3′ |
(Espinosa et al., 2020) | N/A |
| Adapter 3823: 5′-TTCCCTACACGACGCTCT TCCGATCTAGCATANN-3′ |
(Espinosa et al., 2020) | N/A |
| Adapter 3824: 5′-TATGCTAGATCGGAAGA GCGTCGTGTAGGG-3′ |
(Espinosa et al., 2020) | N/A |
| P5 NGS 3457: 5′-AATGATACGGCGACCACC GAGATCTACACTCTTTCCC TACACGACGCTCTTCCGATCTCAGT-3′ |
(Espinosa et al., 2020) | N/A |
| P5 NGS 3825: 5′-AATGATACGGCGACCACCGA GATCTACACTCTTTCCCTACA CGACGCTCTTCCGATCTCGTA-3′ |
(Espinosa et al., 2020) | N/A |
| P5 NGS 3826: 5′-AATGATACGGCGACCACCGA GATCTACACTCTTTCCCTACA CGACGCTCTTCCGATCTACAGT-3′ |
(Espinosa et al., 2020) | N/A |
| P5 NGS 3827: 5′-AATGATACGGCGACCACC GAGATCTACACTCTTTCCCT ACACGACGCTCTTCCGATCTTACTC-3′ |
(Espinosa et al., 2020) | N/A |
| P5 NGS 3828: 5′-AATGATACGGCGACCACC GAGATCTACACTCTTTCCC TACACGACGCTCTTCCGATCTCTAGT-3′ |
(Espinosa et al., 2020) | N/A |
| P5 NGS 3829: 5′-AATGATACGGCGACCAC CGAGATCTACACTCTTTC CCTACACGACGCTCTTCC GATCTTGACTC-3′ |
(Espinosa et al., 2020) | N/A |
| P5 NGS 3830: 5′-AATGATACGGCGACCACCG AGATCTACACTCTTTCCCTA CACGACGCTCTTCCGATCTATGCTA-3′ |
(Espinosa et al., 2020) | N/A |
| P5 NGS 3831: 5′-AATGATACGGCGACCACCGA GATCTACACTCTTTCCCTACA CGACGCTCTTCCGATCTAGCATA-3′ |
(Espinosa et al., 2020) | N/A |
| P7 NGS∗: 5′-CAAGCAGAAGACG GCATACGAGATxxxxxxGTGACTGGA GTTCAGACGT GTGCTCTTCCGATCTTCTA-3′ |
(Espinosa et al., 2020) | N/A |
| Recombinant DNA | ||
| R6Kori mobRP4cat magellan5-MmeI loxP-loxP (kan) | (Espinosa et al., 2020) | pEE22 |
| Software and Algorithms | ||
| Cutadapt | (Martin, 2011) | https://pypi.org/project/cutadapt/ |
| Samtools | (Li and Durbin, 2010) | http://bio-bwa.sourceforge.net |
| BWA | (Li et al., 2009) | http://www.htslib.org/ |
| MATLAB version R2018b | Mathworks | Mathworks.com |
| Other | ||
| Membrane filter 0.45 μm | Millipore | Cat# HAWP02500 |
| Glass Petri dishes of 20 cm diameter | LABO MODERNE | Cat# FS70517 |
| Pippin Prep | SAGE science | https://www.SAGEscience.com |
| DNA LoBind Tubes | Eppendorf | Cat# 0030108051 |
| Nanodrop | Thermo Scientific | https://www.thermofisher.com |
∗ xxxxxx corresponds six-base sequence index for Illumina. Prepare multiple primers with different barcode accordingly to your project.
Materials and Equipment
M9 Minimal Media
M9 2× Stock
| Reagent | Final Concentration | Amount |
|---|---|---|
| KH2PO4 | 20 mM | 3 g |
| Na2HPO4 | 40 mM | 6 g |
| NaCl | 8 mM | 0.5 g |
| NH4Cl | 18 mM | 1 g |
| ddH2O | N/A | 1 L |
Supplemented M9 for Cultures
| Reagent | Final Concentration | Stock Concentration | Amount |
|---|---|---|---|
| M9 2× | 1× | 2× | 50 mL |
| Thiamine | 1 μg/mL | 5 mg/mL | 20 μL |
| MgSO4 | 1 mM | 1 M | 100 μL |
| CaCl2 | 0.1 mM | 100 mM | 10 μL |
| Fructose | 0.2% | 20% | 1 mL |
| ddH2O | N/A | N/A | 48.87 mL |
| Total | N/A | N/A | 100 mL |
Supplements to Media
| Reagent | Final Concentration | Stock Concentration | Abbreviation |
|---|---|---|---|
| Kanamycin | 50 μg/mL | 50 mg/mL in bidistilled H2O | Km |
| Chloramphenicol | 25 μg/mL | 25 mg/mL in 96%C Ethanol | Cm |
| Gentamycin | 50 μg/mL | 50 mg/mL in bidistilled H2O | Gm |
| 2,6-Diaminopimelic acid | 0.3 mM | 50 mM in bidistilled H2O | DAP |
| L-arabinose | 0.02% | 20% in bidistilled H2O | L-ara |
| Isopropyl β-D-thiogalactoside | 0.1 mM | 1 M in bidistilled H2O | IPTG |
10× Hybridization Buffer
| Reagent | Final Concentration | Stock Concentration | Amount |
|---|---|---|---|
| Tris-HCl pH 7.5 | 100 mM | 1 M | 1 mL |
| NaCl | 500 mM | 5 M | 1 mL |
| EDTA | 10 mM | 0.5 M | 200 μL |
| ddH2O | N/A | N/A | 7.8 mL |
| Total | N/A | N/A | 10 mL |
Step-By-Step Method Details
Pilot Experiment of Transposon Insertion Library: Days 1–3
-
1.
Inoculate a single colony of Escherichia coli EE14 into 5 mL of LB broth containing Cm and DAP and a single colony of V. cholerae EEV29 into 5 mL of LB broth containing Gm. Incubate at 37°C with shaking (180 rpm) for 16 h to make overnight culture.
Note: It is important to start cultures from a single colony (rather than directly from a cryostock) and the colony should not be too old. We recommend isolating colonies in a plate 1 or 2 days before performing the conjugations. Furthermore, it is recommended to keep incubation time (i.e., 16 h) consistent among experiments.
Note: For a standard protocol, cultures are grown at 37°C for 16 h, but depending on the recipient (e.g., thermosensitive mutants), growth conditions can be modified.
Alternatives: Here we use E. coli strain β2163 as the host of SC2 reporter delivery plasmid. Any other E. coli strain that is pir+ and able to mobilize mobRP4 plasmids (e.g., SM10 λ pir) can be used. However, if E. coli is your recipient, it is essential to use MFDpir (Ferrières et al., 2010) for the donor. The use of a Mu+ donor results in disastrously biased insertion of transposon (Figure 1), which makes the Hi-SC2 analysis in vain (Problem 1).
Figure 1.

Distribution of Transposon Insertion in E. coli
Top: biased distribution of Mariner transposon at oriC region in E. coli using β2163 as a donor. Bottom: random distribution of Mariner transposon in E. coli using MFDpir as a donor. Chromosomal domains and genomic positions are represented below. Red and blue peaks correspond to direction of SC2 reporter insertions.
-
2.Carry out conjugations.
-
a.Prepare 4 microcentrifuge tubes (1.5-mL), labeled A-D.
-
i.In the two tubes (A and B), mix 250 μL of donor and 250 μL of recipient cultures, add LB up to a final volume of 1 mL.
-
ii.In the other two tubes (C and D), mix 500 μL of donor and 500 μL of recipient.
-
i.
-
b.Centrifuge at 6,000 × g at 21°C for 3 min then discard the supernatant with a pipette (be careful not to discard part of the pellet).
-
c.To wash the cells, add 1 mL of LB broth to the tube and resuspend the pellet by pipetting. Centrifuge at 6,000 × g at 21°C for 3 min.
-
d.Carefully discard the supernatant with a pipette and resuspend the pellet in the residual medium.
-
e.For each of 4 LB agar plate supplemented with DAP, place one filter membrane at the middle. Carefully put the whole amount of donor-recipient cell suspension (step 2d) at the middle of the membrane. Be careful not moving the plate to avoid cells dripping out the membrane. Once the drops got dry (∼10 min with lid closed), incubate conjugations at 37°C for 2 h (for A and C) and 4 h (for B and D).
-
a.
-
3.
After incubation, by using sterile forceps, take a filter membrane and put into a tube (e.g., 14-mL round-bottom test tube, 15-mL centrifuge tube, or 50-mL centrifuge tube). Add 1 mL of LB broth and mix well by vortex to resuspend the cells.
-
4.
Prepare serial dilutions (10, 100, 1000 times) in LB and spread 100 μL of the dilutions into LB agar plates supplemented with Km. Incubate the plates for 16 h at 37°C.
-
5.
Determine the conjugation condition and number of conjugations to carry out for the following Hi-SC2 insertion library construction.
-
6.
Patch 200 Km-R colonies from corresponding condition to LB plate supplemented with Cm to check Cm-sensitivity.
Note: During the resuspension of donor-recipient mixture after washing (step 2d), it is possible to add a small amount of LB broth, but it should not exceed 50 μL.
Note: The insertion mutants should be Km-resistant (Km-R) and Cm-sensitive (Cm-S). However, Cm-R Km-R colonies can be obtained by adverse events such as plasmid integration to the recipient genome (by homologous recombination). A good library should contain less than 1% of plasmid insertions. However, the presence of 2%–3% of Cm-R colonies can be tolerated for further application.
Note: In case of using an alternative donor host that is dap+ (e.g., SM10 λpir), an appropriate selection pressure for the recipient strain is required in addition to Km.
Table 2 shows an example of the results from a pilot experiment. Among four different conjugation conditions, longer incubation time (B and D) would give more transposon insertions than shorter incubation. However, since cells can grow and divide during the incubation, it does not necessarily come with a higher complexity. Furthermore, doubling the volume of reaction would not result in doubling the number of colonies obtained (compare A and C for example). Therefore, it is better to choose shorter incubation time as long as it is feasible, and carry out multiple conjugation reactions (up to 30 conjugations are very much practical) instead of increasing the reaction volume. In this example, we will use condition C, which require 14 conjugation reactions.
Note: The efficiency of the SC2 reporter insertion changes not only in different species but also with different genetic background. The pilot experiment is strikingly important and must be carried out for each mutant. In some cases for certain species and/or mutants, it might be considered to expand the conjugation parameters such as the volume of donor and recipient mixture, the donor and recipient ratio, as well as the incubation time, for the best possible results (Problem 2).
Table 2.
Example of Pilot Experiment
| Condition | Volume of Cells | Hours of Incubation | Number of Colonies per Conjugation | Number of Conjugations Required |
|---|---|---|---|---|
| A | 250 μL | 2 | 17,000 | 24 |
| B | 250 μL | 4 | 44,000 | 9 |
| C | 500 μL | 2 | 28,000 | 14 |
| D | 500 μL | 4 | 55,000 | 7 |
SC2 Reporter Insertion Library: Days 4–6
-
7.
Inoculate a single colony of E. coli EE14 donor into 5 mL of LB broth containing Cm and DAP and a V. cholerae EEV29 into 5 mL of LB broth containing Gm. Incubate at 37°C with shaking (180 rpm) for 16 h to make overnight culture.
Note: Depending on the conjugation conditions defined by the pilot experiment, prepare multiple number of tubes for the donor and/or recipient (for this example, we prepare two tubes each).
-
8.Carry out conjugations
-
a.Prepare 14 microcentrifuge tubes (1.5 mL). In each tube, mix 500 μL of donor and 500 μL of recipient.
-
b.Centrifuge at 6,000 × g at 21°C for 3 min and discard the supernatant with a pipette (be careful not to discard part of the pellet).
-
c.To wash the cells, add 1 mL of LB broth and resuspend the pellet by pipetting. Centrifuge at 6,000 × g at 21°C for 3 min.
-
d.Carefully discard the supernatant with a pipette and resuspend the pellet in the residual medium.
-
e.Place filter membrane(s) to LB agar plate supplemented with DAP, and carefully put the whole amount of donor-recipient cell suspension (step 8d) on the membrane. Up to five filter membranes can be placed on one LB agar plate, and each membrane can hold maximum three conjugations (Figure 2A). Once drops get dry (∼10 min with lid closed), incubate the plate at 37°C for 2 h.
-
a.
-
9.After incubation, collect all the conjugation reactions into one tube.
-
a.Using sterile forceps, introduce two filters per tube (e.g., 14-mL round-bottom test tube, 15-mL centrifuge tube, or 50-mL centrifuge tube) and add 1 mL of LB broth. Vortex to resuspend the cells.
-
b.Mix all the conjugations in a new tube and add LB broth up to a final volume of 6 mL.
-
a.
-
10.Spread conjugation mix to LB plates supplemented with Km.
-
a.Spread 1:1,000 of the volume (6 μL) on a Ø 85 mm plate to estimate the size of library.
-
b.Spread 1.5 mL per a large plate. Let all the liquid completely absorbed (∼10 min). Repeat with each one of the four large plates.
-
c.Incubate plates for 16 h at 30°C.
-
a.
Note: Cell pellet can be very loose after centrifugation. It is suggested to arrange the number of tubes centrifuged together so that the supernatant can be discarded immediately and without losing cells, no more than six tubes should be centrifuged at the same time.
Note: In case the volume of conjugation mix (step 9b) exceeds 6 mL, or if using Ø 85 mm plates instead of large plates, adjust the number of plates and/or volume to spread on each plate in order to obtain isolated colonies.
Note: We suggest to incubate at 30°C for 16 h to avoid colony-crowding in the morning of the next day (Figure 2B). If the colonies are too small, plates can be incubated for a few hours at 37°C before starting the next step.
-
11.Estimate the size of SC2 reporter insertion library
-
a.Count the number of Km-R colonies formed on the Ø 85 mm plate
-
b.Multiply the number by 1,000, to calculate approximate number of Km-R colonies obtained in total.
-
a.
-
12.Recovery of the SC2 reporter insertion library
-
a.Add 5 mL of LB broth to a large plate and scrape cells over the large plate using a cell spreader. Gather cell suspensions with the cell spreader and by tilting the plate and recover them into a 50-mL centrifuge tube by pipetting. Add another 5 mL of LB broth and repeat the procedure to collect remaining of cells. Recovered cell suspensions into the same centrifuge tube.
-
b.Repeat the same procedure for the rest of the large plates. Recover all cells into the same 50 mL tube. Vortex very well to mix and disaggregate the colonies.
-
c.Centrifuge at 3,200 × g at 21°C for 20 min. Discard the supernatant and resuspend the cells in 10 mL of media.
-
d.Add 3.3 mL of sterile 80% glycerol (v/v, final concentration at 20%). Mix thoroughly and make aliquots of 200 μL (10–20 tubes). Store at −80°C.
-
a.
Note: If the number of colonies on the Ø 85 mm plate (11) is somewhat low, even after a few hours of incubation at 37°C, calculate the coverage of library. If it is less than one time the number of possible insertion sites, do not proceed further steps and repeat from step 7 (or step 1).
Note: In case the SC2 reporter library was prepared in Ø 85 mm plates instead of large plates, scrape the cells twice using 1 mL of LB each time (steps 12a and 12b).
Pause Point: SC2 reporter insertion libraries can be stored for a long term at −80°C. However, freeze-thaw cycles should be avoided.
Figure 2.

SC2 Reporter Library Preparation
(A) Fourteen drops of independent conjugation reactions were placed on five membrane filters without touching each other.
(B) Example plate of almost confluent SC2 insertion library. Right: zoom of a section of the plates showing isolated colonies.
Site-Specific Recombination of SC2 Reporters: Days 7 and 8
-
13.Prepare the cells for the recombination assay
-
a.Take one tube of SC2 reporter insertion library from −80°C stock and completely thaw it on ice.
-
b.Mix 10 μL of cells and 990 μL of minimal media (1:100 dilution). Measure the OD600 nm.
-
c.Prepare 100 mL of minimal media supplemented with IPTG in 500-mL flask. Add cells to make a final OD600 nm of 0.05 (∼108 cells).
-
a.
-
14.
Incubate the cells at 30°C with shaking (180 rpm) until they reach early exponential phase (in the case of V. cholerae, 3.5 h to OD600 nm 0.1).
-
15.
Prepare two 50-mL centrifuge tubes and pour 20-mL of cultures each. Sediment cells at 3,200 × g for 10 min at 21°C and carefully discard the supernatant. Dilute the pellets into 10 mL of minimal medium without IPTG in a 125-mL flask. (Record the OD600 nm).
-
16.Cell sampling
-
a.Restart growing cell at 30°C with shaking (180 rpm).
-
b.After 30 min (t = 0), add 10 μL of 20% L-arabinose (final 0.02% w/v) to one sample to induce expression of the recombinase. Keep the flask shaken at 30°C. For the other flask, measure the OD600 nm and transfer the culture into 15-mL centrifuge tube. Sediment the cells by centrifugation for 10 min at 3,200 × g at RT, followed by discarding the supernatant. Snap freeze the cell pellet in liquid nitrogen and keep it at −80°C until next step.
-
c.For the induced sample, measure OD600 nm after 90 min of induction (t = 90) and transfer the culture into 15-mL centrifuge tube. Prepare the snap-frozen cells as written above.
-
a.
Note: Using the OD600 nm you can estimate the number of generations passed during the incubation/induction. It is also possible to calculate it by determining the number of CFU at each step.
Note: We grow cells in a minimal medium at 30°C. Other growth conditions can be applied, which can result in different overall Hi-SC2 profiles (Espinosa et al., 2020).
Note: As shown in Figure 3, the excision frequency increases as a function of the duration of recombinase induction. Growth conditions for the recombination assay should therefore be adjusted depending on the experiment to avoid saturation in certain locus. It is suggested to carry out time course experiment for the first assay (e.g., 0, 30, 60, 90, 120 min of induction).
Figure 3.

Hi-SC2 Analysis Results in V. cholerae
Frequency of excision of SC2 reporter during a time course. No recombination is observed before induction. Chromosome 1 and 2 are represented in blue and green, respectively. Modified from Espinosa et al. (2020).
-
17.
Thaw the frozen cell pellet and extract gDNA using GenElute Bacterial Genomics DNA kit following the manufacturer’s instructions. Perform two elutions of 200 μL each.
-
18.Check recombination state.
-
a.Carry out PCR for each sample as shown below.
Reagent Amount Purified gDNA (from step 17) 1 μL DreamTaq DNA polymerase buffer 10× 1.5 μL 10 mM each dNTPs mix 0.3 μL 10 μM 1514 primer 0.3 μL 10 μM 3459 primer 0.3 μL DreamTaq DNA polymerase 0.075 μL ddH2O 11.525 μL Total 15 μL -
b.Carry out PCR as following
PCR Cycling Conditions
Steps Temperature Time Cycles Initial Denaturation 98°C 30 s 1 Denaturation 98°C 30 s 30 cycles Annealing 55°C 30 s Extension 72°C 30 s Final Extension 72°C 7 min 1 Hold 15°C Forever -
c.Run 5 μL of the PCR product in a 2% agarose gel. Uninduced sample should have unique amplification with 190 bp while induced samples should rise three species of fragments (136, 190, and 244 bp; Figure 4).
-
a.
Alternatives: Any equivalent DNA polymerases can be used for the PCR (step 18a).
Figure 4.

Recombination Status of Uninduced and Induced Samples
Lane 1: size standard (0.5 μg of 100 GenRuler DNA Ladder, Thermo Scientific). Lane 2: uninduced sample containing one single amplified fragment. Lane 3: induced sample containing three amplified fragments. Sizes are indicated.
Hi-SC2 Sequencing DNA Library Construction: Days 9–11
-
19.
Quantify the gDNA concentrations using Nanodrop. A minimum of 20 ng/μL is suggested
-
20.Digest the gDNA with MmeI.
-
a.Prepare reaction as following.
Reagent Amount 2 μg gDNA (from step 17) X μL CutSmart buffer 10× 15 μL 1:10 diluted SAM 2 μL MmeI (2 U/μL) 3 μL ddH2O 130–X μL Total 150 μL -
b.Incubate at 37°C for 4 h.
-
c.Inactivate MmeI enzyme by incubating at 65°C for 20 min.
-
a.
Note: It is important to calculate the MmeI units required for the reaction. We use 6 Units of MmeI as V. cholerae genome contains 2,983 sites. The number of units needed should be adjusted according to the number of MmeI restriction sites in the genome. To calculate the number of units needed we calculated the number of MmeI sites present in 1 μg of ɸX174 which corresponds to the number of sites digested by 1 unit of enzyme. We then calculated the number of MmeI sites in 2 μg of gDNA of V. cholerae to estimate the number of MmeI units needed for restriction digestion.
Note: SAM (S-adenosyl methionine) is thermosensitive. It is recommended to store 1× SAM in aliquots to avoid thawing and freezing several times. 1:10 dilution has to be prepared on ice just before each experiment.
Optional: After step 20c, run 2 μL of purified gDNA and 5 μL of MmeI-digested DNA in 0.8% agarose gel to check the digestion. MmeI digestion should give smear (Figure 5).
Figure 5.

MmeI gDNA Digestion
Lane 1: size standard (0.5 μg of 1 kb DNA Ladder, New England Biolabs). Lanes 2 and 3: uninduced sample. Lanes 4 and 5: induced sample. (-) Not digested gDNA, (+) digested gDNA.
-
21.
Purify and concentrate the DNA with QIAGEN MinElute PURIFICATION KIT. Follow the manufacturer’s instructions, except performing two times for the elution step with 16 μL of Buffer EB (total elution volume ∼30 μL).
-
22.Size-select for the DNA fragments containing SC2 reporter by Pippin Prep.
-
a.Set a 1.5% agarose-Dye free cassette in Pippin Prep, and load the samples accordingly to the manufacturer’s instructions. Program a protocol with “Range” collection mode from (start) 800 to (end) 2,000 bp.
-
b.Recover the samples (∼40 μL) into 1.5 mL LoBind tubes.
-
a.
-
23.
Quantify the DNA concentration using Nanodrop. A minimum of 17 ng/μL is suggested.
-
24.Ligate the double-stranded adapters to the MmeI-digested sample.
-
a.Prepare the following reaction
Reagent Amount 400 ng purified DNA (from step 22) X μL T4 ligase buffer 10× 3 μL Adaptor 0.52 μL High conc. T4 Ligase (2,000,000 U/mL) 2 μL ddH2O 24.48–X μL Total 30 μL -
b.Incubate at 16°C for 16 h.
-
a.
Note: From now on, it is recommended to use low DNA-binding microcentrifuge tubes (e.g., LoBind) when manipulating the DNA for the sequencing library.
Figure 6.

Scheme of Sequencing PCR Products
Not recombined (A) and recombined (B) status are illustrated. Annealing sites for P5 (Adaptor) and P7 (Rd2SP) are shown in orange and green, respectively. Nucleotide sequences important for analysis are indicated. Ns represent the Reporter position. R1 (green) and R2 (orange) lines refer to the sequencing products from P5 and P7, respectively. Binding sites for sequencing primers P5 and P7 are indicated (black arrows).
-
25.
Purify and concentrate the ligated DNA with QIAGEN MinElute PURIFICATION KIT (QIAGEN). Follow the manufacturer’s instructions, except elution with 11 μL of Buffer EB.
-
26.PCR amplification of Illumina libraries.
-
a.Prepare 3 tubes (reactions) of PCR amplification for each sample.
Reagent Amount Ligated DNA (from step 25) 3 μL Phusion DNA polymerase buffer 5× 10 μL 10 mM each dNTPs mix 1 μL 2 μM P5 primer (see Table 1) 5 μL 2 μM P7 primer 5 μL Pushion DNA polymerase 0.5 μL ddH2O 25.5 μL Total 50 μL -
b.Carry out PCR as following.
PCR Cycling Conditions
Steps Temperature Time Cycles Initial Denaturation 98°C 30 s 1 Denaturation 98°C 30 s 17 cycles Annealing 65°C 30 s Extension 72°C 30 s Final Extension 72°C 7 min 1 Hold 15°C Forever -
c.Run 5 μL of the PCR reaction in a 2% agarose gel to check amplification (Figure 7A).
-
a.
-
27.
Mix the three replicates of the PCR reaction and purify using MinElute PURIFICATION KIT. Follow the manufacturer’s instructions, except performing two times for the elution step with 16 μL of Buffer EB (total elution volume ∼30 μL).
-
28.Size-select the PCR products using Pippin Prep.
-
a.Set a 2% agarose-Dye free cassette in Pippin Prep and load the samples accordingly to the manufacturer’s instructions. Program a protocol with “Range” collection mode from (start) 160 to (end) 400 bp.
-
b.Recover the samples (∼40 μL) into 1.5 mL LoBind tubes.
-
a.
-
29.
Check the quality of size-selected DNA by running 5 μL in a 2% Agarose gel (Figure 7B).
Note: It is recommended to perform a pilot PCR amplification with only one tube (instead of three). A good sample should have a single band of 207 bp for uninduced control and 153 and 207 bp amplified bands for induced samples (Figure 7). In case of a low amount of amplification, see Problem 4. In Figure 3, we show the products obtained after 30 cycles of PCR, which permits to detect the recombined products on an agarose gel even if there are in low quantity. In Figure 7, we show the products obtained after 17 cycles of PCR. In that case, recombined products are fainter and more difficult to distinguish in an agarose gel.
Alternatives: A quality check of the library DNA (step 29) can be done with a BioAnalyzer (Aligent Technologies). The quality check can be performed by the next generation sequencing facility.
Figure 7.

Sequencing PCR Products
Hi-SC2 DNA libraries after PCR (A) and purification (B) step. Lane 1: size standard (0.5 μg of 100 GenRuler DNA Ladder). Lane 2: uninduced sample containing one amplified fragment of 207 bp. Lane 3: induced sample containing two amplified fragments of 153 and 207 bp. Smear above 1 kb correspond to the template. Fragments below 100 bp correspond to P5 and P7 primers.
Illumina Sequencing
We outsource Illumina sequencing to next generation sequencing facility. Typically, we use a Nextseq platform with 2 × 75 bp read length, which allows a total of 300–400 M reads per a flow cell. We aim to get ∼20 M reads for each sample. To do so, quantification of the library DNA should be done with Qubit (Thermo Scientific).
Note: The number of reads required for the analysis varies depending on the genome size.
Hi-SC2 Analysis: Days 12 and 13
This section shows how to analyze Illumina sequencing results.
Note: As all paired-end Illumina sequencing, two sequence files in .fastq format will be obtained. The file labeled with “R1” corresponds reporter position while the other file with “R2” contains the reporter status (Figure 6). Since the two files are coupled, they must be treated together.
Note: Here results files are indicated as samplename_R1.fastq and samplename_R2.fastq. In our case P5 and P7 sequences are already removed by the next generation sequencing facility. If not, you need to remove these sequences before succeeding the following steps.
-
30.Extract reporter information by Cutadapt
-
a.Remove the spacer sequence of the adapters. Command:
- cutadapt -g ˆ[spacersequence] -G NNNNNNN --discard-untrimmed -e 0.2 -o samplename_trim1R1.fastq -p samplename_trim1R2.fastq samplename_R1.fastq samplename_R2.fastq
- [spacersequence] corresponds to the specific sequence of each adapter couple (see Table 1)
-
b.Extracting the reporter position information from trim1 files. Command:
- cutadapt -a ACAGGTTGGATGATA -A NNNNNNN --discard-untrimmed -e 0.2 -o samplename_totR1.fastq -p samplename_totR2.fastq samplename_trim1R1.fastq samplename_trim1R2.fastq
-
c.Collecting the reporter position information from those with “recombined” status. Command:
- cutadapt -a ACAGGTTGGATGATA -A TATTCTAGA --discard-untrimmed -e 0.2 -o samplename_recR1.fastq -p samplename_recR2.fastq samplename_trim1R1.fastq samplename_trim1R2.fastq
-
d.From the files samplename_totR1.fastq and samplename_recR1.fastq, remove sequences shorter than 14 bp and longer than 24 bp. Command:
- cutadapt -m 14 -M 24 -o samplename_totR1.fastq samplename_totR1.fastq
- Repeat with the rec file.
-
e.Remove low-quality reads. Command:
- cutadapt -q 35 -o samplename_tot.fastq samplename_totR1.fastq
- Repeat with the rec file.
-
a.
Note: Two resulting files, samplename_tot.fastq and samplename_rec.fastq will be used for further steps. The former represents the reporter position information no matter how the recombination status is for each reporter, and the later contains the reporter position of “recombined” status. Other intermediate files can be discarded (do not discard raw sequencing results files).
-
31.
Using a text edit software, create a reference genome file in multi-FASTA format. Assemble all the chromosome and/or plasmid sequences in the genome in one file with different entries (e.g., > chr1, > chr2 …). Save as refgenome.fa
Note: It is suggested to include the SC2 reporter delivering plasmid in the genome file. It will allow you to detect how often pEE22 was integrated into the host genome and how much those non-informative reads were present in the library. pEE22 was deposited in Addgene.
-
32.Align treated sequence results with the reference genome using BWA.
-
a.Command:
- bwa index refgenome.fa
- bwa aln refgenome.fa samplename_tot.fastq > samplename_tot.sai
- bwa samse refgenome.fa samplename_tot.sai samplename_tot.fastq > samplename_tot.sam
-
b.Repeat step a with the rec file.
-
a.
From now on, the analysis will be performed in MATLAB. The scripts (available at Mendeley, see Key Resources Table) have to be kept in the same folder.
-
33.Extract SC2 reporter insertion site and direction information. Resulting MATLAB file (cell lists) composed of matrices, each of them corresponds to chromosome or plasmid. Similar to .wig file, number of rows equals to the size of chromosome or plasmid.
-
a.Launch MATLAB.
-
b.“Add path” or navigate to the folder containing the scripts.
-
c.In the Command Window, type: sam2wig
-
d.Select the .sam files (samplename_tot.sam and samplename_rec.sam). Multiple .sam files can be processed at the same time.
-
e.New files (e.g., samplename_tot_wig.mat and samplename_rec_wig.mat) will appear in the folder with .sam files.
-
a.
Note: During this process, the script takes into account the reads that map only once in the genome and those mapped more than once in the genome are disregarded.
-
34.Combine two matrices for total reads and recombined reads into one workspace.
-
a.In the Command Window, type:
- combineSC2(‘samplename’)
-
b.Select paired files: samplename_tot_wig.mat and samplename_rec_wig.mat
-
c.Once files are loaded, metadata table will appear in the Command Window. You can register accordingly. Entries include:
-
i.“SSR system” refers to the Site-Specific Recombination system used for the experiment. The default is Cre-loxP.
-
ii.“Growth medium” refers to the growth conditions used in the recombination assay. The default is MM for minimal media.
-
iii.“Temperature” refers to the growth conditions used for the assay. The default is 30.
-
iv.“Drugs” refers to the additions of antibiotics or any other chemical compound supplemented during the experiment. The default is no drugs.
-
v.“Timepoint” refers to the period of time the recombinase was induced. The default is 0.
-
vi.“Inducer” refers to concentration of L-ara used for the induction. The default is ara0.02.
-
i.
-
a.
-
35.Create a workspace “strainname.mat,” which contains the genome information as well as Hi-SC2 data mapped to the reference genome.
-
a.Create a GenBank file of each chromosome. Save as “strainname_chr1_cds.txt.”
-
b.Create a nucleotide sequencing file of each chromosome. You can use .fasta file but the header line (with “>”) must be removed. Save as “strainname_chrX_seq.txt.”
-
c.Repeat steps a and b with all the chromosomes. No matter the replicon is chromosome or plasmid, use chr and numerical order (e.g., chr1, chr2, chr3…)
-
d.In the Command Window, type:
- CreateGenomeFile
-
e.A table will appear in the Command Window. You register as follows:
-
i.“Genome file name” corresponds to the name of strain.
-
ii.“Features” include Hi-SC2 data (shown as SC2 because MATLAB does not allow to use hyphen), CDS information and any other features/genomic region of interest (see Note). Entries should be separated by comma (,).
-
i.
-
f.Select the file(s) “strainname_chrX_seq.txt.” Select all applicable files at the same time.
-
g.Select the file(s) “strainname_chrX_cds.txt.” Select all applicable files at the same time.
-
h.Select the combined Hi-SC2 file created above at step 34.
-
a.
Note: Hi-SC2 plotting function (see below) also allows highlighting certain genomic regions in the figure to facilitate your analysis and/or presentation. To do so, such region(s) of interest must be registered before the step 35d. In the CreateGenomeFile.m, there is a section called “%%Get Feature Data” (line 82–98). You can follow the instructions written in the code.
Note: CreateGenomeFile script also allows to add new Hi-SC2 data in a genome file already generated. To do so, use “strainname.mat” instead of “strainname” at the step 35e i. and only type “SC2” in the Features box at the step 35e ii.
-
36.Analyze and plot Hi-SC2 data.
-
a.In the Command Window, type:
- Plot_HiSC2
-
b.A box will appear in the Command Window.
-
i.“Resolution SC2” defines the size of sliding window for the plotting. The number corresponds to the size of window (bp) from the middle point. For example, the default number 5,000 corresponds to a sliding window of 10 kb.
-
ii.“Data” refers to the Hi-SC2 result(s) to plot. Enter “strainname” followed by “samplename” separated by a comma. To plot multiple Hi-SC2 results at the same time, enter “strainname, samplename” pairs separated by a semicolon.
-
i.
-
c.A box called “Other features to display and figure properties” will appear in the Command Window. In case of adding features in X-axis, each feature must be registered in a fashion with [Feature name, marker style, marker size]. Different features must be separated by semicolon.
-
d.Two different figures will appear:
-
i.“Recombination frequency” Figure shows Hi-SC2 results along the genome.
-
ii.“Comparative Tn hits” Figure shows insertion profile of Hi-SC2 reporter.
-
i.
-
a.
Note: To plot several Hi-SC2 results together, all the samples must be aligned with the same reference genome.
Expected Outcomes
In this protocol, we have described a new methodology to measure sister-chromatid contacts after replication. This method relies on the combination of two very well-established techniques, Tn-seq analysis (van Opijnen et al., 2009) and site-specific recombination assays (Lesterlin et al., 2012).
Concerning the SC2 reporter insertion library, a random distribution along the genome is essential to obtain reliable results in Hi-SC2 (Figure 2 in (Espinosa et al., 2020)). For Hi-SC2 analysis, a tight regulation of recombinase expression is a key step in the experiment. The recombination frequencies of non-induced sample should be almost zero throughout the genome, whereas they should increase progressively over the time of induction (Figure 3).
Limitations
This technique has been developed to study sister-chromatid contacts, and this protocol uses V. cholerae as the organism of interest along with Mariner transposon-based SC2 reporter which targets 5′-TA-3′ dinucleotides. Although we have tested only a few γ-proteobacteria species, Hi-SC2 is applicable to other bacteria and eukaryotic cells. Due to the nature of Mariner transposon, sufficient SC2 reporter insertion throughout the genome might be limited in bacteria with extremely high GC content. It is possible to engineer SC2 reporter with different transposon such as Tn5 or Mu. Furthermore, for eukaryote species, it is possible to use LTR-retrotransposons or gamma-retroviruses for the delivery of SC2 reporter.
The Hi-SC2 method is based on the even insertion of a transposon in the genome of the studied organism. Insertions cannot be obtained into essential genes, impeding the analysis of the recombination frequency at a locus resolution at those positions. However, the analysis of the variations around those positions can be performed using a sliding window. For V. cholerae, we typically used a 10 kb resolution window.
Troubleshooting
Problem 1
SC2 reporter insertion exhibiting not even but extremely enriched at certain region in the chromosome.
Potential Solution
It is especially important to obtain a random distribution of the SC2 reporter insertion in the genome. Before Hi-SC2 analysis, distribution of the SC2 reporter can be analyzed using Artemis (Carver et al., 2012). In case of biased insertions of the SC2 reporter in the genome, a different donor must be used (Figure 1).
Problem 2
Low SC2 reporter insertion.
Potential Solution
Several mutants exhibit slower growth rate or require special growth conditions, e.g., thermosensitive strains. If this is the case, the number of SC2 reporter insertions can be increased by changing conditions during conjugation. Performing a pilot experiment modifying the ratio of donor and recipient cells, the incubation time, or the growth temperature helps to decide the best conditions for conjugation.
Problem 3
Recombination in the non-induced sample (step 18).
Potential Solution
Addition of 0.1 mM IPTG efficiently represses the leaky expression of the arabinose promoter. Recombination in the not induced sample suggests that the PBAD promoter was not completely repressed during the SC2 reporter insertion library preparation. A new conjugation (steps 8–10) should be performed.
Problem 4
Low PCR product (step 26).
Potential Solution
In case of low amount of PCR products, there are several options to increase the quantity of amplified DNA, we suggest to carry out them in the following order: (1) increase the number of PCR reactions, (2) increase the amount of DNA template, (3) increase the number of cycles (up to 19). Also, repetition from the ligation step can be performed (step 24).
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, François Xavier Barre (francois-xavier.barre@i2bc.paris-saclay.fr).
Materials Availability
pEE22 plasmid generated in this study has been deposited in Addgene.
Data and Code Availability
E. coli transposon insertion data are available in Array Express Database (https://www.ebi.ac.uk/arrayexpress/). Codes generated during this study are deposited in Mendeley Database (https://data.mendeley.com/).
Acknowledgments
This research was supported by the ERC program FP7/2007-2013, grant number 28159 (to F-X.B.) and the French National Research Agency, grant numbers ANR-16-CE12-0030-0 and ANR-18-CE12-0012-03 (to F-X.B.), grant number ANR-18-CE35-0008 (to Y.Y.). We thank the high-throughput facility of the I2BC and Jihane Challita, Adrien Camus, and Sandra Daniel for suggestions on the manuscript.
Author Contributions
Conceptualization, F-X.B. and E.E.; Investigation, Y.Y. and E.E.; Formal Analysis, Y.Y., E.E., and F-X.B.; Software, F-X.B.; Writing – Original Draft, Y.Y. and E.E.; Writing – Review & Editing, Y.Y., E.E., and F-X.B.; Funding Acquisition, Y.Y. and F-X.B.
Declaration of Interests
The authors declare no competing interests.
Contributor Information
Elena Espinosa, Email: elena.espinosa@i2bc.paris-saclay.fr.
François-Xavier Barre, Email: francois-xavier.barre@i2bc.paris-saclay.fr.
References
- Carver T., Harris S.R., Berriman M., Parkhill J., McQuillan J.A. Artemis: an integrated platform for visualization and analysis of high-throughput sequence-based experimental data. Bioinformatics. 2012;28:464–469. doi: 10.1093/bioinformatics/btr703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demarre G., Guérout A.-M., Matsumoto-Mashimo C., Rowe-Magnus D.A., Marlière P., Mazel D. A new family of mobilizable suicide plasmids based on broad host range R388 plasmid (IncW) and RP4 plasmid (IncPalpha) conjugative machineries and their cognate Escherichia coli host strains. Res. Microbiol. 2005;156:245–255. doi: 10.1016/j.resmic.2004.09.007. [DOI] [PubMed] [Google Scholar]
- Espinosa E., Paly E., Barre F.-X. High-Resolution Whole-Genome Analysis of Sister-Chromatid Contacts. Mol. Cell. 2020;79:857–869. doi: 10.1016/j.molcel.2020.06.033. [DOI] [PubMed] [Google Scholar]
- Ferrières L., Hémery G., Nham T., Guérout A.-M., Mazel D., Beloin C., Ghigo J.-M. Silent mischief: bacteriophage mu insertions contaminate products of Escherichia coli random mutagenesis performed using suicidal transposon delivery plasmids mobilized by broad-host-range RP4 conjugative machinery. J. Bacteriol. 2010;192:6418–6427. doi: 10.1128/JB.00621-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesterlin C., Gigant E., Boccard F., Espéli O. Sister chromatid interactions in bacteria revealed by a site-specific recombination assay. EMBO J. 2012;31:3468–3479. doi: 10.1038/emboj.2012.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26:589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011;17:10. [Google Scholar]
- Mitra A., Skrzypczak M., Ginalski K., Rowicka M. Strategies for achieving high sequencing accuracy for low diversity samples and avoiding sample bleeding using illumina platform. PLoS One. 2015;10:e0120520. doi: 10.1371/journal.pone.0120520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Opijnen T., Bodi K.L., Camilli A. Tn-seq: high-throughput parallel sequencing for fitness and genetic interaction studies in microorganisms. Nat. Methods. 2009;6:767–772. doi: 10.1038/nmeth.1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
E. coli transposon insertion data are available in Array Express Database (https://www.ebi.ac.uk/arrayexpress/). Codes generated during this study are deposited in Mendeley Database (https://data.mendeley.com/).