

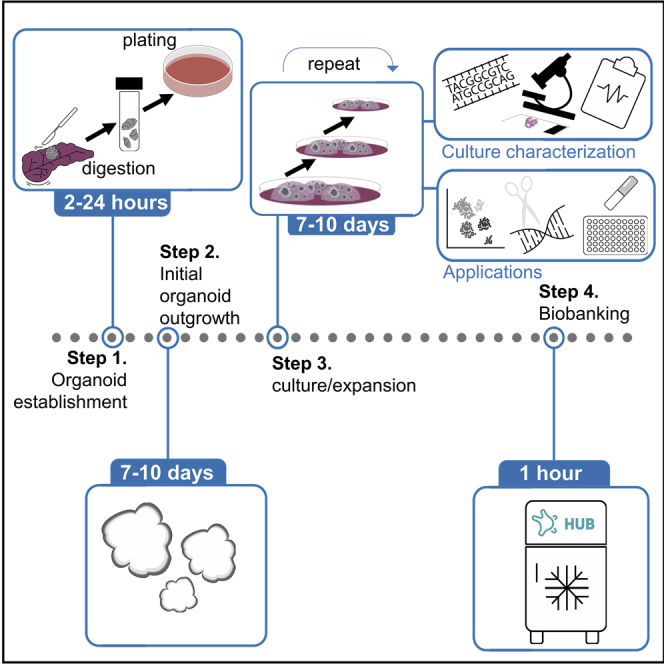
Summary
Establishment of patient-derived adult stem cell-based pancreatic (tumor) organoids was first described in 2015. Since then, multiple laboratories have demonstrated the robustness of this method. We recently described the generation of a pancreatic cancer biobank containing 30 well-characterized tumor organoid models. Here, we describe the applied methods in detail. Use of tumor-selective media prevents contamination with normal cells. Generated organoids can be cryopreserved and can serve as a living biobank of pancreatic cancer for biomarker identification and drug screening.
For complete information on the generation and use of this protocol, please refer to Driehuis et al. (2019).
Subject Areas: Cancer, Organoids, Stem Cells
Graphical Abstract
Highlights
-
•
Protocol to establish pancreatic patient-derived organoids from resections and biopsies
-
•
Detailed procedure to generate and expand both normal and tumor organoid models
-
•
Methods for freezing and thawing organoids to successfully build living biobanks
Establishment of patient-derived adult stem cell-based pancreatic (tumor) organoids was first described in 2015. Since then, multiple laboratories have demonstrated the robustness of this method. We recently described the generation of a pancreatic cancer biobank containing 30 well-characterized tumor organoid models. Here, we describe the applied methods in detail. Use of tumor-selective media prevents contamination with normal cells. Generated organoids can be cryopreserved and can serve as a living biobank of pancreatic cancer for biomarker identification and drug screening.
Before You Begin
Preparation of Organoid Media
Timing: 60 min
For pancreatic organoids, the medium used for culturing differs depending on the origin of the organoids. Organoids established from normal (non-tumor) tissue are grown on a complete medium containing all growth factors known to enhance pancreatic organoid growth (Boj et al., 2015). This medium is called pancreatic normal medium. To grow tumor organoids, a medium lacking specific growth factors present in the pancreatic normal medium is used. These growth factor withdrawals are based on molecular alterations (e.g., caused by genetic driver mutations) known to occur in pancreatic cancer. For example, a high percentage of pancreatic adenocarcinomas is known to harbor a mutation in KRAS (Waters and Der, 2018), resulting in ligand-independent signaling of the EGFR pathways. Therefore, the EGFR ligand EGF can be withdrawn from the culture medium for tumor, but not for normal cells. Another example is the withdrawal of Wnt-ligand. Although the molecular alterations responsible are unknown, it has been described that pancreatic ductal adenocarcinomas are Wnt-independent.
As it is not known beforehand which genetic alterations are present in the tumor that is brought into culture, we recommend the use of two-tumor medium, each lacking different growth factors. As such, the chances that a tumor organoid culture can be established are increased, as both tumor mediums apply a different selective pressure to the plated primary cells. After our initial publication on pancreatic organoids, others establishing pancreas tumor organoids have used comparable, but slightly different culture medium. Importantly, these research groups reported comparable outgrowth efficiencies as us. This suggests that, using these tumor-selective mediums, we do not miss a specific subset of tumors.
To prepare the different medium, a basic medium can be made, from which both of the pancreatic tumor and normal mediums can be made. The medium table in the section ‘Materials and Equipment’ provides an overview of the composition of the different medium.
Note: once prepared, 1× pancreatic normal (or tumor) medium can be used for 10 days when stored at 4°C. Do not prepare more medium than required, to prevent wasting of expensive growth factors. Pancreatic basic medium can be prepared in bulk and stored 4 months at −20°C.
-
1.Pancreatic basic medium (2×)
-
a.Thaw the required B27, noggin, and gastrin aliquots on ice.
-
b.To make 1 L of pancreatic basic medium, add 900 mL Ad-DF+++ to a sterile beaker.
-
c.To the Ad-DF+++, add 40 mL Noggin UPE (final concentration 4%). When using recombinant Noggin, add 2 mL of the 100 μg/mL stock to the Ad-DF+++ (final concentration 200 ng/mL).
-
d.Add 40 mL B27 50× (final concentration 2×).
-
e.Add 0.41 g powder N-Ac (final concentration 2.5 mM) to 15 mL plastic tube containing 5 mL of distilled water, filter the solution using a 0.22 μm filter and add it to the Ad-DF+++.
-
f.Add 2.44 g powder Nic (final concentration 20 mM) to 20 mL of sterile PBS0 in a 100 mL bottle, filter the solution using a 0.22 μm filter and add it into the beaker using a 50 mL sterile plastic pipette.
-
g.Add 200 μL Gastrin 100 μM (final concentration 20 nM).
-
h.Mix carefully by stirring in a circular motion with a 50 mL serological pipette.
-
a.
-
i.Aliquot the pancreatic basic medium in tubes or flasks, considering that the basic medium will be diluted once with its own volume to generate the media used for organoid culture. Therefore, use tubes/flasks that are large enough to allow this.
-
j.Store the aliquots at −20°C until use.
-
j.
-
2.Pancreatic normal medium (1×)
-
a.Thaw a bottle of 50 mL pancreatic basic medium 12–16 h at 4°C or several hours at room temperature (between 18°C and 22°C).Note: do not thaw the basic medium aliquot in the 37°C water bath.
-
b.Thaw the required EGF, FGF10, Primocin, and Rspo-3 aliquots on ice. Thaw the required PGE2 and A83-01 aliquots at room temperature (between 18°C and 22°C).Note: The following amounts are calculated for making 100 mL of pancreatic normal medium (starting with 48.7 mL of pancreatic basic medium).
-
c.Add 50 mL Wnt3a CM to the 50 mL of thawed pancreatic basic medium.
-
d.Add the required amount of Rspo-3 stock (final concentration 250 ng/mL) to the medium.
-
e.Add 100 μL Primocin 50 mg/mL (final concentration 50 μg/mL) to the medium.
-
f.Add 100 μL EGF 50 μg/mL (final concentration 50 ng/mL) to the medium.
-
g.Add 100 μL FGF10 100 μg/mL (final concentration 100 ng/mL) to the medium.
-
h.Add 10 μL PGE2 10 mM (final concentration 1 μM) to the medium.
-
i.Add 10 μL A83-01 5 mM (final concentration 500 nM) to the medium.
-
j.Mix reagents well by pipetting up and down. Store the medium at 4°C, for a maximum of 10 days.
-
a.
-
3.Pancreatic tumor medium 1(1x)
-
a.Thaw a bottle of pancreatic basic medium for 12–16 h at 4°C or several hours at room temperature (between 18°C and 22°C).Note: do not thaw the pancreatic basic medium aliquot in the 37°C water bath.
-
b.Thaw the required FGF10, Primocin, and Rspo-3 aliquots on ice. Thaw the required A83-01 aliquots at room temperature. (between 19°C and 22°C).Note: The following amounts are calculated for making 100 mL of Pancreatic tumor medium 1 (starting with 48.7 mL of Pancreatic basic medium)
-
c.Add 50 mL Wnt3a CM to the 50 mL of thawed Pancreatic basic medium.
-
d.Add the required amount of Rspo-3 stock (final concentration 250 ng/mL) to the medium.
-
e.Add 100 μL Primocin 50 mg/mL (final concentration 50 μg/mL) to the medium.
-
f.Add 100 μL FGF10 100 μg/mL (final concentration 100 ng/mL) to the medium.
-
g.Add 10 μL A83-01 5 mM (final concentration 500 nM) to the medium.
-
h.Mix reagents well by pipetting up and down. Store the medium at 4°C, for a maximum of 10 days.
-
a.
-
4.Pancreatic tumor medium 2 (1x)
-
a.Thaw a bottle of pancreatic basic medium for 12–16 h at 4°C or several hours at room temperature (between 18°C and 22°C).Note: do not thaw the pancreatic basic medium aliquot in the 37°C water bath.
-
b.Thaw the required EGF, FGF10, Primocin, and Rspo-3 aliquots on ice.Note: the following amounts are calculated for making 100 mL of pancreatic tumor medium 2 (starting with 48.7 mL of pancreatic basic medium)
-
c.Add 50 mL Ad-DF+++ to the 50 mL of thawed pancreatic basic medium.
-
d.Add the required amount of Rspo-3 stock (final concentration 250 ng/mL) to the medium.
-
e.Add 100 μL Primocin 50 mg/mL (final concentration 50 μg/mL) to the medium.
-
f.Add 100 μL EGF 50 μg/mL (final concentration 50 ng/mL) to the medium.
-
g.Add 100 μL FGF10 100 μg/mL (final concentration 100 ng/mL) to the medium.
-
h.Mix reagents well by pipetting up and down. Store the medium at 4°C, for a maximum of 10 days.
-
a.
Note: Growth factors need to be aliquoted and stored under the required conditions.
Note: For reagent preparation, DPBS (without Ca2+ and Mg2+) is used and it will be referred through the document as PBS0.
Note: If required, media can be prepared in advance. Keep in mind that, once prepared, full media (containing all the grow factors required) can be stored for a maximum of 10 days at 4°C.
CRITICAL: Cell culture plates need to be pre-warmed at 37°C for 12–16 h (as a minimum, but preferably several days), before they can be used to culture organoids. This will prevent the flatness of the Matrigel/BME domes where the organoids are embedded.
Note: Matrigel/BME needs to be thawed for a minimum of 12–16 h at 4°C and kept on ice for > 30 min before it can be used to culture organoids. Matrigel or BME can both be used to establish and expand pancreatic organoids. In our hands, both extracellular matrices work equally well.
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological Samples | ||
| Human patient material | University Medical Center Utrecht | n/a |
| Chemicals, Peptides, and Recombinant Proteins | ||
| A83-01 | Tocris Bioscience | Cat#2939-10mg |
| Advanced DMEM/F12 | Thermo Fisher | Cat#12634028 |
| B27 supplement (50×), serum free | Thermo Fisher | Cat#17504044 |
| BME, Cultrex RGF Basement Membrane Extract, Type R1 | R&D Systems | Cat#3433-005-R1 |
| BSA, bovine serum albumin | Sigma-Aldrich | Cat#A7030 |
| Collagenase type II from Clostridium histolyticum | Thermo Fisher | Cat#17101015 |
| Dimethyl sulfoxide | Millipore | Cat#1.01900.1000 |
| Dispase II | Sigma-Aldrich | Cat#D4693 |
| Distilled water | Gibco | Cat#15230089 |
| DMEM Glutamax | Thermo Fisher | Cat#31966047 |
| DPBS, no calcium, no magnesium (PBS0) | Thermo Fisher | Cat# 14190250 |
| EGF, recombinant human EGF | Peprotech | Cat#AF-100-15 |
| Fetal bovine serum | Sigma-Aldrich | Cat#F7524 |
| FGF10, recombinant human FGF10 | Peprotech | Cat#100-26 |
| Gastrin, recombinant human gastrin (Leu-15) | Sigma-Aldrich | Cat#G9145 |
| GlutaMAX Supplement | Thermo Fisher | Cat#35050038 |
| Glycerol | Sigma-Aldrich | Cat# G2025-100mL |
| 10× HBSS +calcium/magnesium | Thermo Fisher | Cat#14065049 |
| HEPES (1 M) | Thermo Fisher | Cat#15630056 |
| Matrigel, GFR, phenol free | Corning | Cat#356231 |
| N-Acetyl cysteine | Sigma-Aldrich | Cat#A9165-25G |
| Nicotinamide, cell culture tested, insect cell culture tested | Sigma-Aldrich | Cat# N0636-100G |
| Noggin-Fc Fusion Protein conditioned medium | U-protein express | Cat#N002 |
| Recombinant Human Noggin | Peprotech | Cat#120-10C |
| Noggin UPE | U-Protein Expressed BV | Cat# N002 |
| Penicillin-streptomycin (10,000 U/mL) | Thermo Fisher | Cat#15140122 |
| PGE2, prostaglandin E2 | R&D Systems | Cat#2296-10 mg |
| Primocin | Invivogen | Cat#ANT-PM-2 |
| Recovery cell culture freezing medium | Thermo Fisher | Cat#12648010 |
| Recombinant human R-spondin 3 | R&D Systems | Cat#3500-RS/CF |
| TrypLE Express Enzyme (1×), phenol red | Thermo Fisher | Cat#12605036 |
| Trypsin inhibitor from Glycine max (soybean) | Sigma-Aldrich | Cat# T6522-100 mg |
| Wnt surrogate | U-Protein Expressed BV | Cat# N001 |
| Y-27632 dihydrochloride (ROCK inhibitor) | Abmole | Cat#M1817-100MG |
| Critical Commercial Assays | ||
| OpenArray Barcode Panel 60 SNP Fingerprinting | Executed by Utrecht Sequencing Facility | See http://useq.nl/snp-fingerprinting/ |
| Other | ||
| CELLSTAR, tube, 15 mL | Greiner | Cat#188271 |
| CELLSTAR, tube, 50 mL | Greiner | Cat#227261 |
| CELLSTAR, plate, 6-well | Greiner | Cat#657160 |
| CELLSTAR, plate, 12-well | Greiner | Cat#665180 |
| CELLSTAR, plate, 24-well | Greiner | Cat#662160 |
| CELLSTAR, plate, 48-well | Greiner | Cat#677180 |
| CELLSTAR, plate, 96-well | Greiner | Cat#655180 |
| Coolcell, Corning Coolcell freezer container | Sigma-Aldrich | Cat#CLS432000 |
| Cryovials 1 mL | Greiner | Cat#123263 |
| 0.22 μm pore size sterile Millex syringe filter | Sigma-Aldrich | Cat#SLGL0250S |
| Pipette, 10 mL, graduated 1/10 mL, sterile | Greiner | Cat#607180 |
| Pipette, 25 mL, graduated 2/10 mL, sterile | Greiner | Cat#760180 |
| Pipette, 5 mL, graduated 1/10 mL, sterile | Greiner | Cat#606160 |
| Pipette, 2 mL, graduated 1/100 mL, sterile | Greiner | Cat#710160 |
| Pipette tips, filter, 1,000 μL | Greiner | Cat#740288 |
| Pipette tips, filter, 200 μL | Greiner | Cat#739288 |
| Pipette tips, filter 20 μL | Greiner | Cat#774288 |
| Pipette tips, filter 10 μL | Greiner | Cat#771288 |
| Low retention filter tips, 300 μL, sapphire | Greiner | Cat#738265 |
| Low retention filter tips, 1,250 μL, sapphire | Greiner | Cat#750265 |
| Low retention filter tips, 20 μL, sapphire | Greiner | Cat#773265 |
| Glass Pasteur pipettes, 150 mm | VWR | Cat#612-1701 |
Materials and Equipment
Solutions/media used:
| Ad-DF+++ | Final Concentration | Volume (mL) |
|---|---|---|
| Ad. DMEM/F12 | not applicable | 485 |
| GlutaMAX Supplement (200 mM) | 2 mM | 5 |
| HEPES (1 M) | 10 mM | 5 |
| Pen/strep (10,000 units/mL) | 100 units/mL | 5 |
| Total | 500 |
Once prepared, stored at 4°C for 4 weeks
| D-BSA | Final Concentration | Volume (mL) |
|---|---|---|
| DMEM GlutaMAX | not applicable | 490 |
| BSA (10%) | 0.1% | 5 |
| Pen/Strep (10,000 units/mL) | 100 units/mL | 5 |
| Total | 500 |
Once prepared, stored at 4°C for 4 weeks
| Digestion Buffer for Normal Tissue | Final Concentration | Volume (μL) |
|---|---|---|
| 1× HBSS | 1× | 4,490 |
| Collagenase type II (20 mg/mL) | 1 mg/mL | 250 |
| ROCK inhibitor (Y-27632) (10 mM) | 10 μM | 5 |
| Trypsin inhibitor (100 mg/mL) | 100 μg/mL | 5 |
| Glycerol (100%) | 5% | 250 |
| Total | 5,000 |
Once prepared, stored at 4°C for 1 week
| Digestion Buffer for Tumor Tissue | Final Concentration | Volume (μL) |
|---|---|---|
| Ad-DF+++ | not applicable | 3,745 |
| Collagenase type II (20 mg/mL) | 5 mg/mL | 1,250 |
| ROCK inhibitor (Y-27632) (10 mM) | 10 μM | 5 |
| Total | 5,000 |
Once prepared, stored at 4°C for 1 week
| Pancreatic Basic Medium (2×) | Final Concentration | Volume (mL) |
|---|---|---|
| Ad-DF+++ | not applicable | 920∗ |
| B27 (50×) | 2× | 40 |
| ∗UPE Noggin (100%) or recombinant Noggin (100 μg/mL) | 4% UPE Noggin or 200 ng/mL recombinant Noggin | 40 or 2, respectively |
| N-Acetyl Cysteine | 2.5 mM | 0.41 gram dissolved in 5 mL H20 |
| Nicotinamide | 20 mM | 2.44 gram dissolved in 5 mL H20 |
| Gastrin (100 μM) | 20 nM | 0.2 |
| Total | 1,000 |
∗assuming UPE Noggin is used. If recombinant Noggin is used, supplement with an additional 38 mL of Ad-DF+++.
Once prepared, aliquot 100 mL in plastic 250 mL plastic bottles and stored at −20°C for 3 months
| Pancreatic Normal Medium (1x) | Final Concentration | Volume |
|---|---|---|
| Pancreatic basic medium | 1× | 50 mL |
| ∗Wnt CM or Wnt surrogate | 0.5× or 0.5 nM∗ | 50 mL or X μL∗ |
| Recombinant R-spondin 3 (conc. varies depending on the stock) | 250 ng/mL | Varies depending on the stock |
| Primocin (50 mg/mL) | 50 μg/mL | 100 μL |
| EGF (50 μg/mL) | 50 ng/mL | 100 μL |
| FGF10 (100 μg/mL) | 100 ng/mL | 100 μL |
| PGE2 (10 mM) | 1 μM | 10 μL |
| A83-01 (5 mM) | 500 nM | 10 μL |
| Total | 100 mL |
∗If Wnt surrogate is used as a Wnt source instead of Wnt CM, supplement with an additional 50 mL of Ad-DF+++ to reach the total volume of 100 mL pancreatic normal medium.
Once prepared, stored at 4°C for 1 week
| Pancreatic Tumor Medium 1 (1x) | Final Concentration | Volume |
|---|---|---|
| Pancreatic basic medium | 1 | 50 mL |
| ∗Wnt CM or Wnt surrogate | 0.5× or 0.5 nM∗ | 50 mL or X μL∗ |
| Recombinant R-spondin 3 (conc. varies depending on the stock) | 250 ng/mL | Varies depending on the stock |
| Primocin (50 mg/mL) | 50 μg/mL | 100 μL |
| FGF10 (100 μg/mL) | 100 ng/mL | 100 μL |
| A83-01 (5 mM) | 500 nM | 10 μL |
| Total | 100 mL |
∗Wnt surrogate has varying concentrations depending on the batch. Therefore, the final volume that is required to be added differs between different batches used. If Wnt surrogate is used as a Wnt source instead of Wnt CM, supplement with an additional 50 mL of Ad-DF+++ to reach the total volume of 100 mL pancreatic tumor medium 1.
Once prepared, stored at 4°C for 10 days
| Pancreatic Tumor Medium 2 (1x) | Final Concentration | Volume |
|---|---|---|
| Pancreatic basic medium | 1× | 50 mL |
| Ad-DF+++ | not applicable | 50 mL |
| Recombinant R-spondin 3 (conc. varies depending on the stock) | 250 ng/mL | Varies depending on the stock |
| Primocin (50 mg/mL) | 50 μg/mL | 100 μL |
| EGF (50 μg/mL) | 50 ng/mL | 100 μL |
| FGF10 (100 μg/mL) | 100 ng/mL | 100 μL |
| Total | 100 mL |
Once prepared, stored at 4°C for 10 days
Growth Factor and Solution Preparations
Matrigel or BME
Matrigel or BME can both be used to establish and expand pancreatic organoids. Thaw a −20°C stored bottle of Matrigel or BME for a minimum of 12–16 h at 4°C on ice. Resuspend to homogenize the extracellular matrix solution before use. When purchasing a batch, ensure protein concentration is between 9.5 and 12 mg/mL for Matrigel and >10 mg/mL for BME, respectively, with an endotoxin level equal or lower than 1.5.
PBS0 0.1% BSA for Making Stock Solutions
-
•
To 40 mg BSA, add 40 mL PBS0.
-
•
Mix until is dissolved.
-
•
Filter the solution through a 0.22 μm syringe filter and collect in a sterile 50 mL plastic tube.
-
•
Aliquot and store at −20°C. Can be stored for 2 years at −20°C.
A83-01 (25 mM and 5 mM Stocks)
-
•
Add 0.95 mL DMSO to the bottle containing 10 mg A83-01.
-
•
Mix by pipetting up and down until the powder is dissolved.
-
•
Aliquot 100 μL in dark 1.5 mL micro tubes.
-
•
Store aliquots at −20°C (stable for 6 months).
-
•
For a 5 mM working stock, thaw an aliquot of 100 μL at 25 mM and add 400 μL of DMSO.
-
•
Pipette up and down and aliquot. Store aliquots at −20°C for 3 months.
B27 Supplement (50×)
-
•
Bottles of 100 or 10 mL are stored at −20°C.
-
•
Thaw for a minimum of 12–16 h at 4°C.
-
•
Pipette up and down for resuspending the solution. Make sure there is no precipitate.
-
•
In case of not using all, store at −20°C. Avoid freezing-thawing cycles. If needed, aliquot 1 mL using 1.5 mL dark micro tubes. Expiration date is provided by the supplier. B27 is stable for a year at −20°C.
BSA (10%)
-
•
Transfer 20 g BSA to a 250 mL bottle.
-
•
Add 200 mL PBS0.
-
•
Mix until dissolved on the mixer.
-
•
Filter it through a 500 mL 0.22 μm unit filter bottle.
-
•
Make aliquots of 5 mL in 15 mL plastic tubes. Store at 4°C, stable for 1 year.
Collagenase Type II from Clostridium histolyticum (20 mg/mL)
-
•
Transfer 1 g of Collagenase type II to a 100 mL bottle.
-
•
Add 50 mL Ad-DF+++ and mix.
-
•
Collect the solution with a 50 mL syringe and filter it through a 0.22 μm filter into a sterile 50 mL plastic tube.
-
•
Aliquot 1 mL in 1.5 mL dark micro tubes. Store at −20°C, stable for 3 months.
Dispase II
-
•
Resuspend the powder in 0.1% BSA in PBS0 to reach 25 U/mL.
-
•
Mix by pipetting up and down until dissolved.
-
•
Aliquot 2 mL in 5 mL tubes. Store at −20°C, stable for 1 year or stable for a week at 4°C.
EGF (500 μg/mL and 50 μg/mL)
-
•
For 500 μg/mL stock, add 2 mL 0.1% BSA in PBS0 to a bottle with 1 mg EGF.
-
•
Mix by pipetting up and down until dissolved.
Note: Do not vortex.
-
•
Aliquot 100 μL in 1.5 mL sterile tubes. Store at −20°C, stable for a year.
-
•
For 50 μg/mL working stock, add 900 μL 0.1% BSA in PBS0 to one aliquot with 100 μL at 500 μg/mL stock and mix by pipetting up and down.
Note: Do not vortex.
-
•
Aliquot 50 μL in 1.5 mL sterile tubes. Store at −20°C, stable for 1 year or stable for a week at 4°C.
FGF10 (100 μg/mL and 10 μg/mL)
-
•
For 100 μg/mL stock, add 1 mL of PBS0 0.1% BSA to a bottle with 100 μg FGF10.
-
•
Mix by pipetting up and down until dissolved.
Note: Do not vortex.
-
•
Aliquot 50 μL in 1.5 mL sterile tubes.
-
•
For 10 μg/mL solution, add 450 μL PBS0 0.1% BSA to one aliquot with 50 μL at 100 μg/mL and mix by pipetting up and down.
Note: Do not vortex.
-
•
Aliquot 50 μL in 1.5 mL sterile tubes. Store at −20°C, stable for 3 months or stable for a week at 4°C.
FBS (Fetal Bovine Serum)
-
•
Thaw the 500 mL FBS bottles in the 37°C water bath.
-
•
Mix very well to resuspend possible precipitations.
-
•
Aliquot 30 mL in sterile 50 mL plastic tubes.
-
•
Store at −20°C, stable for 3 years.
Gastrin (100 μM)
-
•
To a bottle containing 1 mg of Gastrin, add 1 mL of PBS0.
-
•
Mix by pipetting up and down until dissolved.
Note: Do not vortex.
-
•
Transfer content to a 15 mL plastic tube containing 3.77 mL PBS0.
-
•
Mix by pipetting up and down until dissolved.
-
•
Aliquot 200 μL in 1.5 mL sterile tubes. Store at −20°C, stable for 6 months.
GlutaMAX Supplement (100× Concentrated)
-
•
Before aliquoting, pipette up and down to resuspend. Make sure there are no precipitates.
-
•
Aliquot 5 mL into sterile 15 mL plastic tubes. Store at 4°C, expiration date is provided by the supplier.
HEPES (1 M)
-
•
Before aliquoting, pipette up and down to resuspend. Make sure there are no precipitates.
-
•
Aliquot 5 mL into sterile 15 mL plastic tubes. Store at 4°C, expiration date is provided by the supplier.
N-Acetyl Cysteine (1.25 MM)
-
•
N-Ac is prepared fresh when making medium. Weigh 0.82 g N-Ac in a 50 mL plastic tube.
-
•
Add 10 mL of distilled water.
-
•
Mix and place incubate at 37°C until dissolved.
-
•
For sterilizing, collect the solution with a 30 mL syringe and filter it through a 0.22 μm filter into a sterile 15 mL plastic tube. Aliquot the reagent in 1.5 mL sterile tubes. Store at −20°C, stable for 3 months.
Note: When using small amounts make more N-Ac solution than required, since the dead volume in the 0.22 μm filter will result in loss of the solution.
Nicotinamide (10 mM)
-
•
Weigh 4.88 g Nicotinamide (Nic) in a 50 mL plastic tube.
-
•
Add 40 mL of PBS0.
-
•
Mix and place incubate at 37°C until dissolved.
-
•
For sterilizing, collect the solution with a 50 mL syringe and filter it through a 0.22 μm filter into a new 50 mL plastic tube. Aliquot the reagent in 1.5 mL sterile tubes. Store at −20°C, stable for 3 months.
Note: When using small amounts make more Nic solution than required, since the dead volume in the 0.22 μm filter will result in loss of the solution.
U-protein Express Noggin (Noggin UPE)
-
•
Thaw the Noggin UPE (stored at −20°C) for a minimum of 12–16 h at 4°C.
-
•
Pipette up and down to resuspend. Make sure there is no precipitate left.
-
•
Use directly or aliquot and store at −20°C. Stable for one year when stored at −20°C.
Noggin Conditioned Medium (CM)
Noggin CM is produced as previously described (Farin et al., 2012).
-
•
Noggin CM collected from separate plates should be pooled and mixed, to assure similar activity for all aliquots.
-
•
Aliquot 20–50 mL (depending on usage) in 50 mL plastic tubes. Store at −20°C. Stable for one year when stored at −20°C.
-
•
When needed, thaw an aliquot in a 37°C water bath. Remove from water bath when a small piece of ice is left.
-
•
Pipette up and down with a 10 mL serological pipette. Use what is required. Remaining thawed Noggin CM can be stored at 4°C for 1 week.
Recombinant Noggin (100 μg/mL Stock)
-
•
Centrifuge the 1 mL tube containing the lyophilized Noggin to assure all product is collected at the bottom of the tube.
-
•
Depending on the amount of lyophilized Noggin, add the required amount of PBS0 0.1% BSA to reach a final concentration of 100 μg/mL. Use or aliquot and store at −80°C. Aliquots can be stored for 6 months at −80°C.
Pen/Strep (10,000 units/mL)
-
•
Thaw Pen/Strep bottle at room temperature (between 18°C and 22°C).
-
•
Pipette up and down to resuspend. Make sure there is no precipitate left. Aliquot 5 mL into sterile 15 mL plastic tubes. Store at −20°C, expiration date is indicated by supplier.
Primocin (50 mg/mL)
-
•
Thaw a Primocin bottle stored at −20°C and thaw at room temperature (between 18°C and 22°C).
-
•
Pipette up and down to resuspend. Make sure there is no precipitate left. Aliquot 100 μL in 1.5 mL sterile tubes. Store at −20°C, expiration date is indicated by supplier.
PGE2 (Prostaglandin E2) (10 mM)
-
•
Add 2.84 mL DMSO to PGE2 bottle containing 10 mg.
-
•
Mix until dissolved. Aliquot 50 μL in 1.5 mL dark micro tubes. Store at −20°C, stable for 3 months.
ROCK Inhibitor (10 mM)
-
•
To a bottle containing 10 mg of ROCK inhibitor, add 312 μL distilled water.
-
•
Mix by pipetting up and down until dissolved. Aliquot 100 μL in 1.5 mL sterile tubes. Store at −20°C, stable for 6 months.
-
•
To make a 10 mM working solution, thaw one tube with 100 μL at 100 mM and add 900 μL of distilled water. Aliquot 50 μL in twenty 1.5 mL sterile tubes. Store at −20°C, stable for 3 months.
Recombinant Rspo3
Each batch R-spondin-3 has different concentration. This information is on the bottle.
-
•
Thaw a bottle (stored at −80°C) for a minimum of 12–16 h at 4°C on ice.
-
•
Prepare aliquots fitting to the batches of organoid medium aliquots that will be used. Store at −80°C, stable for one year after arrival (in original tube), 3 months at −80°C after aliquoting, and one month at 4°C.
Wnt3a CM
Wnt3a CM is produced as previously described (Boj et al., 2017)
Note: Wnt3a CM activity should be measured/tested before using any batch of CM by performing a TOP-luciferase based assay as previously described (Boj et al., 2017).
-
•
Pool the different batches of Wnt3a CM in a sterile 10 L beaker and make 100 mL aliquots in sterile 100 mL bottles. Store at 4°C, stable for 6 months.
Next-Generation Wnt Surrogate (NGS)
Note: Each batch Wnt surrogate has a different concentration and volume. NGS Wnt surrogate has been previously described ((Miao et al., 2020))
-
•
In the product information sheet, the molecular weight of the surrogate is indicated as 56.6 kids, but molarity is calculated with 113.2 kDa, since surrogate is active as a dimer.
-
•
Calculate the volume required to generate a working stock of 250 nM.
-
•
Dilute with Ad-DF+++ to generate a 250 nM stock. Store at −80°C, stable for 1 year. Undiluted Wnt surrogate is stable for one month at 4°C.
A detailed list of reagents and material, including company and reference number is provided as Key Resources Table.
Step-By-Step Method Details
Processing of Normal and Tumor Tissue for Digestion
-
1.Patient-derived material can be obtained from pancreatic resections (partial pancreatic resections, whipple resections etc.), or core/fine needle biopsies (from either primary cancers or metastases)Note: Collection of human material must conform to all relevant institutional/ governmental regulations. Informed consent must be obtained from all subjects prior to collection of the primary human tissue material.
-
a.For resections, if possible, obtain tumor fragments from different tumor areas. As such, the chance that the resulting organoid culture represents the in vivo tumor heterogeneity (at the time of resection) is increased. Transfer the fresh patient material that will be used to generate organoids as soon as possible to collection medium ((Ad-DF+++) containing 50 ng/mL Primocin).
-
b.For needle biopsies, transfer all the material obtained to collection medium (Ad-DF+++ containing 50 ng/mL Primocin). For isolation from biopsies, be aware that establishment efficiency is generally lower for organoids derived from biopsies (+/- 30%) than from resections (+/- 70%) (Driehuis et al., 2019). We hypothesize this is due to the low tumor cellularity present in the biopsied tissue. This might be especially relevant considering that pancreatic tumors generally contain many non-epithelial (stromal) cells. Therefore, try to obtain as much tissue as possible.
-
a.
Pause Point: Patient-derived material can be stored in collection medium for a maximum of 48 h at 4°C. However, the longer tissue is stored before processing, the lower the outgrowth efficiency generally will be.
Optional: 10 μM ROCK inhibitor can be added to the collection medium to prevent cell death by anoikis and enhance organoid outgrowth.
-
2.
Transfer the material to a 10 cm glass petri-dish. This can be done by carefully removing the collection medium from the collection tube and inverting the tube above a petri-dish once all media is removed. Alternatively, tweezers can be used to ‘fish’ material from the collection medium. Make sure that tissue does not dry out in the petri-dish, by covering it in 1–2 mL of collection medium.
-
3.
For resections, remove tissue that is not pancreatic epithelium such as muscle, fat, or intestinal epithelium as much as possible before proceeding to tissue digestion. Do not do this for biopsies, as 1) due to the nature of a needle biopsy, different tissues are difficult to distinguish from each other and 2) too little material will remain.
-
4.
Using scalpels, mince the tissue into small fragments of 1–2 mm3. The tissue can become viscous from this handling; this is not a problem.
Optional: Collect 2–3 pieces from different areas for DNA isolation (store at −20°C), 2–3 pieces from different areas for RNA isolation (store at −80°C), and 2–3 fragments for histology (for fixation). We recommend at least collection of DNA, to allow SNP fingerprinting to confirm the identity of the organoid culture that will be established and exclude the possibility of culture swaps (that can occur during handling cultures derived from different patients). For more information on QC standards, see section ‘Quality control (QC)’ when establishing patient-derived organoid cultures.
-
5.
Transfer the tissue fragments to a 50 mL plastic tube by pipetting 5 mL of D-BSA in the petri-dish. Use a pre-wetted 5 mL serological pipette to transfer the material from the petri-dish to the 50 mL plastic tube. Wash the petri-dish with 5 mL of D-BSA and transfer to the 50 mL plastic tube. Rinse the used scalpels with D-BSA to assure all available material is collected for digestion.
-
6.
Add 30 mL of D-BSA and pipette up and down the tissue fragments ten times using a pre-wetted 10 mL serological pipette.
-
7.
Let this tissue sediment by gravity to the bottom of the tube (this will take 2–3 min).
-
8.
Aspirate the top 30 mL of D-BSA.
-
9.
Repeat steps 6–8 two more times.
-
10.
Resuspend the tissue fragments in the remaining 10 mL D-BSA and transfer them to a 15 mL plastic tube. Let the tissue fragments settle by gravity.
-
11.Remove supernatant by aspiration.
-
a.For normal/wildtype tissue: resuspend the tissue fragments in 5 mL of Digestion buffer for normal tissue (1× HBSS, 1 mg/mL collagenase type II, 10 μM ROCK inhibitor, 100 μg/mL Trypsin inhibitor and 5% glycerol, see section ‘Materials and Equipment’).
-
b.For tumor tissue: resuspend the tissue fragments in 5 mL of Digestion buffer for tumor tissue (Ad-DF+++, 5 mg/mL collagenase type II containing 10 μM ROCK inhibitor, see section ‘Materials and Equipment’).
-
a.
-
12.
Place the 15 mL plastic tube containing material in a 37°C incubator containing a shaker (140 rpm at a low angle (∼15˚)).
Note: If a shaker is not available, place the tube in a 37°C water bath. When using a water bath, make sure to pipette up and down the material every 10 min, using a P1000 pipette with a low-retention tip.
Note: Parafilm can be used to wrap the 15 mL plastic tube, to minimize contamination risk.
Tissue Digestion
-
13.Tissue digestion
-
a.For normal tissue, do not exceed a digestion of 2 h. Monitor the digestion at least every 30 min, using a bright-field microscope. When clusters of 5–10 cells are observed, digestion is complete. It is recommended to pipette up and down every 30 min using a pre-wetted 5 mL serological pipette to aid digestion.
-
b.For tumor tissue, digestion can be performed for a maximum of 24 h. Do not exceed 24 h digestion. Monitor every 30 min for the first 4 h of digestion. After the first 4 h of digestion, monitor at regular intervals (as much as schedule/working hours allow). Similar to the digestion of normal tissue, monitor the digestion of the tissue using a bright-field microscope. When clusters of 5–10 cells are observed, digestion is complete. It is recommended to pipette up and down every 30 min (or post 4 h of incubation, every time digestion is monitored) using a pre-wetted 5 mL serological pipette to aid digestion.
-
a.
-
14.
To stop digestion, add 1 mL FBS directly to the digestion mixture.
-
15.
Add 7 mL of D-BSA using a 5 mL serological pipette.
-
16.
Using the same pipette (that is now pre-wet by the D-BSA), pipette the tissue mixture up and down for 20 times vigorously.
-
17.
Centrifuge at 200 × g for 5 min at 8°C.
-
18.
Carefully aspirate the supernatant (we recommend the use of a P1000 for the last remaining liquid). 500 μL of liquid can be left on the pellet.
-
19.
Add 10 mL D-BSA to the tube and spin at 450 × g for 5 min at 8˚C.
-
20.
Carefully aspirate the supernatant (we recommend the use of a P1000 for the last remaining liquid). 500 μL of liquid can be left on the pellet.
-
21.
Resuspend the pellet in 7 mL D-BSA and filter over a 100 μm pore size filter. Collect flow-through in a clean 50 mL plastic tube.
-
22.
Wash the filter with an additional 7 mL Ad-DF+++ and pool the flow-through with the cell suspension collected in step 21.
-
23.
Centrifuge at 200 × g for 5 min at 8°C.
Note: Material that remains on the filter can be collected and stored, for example, to perform SNP validation of the established organoid culture later in time.
-
24.
Carefully aspirate the supernatant (we recommend the use of a P1000/P200 for the last remaining liquid).
Establishment of Pancreas Organoid Culture
-
25.
Resuspend the pellet in 25–30 μL of pancreatic normal (or tumor) medium and the appropriate amount of Matrigel/BME (as a rule of thumb, 200 μL Matrigel/BME for a cell pellet of approximately 50 μL can be used). Resuspend carefully to prevent the formation of air bubbles. Assure that cell pellet is mixed well with the Matrigel/BME. Assure that the percentage of Matrigel/BME stays > 80%.
-
26.
Make small droplets (5 to 10 μL each) of the Matrigel/organoid or BME/organoid mixture that was generated in the step above per well of a pre-warmed 24-well plate. Plate the droplets on the bottom of the plate. You can make 4–6 drops per well (for a total of ∼50 μL per well).
-
27.
Invert the plate and leave the plate for 5 min in the flow cabinet. The solidification of the Matrigel/BME will commence, which will make the Matrigel/BME droplets more stable before transporting the plate to the incubator.
-
28.
After 5–10 min, transfer the plate to a 37°C and 5% CO2 incubator. Leave the plate for 30 min, allowing the Matrigel/BME to solidify.
-
29.
Add 500 μL organoid culture medium (pancreatic normal medium for normal tissue or the two different pancreatic tumor mediums for tumor samples) containing 10 μM ROCK Inhibitor to each of the wells that contain cells. Carefully add the media to the wells, using the side of the wells, to not disturb the formed Matrigel/BME droplets. For tumor samples, use one tumor medium for half of the plated wells, the other tumor medium for the other half of the wells.
Note: In case both tumor medium (1 and 2) support organoid growth, authors recommend expanding and characterizing organoid established with both mediums to identify different tumor clones from the same tumor (Figure 3D).
Figure 7.

Troubleshooting: Pancreatic Tumor Organoids
(A) Representative bright-field images showing the result of culturing organoids with old media. Both panels show the growth of the same tumor organoid culture, that was either expanded for 8 days on 4-week-old medium (left panel), or expanded on freshly prepared medium (right panel). Scale bar, 500 μm.
(B) When freezing organoids, the organoids should have the correct size and not be too large. Representative bright-field microscopy images show the result of thawing a culture that was frozen when the organoids reached a large size. As can be seen, even after 11 days, no new organoids grew out from the thawed cells. This culture could not be recovered and was lost. Scale bar, 500 μm.
(C) Representative bright-field image of an example showing an organoid culture that recovered only poorly from thawing and was plated at a density that was too low (first panel) to result in good outgrowth of the culture (second panel). In this case, the culture could be saved by pooling the organoids that grew out at day 10 (second panel), passaging them and plating them in a decreased volume of BME/Matrigel. Scale bar, 500 μm.
(D) Representative bright-field image of an example showing an organoid culture derived from breast tissue that is attaching to the bottom of the culture plate. The surface where the 2D cells are growing is indicated with arrows. Scale bar, 500 μm.
Figure 3.

Epithelial Cells Isolated from Tumor Resections Show Variable Outgrowth on the Different Tumor Organoid Media
(A–C) Representative bright-field images showing the outgrowth of three samples (isolated from tumors of three different patients), when cultured in either tumor media 1 (TM1, left panels) or tumor media 2 (TM2, right panels). While sample 1 (S1) seemed to grow out equally well in both media, sample 2 (S2) showed a clear preference for TM2. Sample 3 (S3) did not grow in any of the two-tumor media. Scale bars, 100 μm.
(D) Flowchart for helping decision taking when establishing organoid models using the different tumor media. S1, S2, and S3 indicated in the flowchart refer to the representative images shown in (A)–(C).
Figure 1.

Bacterial and Fungal Infections in Organoid Cultures
(A) Bacterial infection in BME/Matrigel drop. Representative bright-field microscopy images of bacterial colonies (indicated by arrows) that grew inside the BME/Matrigel drops. Because of their shape, the round colonies can be interpreted by some as organoids if only look at briefly when quickly scanning a culture plate. In the BME droplet shown here, an organoid was also present (indicated by an asterisk). Scale bar, 500 μm left panel, 100 μm right panel.
(B) Bacterial infection in organoid culture medium. Representative bright-field microscopy images of bacteria growth in culture organoid medium. In this case, a darker “halo” can often be observed around the BME/Matrigel droplet. This is where the bacteria accumulate. The media also can appear cloudy (because of the presence of bacteria). Scale bar, 500 μm left panel, 100 μm right panel.
(C) Fungal (yeast) infection in an organoid culture. Representative bright-field microscopy images of yeast cell, which is much larger than a bacterium. Often, white “colonies” can be observed by the naked eye in the BME/Matrigel drops. Here, the yeast networks (arrows) mainly grew from organoids (asterisk), which can still be seen present hidden under the yeast. Scale bar, 100 μm left panel, 50 μm right panel.
-
30.
Organoids will usually be visiable 2–4 days after plating. Normal organoids appear as cystic structures, whereas tumor organoids can appear as either cystic or dense structures. Figure 2 shows examples of these two different morphologies as seen through a bright-field microscope. Two cystic tumor organoid cultures are shown in Figure 2A. Figure 2B depicts two-tumor organoid cultures that display a dense phenotype. Some organoid cultures can show a mixed phenotype, showing both dense and cystic organoids. Refresh organoid media every 2–3 days to assure sufficient growth factors are supplied to the organoids. Keep ROCK inhibitor in the media for the first week after isolation. When removing media, carefully aspirate using a P1000 pipette without disturbing the Matrigel/BME droplets. Be aware that, over time, Matrigel/BME can disintegrate, making the droplets less stable and detach more easily from the well
Note: If Matrigel/BME drops detach and start to float, media changes become more difficult due to the increased risk of aspirating the organoids. In that case, re-seeding the organoids is highly recommended. For this, collect material from the culture plate and pool it in 15 mL plastic tube. Centrifuge at 450 × g for 5 min at 8˚C. Carefully aspirate the supernatant and repeat steps 25–29, using a fresh pre-warmed 24-well plate.
Figure 2.

Examples of the Cystic and Dense Morphologies of Pancreatic Organoid Cultures
(A) Representative bright-field images of two patient-derived pancreatic tumor organoid cultures with cystic morphology. The top two panels show an organoid culture that has a cystic appearance similar to a normal culture; forming thin-walled large cysts. The bottom two panels show a cystic culture with a phenotype that is only observed in tumor cultures and not in normal (non-tumor) cultures. These cysts are thick-walled, appear dark, and often contain dead cells in the center of the structures. Although these organoids do not show an empty lumen, they are still clearly cystic, containing a (single- or multi-) layered row of epithelial cells that surround an internal compartment that does not contain epithelial cells. Scale bar left panels, 500 μm. Scale bar right panels, 100 μm.
(B) Representative bright-field microscopy images of two patient-derived pancreatic tumor organoid cultures that show an overall dense morphology. In the dense structures, no lumen can be observed. However, in both cultures shown here, a fraction of the organoids of the culture showed a lumen, thereby representing a mixed phenotype. Organoids that form a lumen are indicated by an asterisk, whereas examples of dense organoids are indicated using an arrow. Such mixed phenotype cultures are commonly observed. Scale bar left panels, 500 μm. Scale bar right panels, 100 μm.
Passaging Pancreas Organoid Cultures
Depending on the amount of starting material and on the percentage of viable epithelial cells in the culture, outgrowth efficiency can vary. Some tumor samples grow equally well on both tumor mediums (as the culture shown in Figure 3A). However, others grow better in only one of the tumor medium (such as the organoid culture sample 2 shown in Figure 3B) or in neither of the tumor mediums (Figure 3C). For a flowchart, for decision taking, on expansion of cultures on different mediums, see Figure 3D. Growth rates between samples derived from different patients vary. For the first passage, wait until 80% of the organoids are larger than 50–70 μm before processing them for next passage. This usually takes around 14–21 days (Figure 3 shows organoids grown from isolated primary material that are ready to be passaged). To determine the split ratio and frequency of a newly established culture, as a rule of thumb, pancreatic normal organoids can generally be passaged 1:2–1:3 every 7–10 days. Pancreatic tumor organoids can be passaged 1:2–1:6 every 7–10 days. Work in a conservative way, rather plating the organoids dense (with a low split ratio) than seeding them too sparse. You can always dilute further once the organoids start growing and the density turns out to be too high. In general, organoid cultures (especially normal cultures) do not take it well when they are diluted too much. In newly established cultures, from which the passage rate is not yet determined, closely monitor the culture for the first days after passaging. If organoids in the center of Matrigel/BME drops are dying, this is an indication that the organoids were seeded too densely. Two examples of this are shown in Figure 4A. If this phenomenon is observed, re-seed organoids in a larger volume of Matrigel/BME without passaging the organoids. Let the organoids grow until they reach the desired size before being disrupted for passaging. As mentioned above, organoid cultures derived from different patients differ in their growth rate and optimal passage density. However, to get a general idea of suitable passaging densities, Figures 4B–4D show representative bright-field images of three different pancreatic tumor organoid cultures right after passaging. The second panel then shows the result of the plating after passaging. Figure 4B shows a cystic culture that was plated at a density that was too low. Figures 4C and 4D show examples of correct seeding densities for a cystic culture and a compact culture, respectively.
-
31.
For passaging, pipette the culture medium present in the wells up and down to disrupt the Matrigel/BME drops.
Note: If the organoid medium contains cell debris, do not resuspend using the culture medium. Carefully aspirate the culture medium and add fresh D-BSA to resuspend the Matrigel/BME drops in.
-
32.
Collect all material in a 15 mL plastic tube. Top off to a total of 12 mL with ice-cold D-BSA.
Note: A maximum of a half plate (organoids embedded in ∼600 μL of Matrigel/BME) can be pooled in one 15 mL plastic tube. If more material is collected for passaging, divide it over multiple tubes.
-
33.
Pipette up and down 10 times using a serological 10 mL pipette. Centrifuge at 85 × g for 5 min at 8°C. After centrifugation, check if the organoid pellet is clean (clean meaning that no Matrigel/BME remnants are present on top of the organoid pellet and that all organoids are contained in the pellet, therefore organoids are not scattered in a layer of Matrigel/BME present above the organoid pellet). If clean, proceed.
Note: If the pellet border is not clean and contains Matrigel/BME (containing or not organoids), remove supernatant only up to the border and wash the pellet again (adding 10 mL of D-BSA) and repeat centrifugation, now at 450 × g for 5 min at 8°C. If the pellet still contains a layer of Matrigel/BME, but this Matrigel/BME is devoid of organoids, this layer can be removed carefully by aspiration.
-
34.
Once all organoids are collected in a clean pellet, remove supernatant without disturbing the pellet. If organoids of the same organoid culture were collected in more than one tube (see step 32), pool the pellets by resuspending each pellet in 4 mL D-BSA, pooling the suspension in one tube and centrifuging at 85 × g for 5 min at 8°C. If organoids are not pelleted, repeat the spin at 1,500 rpm (450 × g) 5 min at 8°C.
-
35.
Remove supernatant without disrupting the pellet.
Note: For passaging organoids, the method applied depends on the morphology of the organoid culture. For passaging cystic (normal or tumor) organoids, follow steps 36a–36e. For dense tumor organoids, follow steps 37a–37i.
Note: Normal organoids should not appear dense. A dense morphology is an indication that you are growing tumor cells.
-
36.Cystic organoids: When passaging pancreatic cystic organoids, the new passage will be generated from clumps of cells. Do not make the pancreatic cystic organoids too small (or shear to a single-cell level), since this will negatively impact outgrowth efficiency. Pancreatic cystic pancreatic organoids should be split by mechanical shearing (using a pre-narrowed glass Pasteur pipette) and can be passaged every 7–10 days. For cystic organoids, splitting ratio ranges between 1:2 and 1:3.
-
a.Add Ad-DF+++ up until 1 mL and resuspend the organoid pellet very carefully.
-
b.Using a flame, prepare a pre-narrowed and pre-wet (with Ad-DF+++) sterile glass Pasteur pipette. For this, keep the tip of the glass Pasteur pipette in the flame and roll the pipette in your hand, assuring all sides of the pipette are heated equally. This will narrow the opening of the pipette (Figure 5A). Alternatively, a P1000 with a 1,250 μL retention filter tip fitted with either a 200 (yellow) μL or 10 (white) μL tip on top can be used (Figure 5B).
-
c.Shear the resuspended organoids by pipetting up and down 3 times. Avoid creating foam. Assure that the tip of the (glass) pipette touches the wall of the plastic tube. This generates a mechanical force that will aid to shear the organoids. Monitor shearing of organoids using a bright-field microscope. If sheared, continue the protocol. If not sheared, repeat to pipette up and down for a maximum of 20 times. Check the organoids after every 5 times of shearing (Figure 5C). If still not disrupted after 20 times, centrifuge at 450 × g for 5 min at 8°C, remove supernatant and proceed to the passaging of the organoids as described for dense organoids (see section below).
-
d.Top up to a total of 12 mL using D-BSA. Pipette up and down several times and centrifuge at 85 × g for 5 min at 8°C.Note: If organoids are not pelleted, repeat the spin at 1,500 rpm (450 × g) 5 min at 8°C.
-
e.Remove supernatant carefully, without disturbing the pellet.
-
a.
-
37.Dense organoids: The first time disrupting a pancreatic dense organoid culture after isolation, always try to disrupt it with mechanical shearing as described for cystic organoids. If the organoids are not disrupted after pipetting them 20 times (as described in the section above), then use enzymatic shearing as described in this section. Dense organoids are generated from tumor samples. For most dense tumor cultures, organoids can be disrupted to a single-cell level without compromising the growth of organoids in the next passage, however, it is always recomended not to reach single-cell level. Pancreatic dense organoids can usually be split every 7–10 days. For dense organoids, splitting ratio ranges usually between 1:2 and 1:5.
-
a.Add 750 μL of Ad-DF+++ to the cell pellet (see step 35) and resuspend. Add 250 μL of TrypLE. Add ROCK inhibitor to a final concentration of 10 μM. Place the tube for 30 s in the 37°C water bath. Vortex and check organoid shearing using a bright-field microscope. If organoids are disrupted, proceed to step 37g.Note: Both Ad-DF+++ and TrypLE need to be at room temperature (between 18°C and 22°C).Note: TrypLE is diluted with Ad-DF+++ to decrease its activity, thereby preventing that digestion occurs too quickly and results in a single-cell suspension.
-
b.If organoids are not sheared after the TrypLE incubation, prepare a pre-narrowed and pre-wet sterile Pasteur pipette (Figure 5A), or a P1000 with a 1,250 μL retention filter tip with a 200 (yellow) μL or 10 (white) μL tip (Figure 5B). Shear the organoids by pipetting the 1 mL suspension up and down at least five times. Avoid creating foam by keeping the tip immersed under liquid and pipetting up and down slowly.
-
c.Check organoid shearing using a bright-field microscope (Figure 5C). When sheared sufficiently, proceed to step 37g. If not, pipette up and down a maximum of 20 times. Check shearing after each five times of passaging. When sheared sufficiently, proceed to step 37g.
-
d.If the culture is difficult to disrupt and organoids are not sufficiently sheared, place the organoid suspension in a 37°C water bath and incubate for a maximum of 5 min, checking the organoids every 2 min.
-
e.If organoids are still not disrupted, add 250 μL of TrypLE and place the tube for 30 s in the 37°C water bath. Vortex and check organoid shearing using a bright-field microscope. If organoids are disrupted, proceed to step 37g.
-
f.If organoids are not sheared sufficiently, place organoid suspension in TrypLE solution in a water bath at 37°C. Incubate while revising the status of the organoids every 2 min under the bright-field microscope. Vortex the tube before checking the organoids.
-
g.Once organoids are disrupted, add Ad-DF+++ to a total of 12 mL.
-
h.Pipette up and down several times with a 5 mL plastic pipette and centrifuge at 85 × g for 5 min at 8°C.Note: If organoids are not pelleted, repeat the spin at 1,500 rpm (450 × g) 5 min at 8°C.
-
i.Remove supernatant carefully, without disturbing the pellet.
-
a.
Figure 4.

Passaging of Organoid Cultures
(A) Representative bright-field images of two organoid cultures that were plated too densely after passaging, resulting in death of organoid structures. Death can be observed mostly in the center of the BME/Matrigel dome, most likely because of lack of nutrients (that diffuse into the droplet from the media) and oxygen. Live organoids (enriched on the side of the drops) are indicated by arrows, whereas dead organoids (enriched on the center of the drops) are indicated with asterisks. Scale bar, 500 μm.
(B–D) Representative bright-field microscopy images of three different organoid cultures split for passaging. The left panels show the appearance of the cultures right after shearing (d0). The right panels show the appearance of the cultures few days later (7–8 days) and right before they will be passaged.
(B) Representative bright-field image of a culture that was plated with a density that was too low, as can be seen by the limited number of organoids that grew out after 7 days in culture. Here, the organoid culture was pooled (the material was collected and replated in a smaller volume of BME) to assure that organoids were in close proximity to each other. After that, when organoids grew big, the culture could be passaged again.
(C and D) Representative images of a cystic (C) and dense (D) organoid culture that were split and subsequently passaged at a correct density. This density allowed efficient outgrowth of the organoids, without the cell death shown in Figure 4A as can be observed in the right-side panels. Scale bar, 500 μm.
Figure 5.

Technical Aspects of Passaging Organoid Cultures
(A) Representative images from glass Pasteur pipettes before (indicated by an asterisk) and after (indicated by arrowheads) narrowing of the opening by a flame. The second panel shows a side view of a glass pipette before (asterisk) and after (arrowhead) narrowing by flame.
(B) Representative image of a P1000 pipette with a 1,250 μL tip that holds a 10 μL pipette tip on top, which can be used as an alternative to the narrowed glass pipette to shear organoids during passaging. The smaller volume tip on top of the larger volume tips narrows the opening of the tip, allowing the shearing of organoids.
(C) Representative image indicating how a 15 mL plastic tube containing the organoid culture suspension can be placed under the bright-field microscope to monitor the size of organoids during passaging.
Plating Organoids
Pancreatic organoid cultures derived from different donors can vary in growth rates. Even when properly cultured, tumor cultures can contain cells that show signs of cell death, even though the culture as a whole will survive over time. This might potentially be due to an increase in genomic instability (e.g., caused by loss of the tumor suppressor TP53) that can be observed in tumor cells.
-
38.
Resuspend the organoid pellet in 12 mL Ad-DF+++ to wash. Pipette up and down three times.
-
39.
Centrifuge at 85 × g for 5 min at 8°C. After centrifugation, check (using a bright-field microscope) if the border between the pellet and supernatant is clean.
Note: If organoids are not pelleted, repeat the spin at 450 × g 5 min at 8°C.
-
40.
Remove supernatant as much as possible without disturbing the organoid pellet. Add a small amount of specific organoid medium (<30% of the pellet volume) and resuspend the cell pellet.
-
41.
Estimate the volume of the sheared organoids and the medium remaining in the tube. Resuspend the contents of the tube in 80% Matrigel/BME, dissolved in the organoid medium of choice (normal medium or tumor medium 1 or 2). As a rule of thumb 50 μL of cell suspension can be plated in 200 μL of Matrigel/BME.
-
42.
Plate a 5 μL drop in pre-warmed cell culture plate and check the organoid density. Dilute further if the density is too high (see Figure 4A as example). If density is too low (Figure 4B), wash and centrifuge to pellet the organoid fragments. After removing the supernatant, organoids can be plated in the required (smaller volume) of Matrigel/BME.
-
43.
Once the correct organoid density has been reached, plate a maximum of 200 μL Matrigel/BME per well for a 6-well plate, 100 μL Matrigel/BME per well of a 12-well plate, or 50 μL per well of a 24-well plate in small drops (5–10 μL/drop). Making smaller droplets will improve the diffusion of growth factors into the Matrigel/BME. Avoid creating air bubbles while plating.
-
44.
Invert the plate, in one swift motion, and leave it in the hood for 5–10 min.
Note: This will prevent the bigger organoids from sinking to the bottom of the well.
-
45.
Incubate the plate upside down in the corresponding incubator for 30–40 min.
-
46.
Add 2 mL (6-well plate), 1 mL (12-well plate), 500 μL (24-well plate) of the corresponding culture medium containing 10 μM ROCK inhibitor and place the plate in the incubator at 37°C and 5% CO2.
-
47.
Refresh medium every 2–3 days.
-
48.
Pancreatic organoids are usually passaged every 5–10 days depending on their growth and density. Repeat passaging cycles until the desired amount of material is obtained for freezing.
Freezing of Organoids
-
49.
Passage organoids according to their morphology, as described above. Be mindful that, when passaging for freezing, organoids should never be passaged until single-cell level.
-
50.
Plate the sheared organoids in 50% Matrigel/BME (1:1 diluted with the corresponding organoid culture medium) in a per-warmed culture plate. Plate a maximum of 200 μL Matrigel/BME per well for a 6-well plate, 100 μL Matrigel/BME per well of a 12-well plate, or 50 μL per well of a 24-well plate in small drops (5–10 μL per drop). Making smaller droplets will help improve the diffusion of growth factors into the Matrigel/BME. Avoid creating air bubbles while plating.
-
51.
Invert the plate and leave it in the hood for 5–10 min.
Note: This will prevent the bigger organoids from sinking to the bottom of the well.
-
52.
Incubate the plate upside down in the corresponding incubator for 30–40 min.
-
53.
Add 2 mL (6-well plate), 1 mL (12-well plate), 500 μL (24-well plate) of the corresponding culture medium containing 10 μM ROCK inhibitor and place the plate in the incubator at 37°C and 5% CO2.
-
54.
Monitor the organoid culture daily. Organoids can be frozen when a minimum of 80% of the culture has reached a size > 30 μm. Generally, this occurs between 1 and 3 days after passaging, depending on the growth speed of the organoid culture. Figure 6 shows representative bright-field microscopy images of organoid cultures that are passaged in preparation of freezing (first panel), and their morphology on the day of freezing (second and third panel). Refresh medium every other day until the organoids are ready for freezing. ROCK inhibitor only needs to be added directly after passaging.
-
55.
When organoids have reached the correct size for freezing (see Figure 6 for examples), place the cryovials, freezing medium and 2 mL serological pipettes (in the original wrapping) that will be required on ice.
-
56.
Use a D-BSA pre-wetted P1000 pipette to collect organoids from the culture plate by resuspending the medium in the well and using it to break up the Matrigel/BME drops. Transfer the collected material to a 15 mL plastic tube.
-
57.
Wash the emptied wells with 1 mL of Ad-DF+++ and transfer this to the 15 mL plastic tube to assure all material is collected.
-
58.
Fill up the tube to a total of 12 mL with Ad-DF+++. Pipette up and down 5 times using a 10 mL serological pipette.
-
59.
Centrifuge the organoid suspension at 85 × g for 5 min at 8°C.
Note: If organoids are not pelleted, repeat the spin at 450 × g for 5 min at 8°C.
-
60.
Carefully aspirate the supernatant without disturbing the organoid pellet.
-
61.
Resuspend the pellet in 12 mL of Ad-DF+++ using a 10 mL serological pipette. Pipette up and down 5 times to wash the organoids.
-
62.
Centrifuge at 450 × g for 5 min at 8°C.
-
63.
Confirm organoids are pelleted using a bright-field microscope. If they are, carefully aspirate the supernatant, without disturbing the pellet. If not, repeat centrifugation.
-
64.
Add the corresponding volume of freezing medium with a 10 mL serological pipette while keeping the organoid pellet on ice. Use 500 μL of freezing medium for each 100 μL of original volume of Matrigel/BME collected in step 56.
-
65.
Once all the freezing medium is added, pipette up and down carefully with a precooled 5 mL serological pipette to resuspend all organoids in the freezing medium. Keep the tube(s) on ice.
-
66.
Aliquot 0.5 mL organoid suspension in each cryovial, using the precooled 2 mL serological pipette. To assure equal distribution of the collected organoids over the different cryovials, do not use larger volume pipettes at this point. Work quickly.
-
67.
Transfer the cryovials from ice into the appropriate precooled (precooled at least 12–16 h at 4°C) Coolcell and transfer the Coolcell to −80°C.
-
68.
Place the Coolcell in a −80°C freezer.
-
69.
After >24 h, transfer the cryovials to the liquid nitrogen for long-term storage. Do not leave the cryovials in −80°C for >15 days.
Figure 6.

Freezing of Pancreatic Tumor Organoid Cultures
Representative bright-field images showing the morphology of three different pancreatic tumor organoid cultures in the passage for freezing. The left panels depict how the cultures looked directly after performing 1:1 passaging in preparation of the freezing of the culture. The panels on the right side show the morphology of the same cultures on the day they were frozen. The day of freezing is indicated on the left corner of the image. As can be seen in these images, different cultures (with different growth speeds) require a different amount of days before they reach the right size for freezing. Scale bar left and central panels, 500 μm. Scale bar right panels, 100 μm.
Thawing of Cryopreserved Organoids
-
70.
Pre-warm 30 mL of Ad-DF+++ at 37°C in a 50 mL plastic tube. Place Matrigel/BME on ice at least 30 min before being used for plating organoids.
-
71.
Collect the cryovial that will be thawed from the liquid nitrogen container and transfer it to dry ice.
-
72.
Thaw the cryovial rapidly by agitation in a 37°C water bath until only small fragments of frozen material remain. Thawing should not take longer than 2–3 min.
-
73.
Wipe the cryovial with a tissue paper sprayed with 70% ethanol.
-
74.
Dropwise, add 0.5 mL of the pre-warmed Ad-DF+++ to the contents of the cryovial.
-
75.
Carefully mix the content of the cryovial and transfer the organoid suspension to a 15 mL plastic tube.
-
76.
Dropwise, add an additional 1 mL of warm Ad-DF+++ to the organoid suspension, while gently shaking the plastic tube (by tapping it with your pinky finger).
-
77.
Add pre-warmed Ad-DF+++ up to a total of 12 mL dropwise, using a 10 mL serological pipette and resuspend organoids gently by pipetting up and down with a 10 mL sterile pipette.
-
78.
Centrifuge at 85 × g for 5 min at 8°C.
-
79.
Using a bright-field microscope, check if the organoid pellets contain remnants of Matrigel/BME. If there is either no Matrigel/BME, or the Matrigel/BME is free of organoids, aspirate the supernatant including the Matrigel/BME carefully, without disturbing the pellet. If there is Matrigel/BME present that contains organoids, centrifuge again at 450 × g for 5 min at 8°C and carefully remove the supernatant.
-
80.
Add 10 mL of pre-warmed Ad-DF+++ and gently resuspend the organoid pellet.
-
81.
Centrifuge at 450 × g for 5 min at 8°C.
-
82.
Carefully aspirate the supernatant, without disturbing the pellet, and place the tube on ice.
Note: Remove the remaining Ad-DF+++ using a P200 pipette with 200 μL filter tip.
Note: Try to remove any remaining Matrigel/BME if it does not contain a significant number of organoids.
-
83.
Resuspend the organoid pellet in the desired amount of ∼80% Matrigel/BME. For this, take the volume of the organoid pellet into account, when calculating how much of the corresponding organoid medium needs to be added to reach a ∼80% Matrigel/BME solution.
Note: Resuspend the organoid pellet in the desired amount of organoid medium, before adding the Matrigel/BME. Mixing the pellet directly with 100% Matrigel/BME will be difficult, due to the viscosity of undiluted Matrigel/BME.
-
84.
Make a droplet of 5 μL Matrigel/BME and check if the organoid density is correct using a bright-field microscope.
-
85.
If density is appropriate, continue plating. If density is too high, add more 80% Matrigel/BME.
Note: Directly after thawing, organoid density should be high. This will help the organoids recover from the thawing process.
-
86.
Plate the organoid/Matrigel/BME mixture in a pre-warmed culture plate. Plate a maximum of 200 μL Matrigel/BME per well for a 6-well plate, 100 μL Matrigel/BME per well of a 12-well plate, or 50 μL per well of a 24-well plate in small drops (5–10 μL per drop). Making smaller droplets will help improve the diffusion of growth factors into the Matrigel/BME. Avoid creating air bubbles while plating.
-
87.
Invert the plate, in one swift motion, and leave it in the hood for 5–10 min.
Note: This will prevent the bigger organoids from sinking to the bottom of the well.
-
88.
Incubate the plate upside down in the corresponding incubator for 30–40 min.
-
89.
Add 2 mL (6-well plate), 1 mL (12-well plate), 500 μL (24-well plate) of the corresponding culture medium containing 10 μM ROCK inhibitor and place the plate in the incubator at 37°C and 5% CO2.
-
90.
Refresh the medium every 2–3 days. Once organoids have reached the correct size, passage them as previously described.
-
91.
If organoids recover properly from thawing, passage them when they reach the right size to be processed (see relevant protocol).
Note: This procedure can take place either 5 up to 15 days after thawing. This depends on the organoid culture used (cultures derived from different patients show different growth speeds) and size of the organoids when frozen.
Expected Outcomes
If this protocol is followed, organoid cultures can be established from primary patient material with an efficiency higher than 70%, when using resected tissue. Organoids can be maintained and expanded in culture, although expansion rates vary between organoid cultures derived from different patients. On average, pancreatic tumor organoids can be passaged weekly with a split ratio ranging from 1:2 to 1:5. Pancreatic normal organoids can usually be passaged every 7–10 days with a split ratio ranging from 1:2 to 1:3.
After digestion of primary tissue, organoids usually appear between 2 and 4 days after initial seeding. They will appear as small round or folded structures. It usually takes 14–21 days after initial plating before the culture can be passaged for the first time. After the initial passage, split frequency can usually be increased.
It is highly recommended to freeze organoids as soon as the culture is established. As such, a back-up is available if something goes wrong later on while working with the culture. Moreover, pancreatic normal organoids have a limited expansion span (usually allowing expansion up to passage 18–20) after which the organoids cease proliferation. Tumor organoids generally do not show this senescence.
Limitations
Success Rate of Organoid Establishment
When established from resections, the success rate of pancreatic tumor organoid establishment is higher than 70% (Boj et al., 2015; Driehuis et al., 2019; Hou et al., 2018; Seino et al., 2018). By comparing frequency of detected genetic alterations in the established organoid cultures to those reported to be detected in primary patient material, there does not seem to be a particular (genetic) subset of pancreatic ductal adenocarcinomas that cannot be cultured. Several factors can influence outgrowth efficiency. First, the quality of the starting material. For example, others have reported that, quite intuitively, starting material containing a higher percentage of (tumor) epithelial cells has a higher chance to result in effective organoid outgrowth (Ooft et al., 2019; Vlachogiannis et al., 2018). Moreover, storage of the tissue also affects success rates. Tissue should be stored in culture medium as soon as possible and be kept at 4°C until processed. We have successfully established organoids from tissue that was stored for up to 72 h in collection medium before it was processed. However, it is recommended to process the obtained material as soon as possible, to increase both the chance and frequency of organoid outgrowth. Second, the quality of the medium used. From our experiences, issues to set-up Organoid Technology in laboratories mostly result from the use of suboptimal growth factors (e.g., Wnt conditioned medium that lacks Wnt activity). Quality of growth factors and media component should therefore be assured by following manufacturer’s recommendation for storage location and expiration of the product. Figure 7A shows an example of growth of an organoid culture on either freshly prepared culture medium, or culture media stored for 4 weeks at 4°C. Moreover, when conditioned media are produced in-house, a test to confirm sufficient signaling activity should be performed prior to its use in organoid cultures.
The Risk of Normal (Non-tumor) Cell Contamination and Clonal Selection In Vitro
As pancreatic cancer usually presents as a diffuse tumor, contamination with normal cells is a notorious problem when establishing tumor organoids. This problem is not unique to pancreatic organoids and has been reported for different organoid cultures derived from different organs (Drost and Clevers, 2018). The use of tumor-selective media (lacking growth factors required by normal, but not tumor cells) overcomes this risk of normal-contamination and should therefore be applied when establishing tumor organoids from primary tumor resections.
It has been reported that tumor heterogeneity can be retained in organoid cultures (Weeber et al., 2015). Regardless, it is important to consider that clonal selection might occur over time in culture, especially if tumor-selective medium are applied. If, in a heterogeneous culture, a specific tumor clone has a growth advantage under the culture conditions applied, this might result in expansion of this clone at the costs of others. As such, culture heterogeneity might decrease over time. The alternative – the use of complete medium – will likely result in overgrowth of normal cells as discussed prior. A compromise between these two situations must be made to 1) assure expansion of tumor and not normal cells, and 2) assure minimized selection pressure in vitro.
The use of such tumor-selective media is more often applied to enrich for tumor cells in patient-derived organoid cultures. This is an approach not taken only to enrich for pancreatic tumor organoids, but also for tumor organoids from different origins (Drost et al., 2015; Fujii et al., 2015; Kim et al., 2019; Sachs et al., 2018; Van De Wetering et al., 2015). To minimize selection for specific mutational profiles using growth factor withdrawal, in this protocol, we recommend that tumor samples are cultured on two different tumor media that both select for tumor-specific signaling pathway independencies and thus, might select different tumor types in vitro. Since at the time of establishment the genetic background of the tumor is commonly unknown, it is hard to predict which one of these mediums might be best fitting to establish tumor organoids from the tumor of interest. Moreover, cultures established on tumor medium 1 and tumor medium 2 were found to share genetic driver mutations, but also carry distinct genetic alterations (Driehuis et al., 2019). This could be seen as an indication for clonal outgrowth or selection under these different culture conditions. Since the effect of such selection cannot be predicted, it is essential to perform certain quality control before applying the established organoids as a model to recapitulate the in vivo tumor from which it was derived.
Lastly, it is important to realize that, apart from potential clonal selection or genetic drift of organoid cultures once they are established, the first potential selection occurs at the pathology department or even during a biopsy procedure, where the material that will be used to obtain the organoid culture is selected. If this starting material does not represent the tumor in vivo, it is needless to say that the resulting organoid culture will also not. This important aspect is commonly overlooked. For this reason, if possible, samples should be collected from different areas of a resected tumor.
Quality Control (QC) when Establishing Patient-Derived Organoid Cultures
For the reasons mentioned above, it is important to assess if an established organoid culture recapitulates the tumor tissue from which it was derived. DNA sequencing of organoid cultures can be used for this purpose. Comparison of the detected genetic alteration in both the organoid culture and the primary tissue, will aid to evaluate if the organoids can be used as a representative model for the patient tumor in vivo. Here, it is important to note, that the variant allele frequency (VAF) of detected mutations will differ between the organoids and the corresponding primary tissue. Most often, the primary tumor tissue contains different cell types, including fibroblasts and immune cells, in addition to the epithelial tumor cells that carry the tumorigenic genetic alterations. Although these cells likely contribute to the tumor pathogenesis, they do not carry these genetic alterations and therefore lower the VAF of mutations detected in primary patient tissue. Since these non-epithelial tumor compartments are not maintained in organoid cultures, the VAF of mutations detected in organoids will often be higher than those detected on the same mutation in primary tissue.
An essential step in the quality control of organoid cultures, is the confirmation that the established organoid cultures match the tumor of origin. Especially when working with multiple cultures derived from different donors, sample swaps might occur. Therefore, we recommend confirming that the organoid culture still matches the primary tissue, for example, by SNP fingerprinting.
In summary, these are important steps to consider before organoids can be applied to perform, for example, drug screening to assess the predictive potential of the organoids for therapy response. Depending on the application at hand, additional validation of the organoid models might be required, such as histology to detect specific cell types.
Troubleshooting
Problem 1
Matrigel/BME gets loose (steps 30 and 47). This could be due to Matrigel/BME being diluted too much upon plating, or culture has been maintained for > 14 days without replating/passaging.
Potential Solution
Replate material in fresh Matrigel/BME, by collecting material from the plate, centrifugation at 450 × g for 5 min at 8°C. Replate the resulting pellet in fresh Matrigel/BME. If the culture has been maintained for > 14 days without passaging, consider passaging of the culture. If organoids are too small to passage, only replate as described above.
Problem 2
Matrigel/BME digested overnight after plating of primary tissue (step 29). This could be due to (remnants and/or content of) acinar cells present in the primary tissue that is plated and might release enzymes that will cause digestion of the Matrigel/BME.
Potential Solution
Always check plated primary material one day after initial plating for this phenomenon. If it occurs, collect material from the plate, centrifuge at 450 × g for 5 min at 8°C. Replate the resulting pellet in fresh Matrigel/BME.
Problem 3
The organoid culture is contaminated (steps 29 and 47) and bacteria or fungi overgrow the plate. This can be due to microorganisms that are not sensitive to the antibiotics added to the culture (penicillin, streptomycin, and the antifungal cocktail Primocin). Alternatively, it can also be because organoid medium was not refreshed frequently (every other day), therefore resulting in lower levels of this active anti-bacterial and -fungal agents (see Figure 1 for examples of contamination in organoid cultures).
Potential Solution
Do not work with the contaminated culture plate in a hood that will be used later for maintaining other cultures. Do not use a pipette or a box of pipette tips that will be used later to culture any other organoid culture. If the contamination is not present in all wells, the contaminated wells can be disinfected. Carefully remove the lid of the culture plate and pipette an excess 5 M NaOH to the content of the wells. Make sure the wells do not overflow. Place the lid back and leave in the culture hood for 30 min. Carefully remove the content of the contaminated wells by pipetting it out of the culture plate. Wash the emptied wells with 70% ethanol solution. Change the lid of the culture plate to minimize the chance that the contamination will spread to the uninfected wells. Place the plate back in the incubator. If possible, keep it separate from other culture plates (e.g., by placing it into a “dirty” or “quarantine” incubator if available in the lab facility). Monitor daily to see if infection was removed.
Problem 4
Organoids that grow out are diluted and should be seeded in less Matrigel/BME than their previous passage (steps 41 and 42). Outgrowth capacity of the plated material was limited. Therefore, only a small fraction of the initially plated cells resulted in organoid formation. Now, the culture is ready for the first passage but should be re-seeded after passaging in a smaller amount of Matrigel/BME. Excess Matrigel/BME can be a problem.
Potential Solution
If excess Matrigel/BME remains and should be removed, Dispase can be used. Add 1 U/mL Dispase II to the culture wells containing the organoid culture. Matrigel droplets do not need to be mechanically disrupted. Place the plate back in the incubator for 40 min. Collect well content and proceed as usual with passaging. Dispase will digest the Matrigel/BME while leaving the organoids unaffected.
Problem 5
Organoids recover poorly after thawing (step 91). Organoids were not frozen when they had the right size (Figure 7B shows an example of organoids that were frozen too large and did not recover after thawing). Alternatively, organoid thawing was performed too slow, keeping organoids too long in freezing medium after thawing. Alternatively, organoids were plated at a too low density after thawing, for example, because organoids grew out with a lower efficiency as expected (Figure 7C shows an example of poor culture recovery post thawing, resulting in outgrowth of only a small number of organoids).
Potential Solution
Strictly follow protocols for freezing and thawing. Always take pictures on the day of freezing/day of thawing of the culture to get an idea of the best day of freezing of that culture and the density it was thawed at. If the poor recovery is likely due to an organoid density that is too low, organoids can be collected, washed (to get rid of dead cell remnants which might negatively affect outgrowth of the remaining viable cells) and re-seeded in a smaller volume of Matrigel/BME. As such, organoid density can be increased. Keep ROCK inhibitor in the medium until the organoid culture seems to be recovered.
Problem 6
Organoids attach to the bottom of the plate and start growing in 2D (steps 26, 43, and 86). The culture plate was not inverted after plating of BME/Matrigel drops. Large organoids sink and attach to the bottom of the plate, allowing the growth of epithelial cells in 2D (Figure 7D shows an example of a breast tissue organoid culture that grows partly as a 2D culture at the bottom of the culture plate). This phenomenon, although it can be rarely observed, is not frequently observed when working with pancreatic tumor cultures.
Potential Solution
If not yet performed, invert the plate after plating of the BME/Matrigel domes to assure organoids do not sink to the bottom of the plate and attach. If plate was already inverted after plating and this did not solve this problem, the use of suspension culture plates can be considered. This will decrease the number of cells that can attach. However, when using such plates, be mindful that BME/Matrigel domes can more easily detach from bottom of the plate. This will result in floating of the domes in the culture medium. This will not affect organoid growth. However, it will necessitate more careful medium changes, to prevent the floating drops from being accidentally removed from the plate.
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Sylvia F. Boj (HYPERLINK "mailto:s.boj@huborganoids.nl" \o "mailto:s.boj@huborganoids.nl"s.boj@huborganoids.nl)
Materials Availability
Pancreatic patient-derived organoids are cataloged and available for commercial and academic partners via HUB (www.huborganoids.nl). More information can be obtained via techtransfer@huborganoids.nl.
This study did not generate new unique reagents.
Data and Code Availability
This study did not generate any novel data or codes that are not presented in this published article directly.
Acknowledgments
We thank all employees of U-PORT UMC Utrecht and Onno Kranenburg for their assistance with patient inclusion and tissue acquisition. We want to thank Johan Offerhaus and Lodewijk Brosens of the Pathology Department UMCU for valuable discussions while setting up this protocol. We would like to thank Farzin Pourfarzad, Tamana Mehraban, and Emilio Ramos for their contribution to the validation of the protocol. We thank Johan van Es for his contribution to establishing the pancreatic organoid biobank. The contribution of E.D. and H.C. in this work was funded by the Oncode Institute (partly financed by the Dutch Cancer Society), by a Stand Up to Cancer International Translational Cancer Research Grant, a program of the Entertainment Industry Foundation administered by the American Association for Cancer Research (SU2CAACR-DT1213) and a ZonMw grant (116.006.103).
Author Contributions
E.D. and S.F.B. drafted and wrote the manuscript. A.G. performed practical work. R.G.J.V. and H.C. reviewed and proofread the manuscript.
Declaration of Interests
H.C. is a co-founder and SAB member of Surrozen, a start-up in Silicon Valley, and an SAB member of Merus (Utrecht), Volastra (New York), Decibel (Boston), DImed Inc. (Shanghai), Xilis Inc. (North Carolina). He is a non-executive board member of Roche (Basel) and a board/SAB member of the Roche subsidiary Genentech (San Francisco). He is a scientific advisor for and investor in Life Sciences Partners, a biotech venture capital firm located in Amsterdam. H.C. and S.F.B. are inventors on Organoid Technology patent applications relevant for this manuscript.
Contributor Information
Hans Clevers, Email: h.clevers@hubrecht.eu.
Sylvia Fernández Boj, Email: s.boj@huborganoids.nl.
References
- Boj S.F., Hwang C.-I. Il, Baker L.A., Chio I.I.C., Engle D.D., Corbo V., Jager M., Ponz-Sarvise M., Tiriac H.H., Spector M.S. Organoid models of human and mouse ductal pancreatic cancer. Cell. 2015;160:324–338. doi: 10.1016/j.cell.2014.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boj S.F., Vonk A.M., Statia M., Su J., Vries R.R.G., Beekman J.M., Clevers H. Forskolin-induced swelling in intestinal organoids: An in vitro assay for assessing drug response in cystic fibrosis patients. J. Vis. Exp. 2017;2017 doi: 10.3791/55159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driehuis E., Van Hoeck A., Moore K., Kolders S., Francies H.E., Gulersonmez M.C., Stigter E.C.A., Burgering B., Geurts V., Gracanin A. Pancreatic cancer organoids recapitulate disease and allow personalized drug screening. Proc. Natl. Acad. Sci. U S A. 2019;116:26580–26590. doi: 10.1073/pnas.1911273116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drost J., Clevers H. Organoids in cancer research. Nat. Rev. Cancer. 2018;18:407–418. doi: 10.1038/s41568-018-0007-6. [DOI] [PubMed] [Google Scholar]
- Drost J., van Jaarsveld R.H., Ponsioen B., Zimberlin C., van Boxtel R., Buijs A., Sachs N., Overmeer R.M., Offerhaus G.J., Begthel H. Sequential cancer mutations in cultured human intestinal stem cells. Nature. 2015;521:43–47. doi: 10.1038/nature14415. [DOI] [PubMed] [Google Scholar]
- Farin H.F., Van Es J.H., Clevers H. Redundant sources of Wnt regulate intestinal stem cells and promote formation of paneth cells. Gastroenterology. 2012;143 doi: 10.1053/j.gastro.2012.08.031. [DOI] [PubMed] [Google Scholar]
- Fujii M., Matano M., Nanki K., Sato T. Efficient genetic engineering of human intestinal organoids using electroporation. Nat. Protoc. 2015;10:1474–1485. doi: 10.1038/nprot.2015.088. [DOI] [PubMed] [Google Scholar]
- Hou S., Tiriac H., Sridharan B.P., Scampavia L., Madoux F., Seldin J., Souza G.R., Watson D., Tuveson D., Spicer T.P. Advanced development of primary pancreatic organoid tumor models for high-throughput phenotypic drug screening. SLAS Discov. Adv. Life Sci. R D. 2018;23:574–584. doi: 10.1177/2472555218766842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M., Mun H., Sung C.O., Cho E.J., Jeon H.J., Chun S.M., Jung D.J., Shin T.H., Jeong G.S., Kim D.K. Patient-derived lung cancer organoids as in vitro cancer models for therapeutic screening. Nat. Commun. 2019;10 doi: 10.1038/s41467-019-11867-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao Y., Ha A., de Lau W., Yuki K., Santos A.J.M., You C., Geurts M.H., Puschhof J., Pleguezuelos-Manzano C., Peng W.C. Next-Generation Surrogate Wnts Support Organoid Growth and Deconvolute Frizzled Pleiotropy In Vivo. Cell Stem Cell. 2020;27:840–851.e6. doi: 10.1016/j.stem.2020.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ooft S.N., Weeber F., Dijkstra K.K., McLean C.M., Kaing S., van Werkhoven E., Schipper L., Hoes L., Vis D.J., van de Haar J. Patient-derived organoids can predict response to chemotherapy in metastatic colorectal cancer patients. Sci. Transl. Med. 2019;11:eaay2574. doi: 10.1126/scitranslmed.aay2574. [DOI] [PubMed] [Google Scholar]
- Sachs N., de Ligt J., Kopper O., Gogola E., Bounova G., Weeber F., Balgobind A.V., Wind K., Gracanin A., Begthel H. A living biobank of breast cancer organoids captures disease heterogeneity. Cell. 2018;172:373–386. doi: 10.1016/j.cell.2017.11.010. [DOI] [PubMed] [Google Scholar]
- Seino T., Kawasaki S., Shimokawa M., Tamagawa H., Toshimitsu K., Fujii M., Ohta Y., Matano M., Nanki K., Kawasaki K. Human pancreatic tumor organoids reveal loss of stem cell niche factor dependence during disease progression. Cell Stem Cell. 2018;22:454–467.e6. doi: 10.1016/j.stem.2017.12.009. [DOI] [PubMed] [Google Scholar]
- Vlachogiannis G., Hedayat S., Vatsiou A., Jamin Y., Fernández-Mateos J., Khan K., Lampis A., Eason K., Huntingford I., Burke R. Patient-derived organoids model treatment response of metastatic gastrointestinal cancers. Science. 2018;926:920–926. doi: 10.1126/science.aao2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters A.M., Der C.J. KRAS: the critical driver and therapeutic target for pancreatic cancer. Cold Spring Harb. Perspect. Med. 2018;8:a031435. doi: 10.1101/cshperspect.a031435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weeber F., van de Wetering M., Hoogstraat M., Dijkstra K.K., Krijgsman O., Kuilman T., Gadellaa-van Hooijdonk C.G.M., van der Velden D.L., Peeper D.S., Cuppen E.P.J.G. Preserved genetic diversity in organoids cultured from biopsies of human colorectal cancer metastases. Proc. Natl. Acad. Sci. U S A. 2015;112:13308–13311. doi: 10.1073/pnas.1516689112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van De Wetering M., Francies H.E., Francis J.M., Bounova G., Iorio F., Pronk A., Van Houdt W., Van Gorp J., Taylor-Weiner A., Kester L. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell. 2015;161:933–945. doi: 10.1016/j.cell.2015.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This study did not generate any novel data or codes that are not presented in this published article directly.