

Abstract
The Hippo pathway plays critical roles in cell growth, differentiation, organ development, and tissue homeostasis whereas its dysregulation can lead to tumorigenesis. YAP/TAZ are transcription co-activators and represent the main downstream effectors of the Hippo pathway. Here we show that heat stress induces a strong and rapid YAP dephosphorylation and activation. The effect of heat shock on YAP is dominant to other signals known to modulate the Hippo pathway. Heat shock inhibits LATS kinase by promoting HSP90-dependent LATS interaction with and inactivation by PP5 phosphatase. Heat shock also induces LATS ubiquitination and degradation. YAP/TAZ are crucial for cellular heat shock responses, including the heat shock transcriptome and cell viability. This study uncovers previously unknown mechanisms of Hippo regulation by heat shock as well as physiological functions of YAP in heat stress response. Our observations also reveal a potential combinational therapy involving hyperthermia and targeting of the Hippo pathway.
Keywords: Hippo, YAP, LATS, phosphorylation, heat shock, HSP90, cancer
INTRODUCTION
The Hippo pathway, originally discovered in Drosophila, plays an evolutionarily conserved role in the regulation of cell growth, cell fate, organ development, and tissue homoeostasis1, 2. Dysregulation of the Hippo pathway contributes to tumorigenesis3, 4. Core components of the mammalian Hippo pathway consist of a kinase cascade, in which mammalian Ste20-like kinases 1/2 (MST1/2) or mitogen-activated protein kinase kinase kinase kinase family members (MAP4Ks) phosphorylate and activate the large tumor suppressor 1/2 (LATS1/2), and two downstream transcription co-activators, Yes-associated protein (YAP) and WW domain–containing transcription regulator protein 1 (WWTR1; also known as TAZ)5. LATS inhibits YAP and TAZ through direct phosphorylation. Inactivation of the Hippo kinase cascade leads to dephosphorylation, nuclear translocation, and activation of YAP/TAZ, which then induce gene expression mainly through binding to the TEA domain (TEAD) family of transcription factors6, 7.
Heat shock is a common physiological and pathological stress that has been extensively studied. Activation of the heat shock factor (HSF) plays a critical role in the induction of heat shock proteins (HSPs) to protect cells from heat stress8. Hyperthermia is a therapeutic procedure in which cancer is treated by raising temperature in the tumor tissue. In certain cancers, hyperthermia is applied as an adjunct treatment with various established cancer therapy strategies, such as radiotherapy and chemotherapy9. The rationale for hyperthermia is based on the cell-killing effects of high temperature. Hyperthermia can inhibit cancer cell viability by inducing unique gene expressions, inhibiting angiogenesis, and activating immune response10, 11. However, many key questions remain to be answered for hyperthermia therapy. For example, not only are the predictors of cancer sensitivity to hyperthermia unknown, but the signal pathways involved are also poorly understood9.
In the last decade, many signals have been discovered to regulate the Hippo pathway5, 12. Most signals are known to act upstream of MST1/2 and MAP4Ks to control YAP/TAZ activity. In this study, we discovered that heat shock potently induces YAP dephosphorylation and activation. Heat shock inhibits LATS by two mechanisms, rapid dephosphorylation followed by protein degradation, the former of which is likely mediated through HSP90 dependent interaction with PP5. YAP/TAZ play an important role in heat shock response by enhancing the heat shock transcriptome and cell survival.
RESULTS
YAP is activated by heat shock
To identify new signals that may control the Hippo pathway, we tested many stress conditions and found that heat shock induced YAP dephosphorylation (Fig. 1a). Shifting the high density culture, which is known to induce YAP phosphorylation13, of HEK293A and A549 cells to 43°C induced rapid and complete YAP dephosphorylation, as shown by pYAP(S127) phosphorylation and mobility shifts in phos-tag gels. Heat shock also reduced LATS1 protein whereas the levels of MST1/2 and MAP4K4/7 were unaffected (Fig. 1a). Notably, the reduction of LATS protein was slower than YAP dephosphorylation, indicating that LATS might be rapidly inactivated upon heat shock.
Figure 1. Heat shock activates YAP.

(a) Heat shock induces YAP dephosphorylation. 8×105 HEK293A and A549 cells (human lung cancer) per well were seeded onto six well plates. After 24 hrs, the cells were heat shocked at 43°C for the indicated times and cell lysates were collected for immunoblot and YAP phos-tag gel analyses.
(b) YAP dephosphorylation by heat shock is rapidly reversible. High density HEK293A and A549 cells were heat shocked at 43°C for 30 min or 1 h, then were allowed to recovered at 37°C for the indicated times. Samples were collected for YAP phos-tag gel analyses.
(c) Heat shock induces YAP/TAZ nuclear localization. HEK293A and A549 cells were heat shocked at 43°C for 30 min and then fixed and stained for immunofluorescent microscopy with an anti-YAP/TAZ antibody. Representative pictures from three independent samples are shown. Scale bars, 10 μm.
(d) Quantification of YAP/TAZ nuclear and cytosolic localization. Data were mean ± s.d.; n = 3 biologically independent samples. Two-way ANOVA test.
(e) Heat shock induces YAP target gene expression. High density HEK293A and A549 cells were subjected to heat shock for the indicated times. CTGF and CYR61 mRNAs were measured by RT-PCR. Data are presented as mean ± s.d.; n = 3 biologically independent samples. Two-way ANOVA test.
(f) Heat shock overrides other Hippo activating signals to induce YAP phosphorylation. HEK293A cells under medium confluence (4×105 cells per well seeded onto six well plates 24 h before the treatment) were treated with various YAP-inhibitory signals and environmental stresses for 1 or 2 h and then subjected to heat shock for the indicated times. YAP dephosphorylation was determined by phos-tag gel analyses. Immunoblotting in panels a, b and f has been performed two times with similar results. Source data are available online.
The heat shock transcription factor 1 (HSF-1) is a master heat responsive transcription factor and plays a key role in heat stress response 8. Heat shock caused HSF-1 mobility shift (Fig. 1a), consistent with HSF-1 phosphorylation and activation14. Notably, YAP dephosphorylation occurred slightly earlier than HSF-1 phosphorylation, suggesting that YAP dephosphorylation is not due to HSF-1 activation. Heat shock induced YAP dephosphorylation in many cell types, including HCT116, MEL270, NIH3T3, SCC7 and B16-OVA (Extended Data Fig. 1a).
We found that YAP dephosphorylation induced by heat shock was rapidly reversible when cells were shifted to 37°C, though different cell types showed different recovery kinetics (Fig. 1b). Longer exposure to heat shock caused slower recovery, possibly due to the downregulation of LATS protein. Moreover, the heat shock-induced YAP dephosphorylation sustained up to 24 hours (Extended Data Fig. 1b). Mild heat shock at 40°C induced moderate YAP dephosphorylation in A549 cells but not in HEK293A cells (Extended Data Fig. 1c). Cold temperature of 25°C or 4°C did not induce YAP dephosphorylation (Extended Data Fig. 1d).
The LATS-dependent phosphorylation of YAP/TAZ results in 14-3-3 binding and cytoplasmic localization13. As expected, immunofluorescence staining revealed that heat shock induced YAP/TAZ nuclear accumulation (Fig. 1c, d). Consistent with YAP/TAZ activation, heat shock increased the expression of YAP/TAZ target genes CTGF and CYR61 (Fig. 1e).
In addition to high density, we queried whether heat shock could activate YAP under other inhibitory conditions. Surprisingly, heat shock was able to activate YAP under all inhibitory signals tested, including serum starvation, energy stress by 2-deoxyglucose (2DG), cAMP elevation by forskolin and IBMX, F-actin disruption by latrunculin B or cytochalasin D, osmotic stress by sorbitol, and cell detachment (Fig. 1f). These observations demonstrate that heat shock universally induces a rapid and dramatic YAP activation and likely regulates the Hippo pathway at a step proximal to YAP.
Heat shock induces LATS dephosphorylation and degradation
We tested whether heat shock inhibited the Hippo kinase cascade. We found that 15 min of heat shock strongly reduced LATS phosphorylation in the hydrophobic motif (Thr1079 for LATS1) (Fig. 2a), which is an activating phosphorylation by MST or MAP4Ks15, 16. LATS1 protein levels were also decreased although slower than LATS dephosphorylation (Fig. 2b). Notably, LATS1(Thr1079) phosphorylation was still dramatically reduced even when LATS1 protein levels were normalized (Fig. 2a), indicating that heat shock inhibits LATS by two mechanisms, a rapid dephosphorylation and a slower degradation.
Figure 2. Heat shock induces LATS inactivation and degradation.

(a) Heat shock induces LATS1 dephosphorylation and degradation. High density HEK293A cells were heat shocked at 43°C. LATS1 protein and phosphorylation (Thr1079) were detected by Western blot. The lower panels were normalized against LATS1 protein.
(b) Time course of LATS dephosphorylation and degradation. Date of triplicated experiments from (a) are quantified by ImageJ and presented as mean ± s.d., two-way ANOVA test.
(c) Heat shock decreases LATS1 kinase activity. LATS1 was immunoprecipitated and assayed using GST-YAP as a substrate. The lower panels were normalized against LATS1 protein levels used in the kinase assay.
(d) LATS1 is rapidly reactivated upon recovery post heat shock. HEK293A cells were heat shocked at 43°C for 30 min and allowed to recover at 37°C for the indicated times.
(e) Heat shock has no effect on LATS1-MOB1 interaction. Cell lysates were precipitated with IgG control or LATS1 antibody. Co-immunoprecipitation of MOB1 was detected by Western blot.
(f) Time course of LATS degradation upon heat shock. LATS proteins were quantified by ImageJ and presented as mean ± s.d.; n = 3 biologically independent samples.
(g) Heat shock induces proteasome dependent LATS1 degradation. HEK293A cells were pretreated with 10 μM proteasome inhibitor (MG132) or 200 nM lysosome inhibitor (BafA1) for 6 h, followed by heat shock for the indicated times.
(h-i) Heat shock induces LATS1 ubiquitination. (h) HEK293A cells were heat shocked at 43°C. LATS1 was immunoprecipitated with control IgG or LATS1 antibody, and ubiquitin was detected by Western blot. (i) HEK293A cells were transfected with the indicated plasmids. Cells were treated with 10 μM MG132 for 6 hours and then subjected to heat shock. LATS1 was immunoprecipitated with FLAG antibody, and ubiquitination was detected with HA antibody.
(j) LATS is primarily responsible for YAP phosphorylation. HEK393A WT, YAP/TAZ DKO and LATS1/2 DKO cells were used for analyses.
(k) Heat shock selectively decreases the LATS-dependent YAP phosphorylation. HEK393A cells under median confluence were pretreated with 2-deoxyglucose (2-DG) or sorbitol, then subjected to heat shock and followed by YAP Phos-tag analysis. Immunoblotting in panels a-k has been performed two times with similar results. Source data are available online.
To directly ascertain LATS inactivation by heat shock, we measured LATS1 activity using GST-YAP as a substrate. LATS1 immunoprecipitated from heat shocked HEK293A cells exhibited a rapid decrease of kinase activity (Fig. 2c). Moreover, LATS1(Thr1079) phosphorylation was restored quickly after shifting cells to 37°C (Fig. 2d). We conclude that LATS1 phosphorylation and activity are potently and reversibly regulated by heat shock, and likely be responsible for the corresponding changes in YAP phosphorylation. The scaffold protein MOB1 interacts with and facilitates LATS phosphorylation/activation by MST1/216, 17. Co-immunoprecipitation (Co-IP) indicated that heat shock had no effect on MOB1-LATS1 interaction (Fig. 2e, Extended Data Fig. 2d).
Heat shock not only induced dephosphorylation but also reduced protein levels of LATS1 and LATS2 (Fig. 2f). To determine whether proteasome and/or lysosome is responsible for LATS1 degradation, cells were treated with the proteasome inhibitor MG132 or the lysosome inhibitor Bafilomycin A1. We found that LATS1 levels were stabilized by MG132, but not Bafilomycin A1 (Fig. 2g), suggesting that heat shock induces LATS degradation via proteasome. Consistently, heat shock increased LATS1 ubiquitination (Fig. 2h, i). Neither MG132 nor Bafilomycin A1 affected the dephosphorylation of LATS1 and YAP (Fig. 2g), indicating that heat shock induces LATS1 dephosphorylation and degradation via independent mechanisms, and LATS inactivation by dephosphorylation is largely responsible for the reduced YAP phosphorylation.
To further support our model, we examined YAP phosphorylation in LATS1/2 DKO cells. LATS1/2 knockout abolished the vast majority of YAP phosphorylation (Fig. 2j), as predicted by the current dogma that LATS is the major YAP kinase. However, energy starvation by 2-DG or osmotic stress by sorbitol could still induce some YAP phosphorylation in LATS1/2 DKO cells (Fig. 2k), consistent with previous reports that AMPK and NLK could directly phosphorylate YAP under energy starvation and osmotic stress, respectively18-20. However, the LATS independent YAP phosphorylation was largely insensitive to heat shock (Fig. 2k), supporting the notion that heat shock acts through LATS to modulate YAP phosphorylation.
MST and MAP4Ks are not key mediators from heat shock to LATS inactivation
We investigated whether MST1/2 and MAP4Ks are involved in heat shock-induced YAP regulation by comparing HEK293A wild-type (WT), MST1/2 double-knockout (DKO), and MAP4K4/6/7 triple-knockout (TKO) cell lines. Deletion of MAP4K4/6/7 caused a moderate delay in the dephosphorylation of YAP and LATS1 in response to heat stress (Fig. 3a). In contrast, MST1/2 deletion slightly accelerated these effects on both YAP and LATS1. Deletion of MST1/2 or MAP4Ks did not affect LATS1 protein levels (Fig. 3a). In response to heat shock, MAP4K4/6/7 TKO cells showed similar delays in YAP dephosphorylation under high cell density or serum starvation conditions (Extended Data Fig. 2a). Consistently, YAP/TAZ nuclear accumulation by heat shock was slower in the MAP4K4/6/7 TKO cells. However, YAP still translocated into the nucleus when exposed to heat shock for longer durations (Fig. 3b, c). Upon recovery at 37°C following 1 hour of heat shock, YAP re-phosphorylation was moderately delayed in MST1/2 DKO, but not in the MAP4K4/6/7 knockout cells (Fig. 3d). These observations indicate that neither MST1/2 nor MAP4K4/6/7 are absolutely required for heat shock-induced YAP regulation.
Figure 3. MST1/2 and MAP4Ks are not required for heat shock-induced YAP regulation.

(a) Deletion of MAP4Ks, but not MST, slightly delays heat shock-induced YAP dephosphorylation. HEK393A WT, MST1/2 DKO, and MAP4K4/6/7 TKO cells under medial confluence were pretreated with 2-DG (left panels) or sorbitol (right panels) and then subjected to heat shock. The phosphorylation of YAP and LATS1 were analyzed by Western blot.
(b) MAP4K4/6/7 knockout delays YAP nuclear localization. Cells were stained for YAP/TAZ for immunofluorescent microscopy. Representative pictures from three independent samples are shown. Scale bars,10 μm.
(c) Quantification of YAP/TAZ nuclear and cytosolic localization. Data are mean ± s.d.; n = 3 biologically independent samples. Two-way ANOVA test.
(d) MST1/2 knockout delays recovery after heat shock. HEK293A WT, MST1/2 DKO, and MAP4K4/6/7 TKO cells were pretreated with 2-DG, then subjected to heat shock for 1 h followed by recovery at 37°C for the indicated durations.
(e) Heat shock does not affect MST1 and MAP4K4 kinase activity. Endogenous MST1 (left panel) or MAP4K4 (right panel) immunoprecipitated from heat shocked HEK293A cells was assayed using GST-LATS2 as a substrate. LATS2 phosphorylation was determined with pLATS (Thr1079) antibody.
(f) Heat shock increases LATS and MAP4K4 interaction. HEK293A cells were co-transfected with FALG-MAP4K4 and HA-LATS1. 24 h after transfection, cells were subjected to heat shock. HA (left panel) and FLAG (right panel) antibodies were used for immunoprecipitation and the co-precipitated proteins were detected by Western blot.
(g) Heat shock increases LATS and MST1 interaction. Experiments were similar to panel f except cells were co-transfected with FLAG-MST1 and HA-LATS1 (left panel) or 3×HA-LATS2 (right panel).
(h) YAP re-phosphorylation time course in MST1-rescued or MAP4K4-rescued MM8KO cells after shifting back to 37°C. Plasmids for HA-YAP and FLAG-MST1 or FLAG-MAP4K4 were co-transfected into HEK293A MM8KO cells. One day after transfection, cells were subcultured to new plates and reached medial confluence the next day, serum starved for 2 h, then subjected to heat shock at 43°C for 1 h followed by recovery at 37°C for the indicated durations. Immunoblotting in panels a and d-h has been performed two times with similar results. Source data are available online.
We measured kinase activity of MST1 and MAP4K4 and found that heat shock had no detectable effect on either (Fig. 3e). Consistently, heat shock had little effect on the phosphorylation of GST-MST1 (Extended Data Fig. 2b). Intriguingly, heat shock elevated the interaction of LATS-MAP4K4 or LATS-MST1 (Fig. 3f, g). The functional implication of these increased interactions is currently unclear. To further investigate the role of MST1/2 or MAP4Ks, we rescued the MM8KO (MST1/2-MAP4K1/2/3/4/6/7 KO) cells with either MST1 or MAP4K4. The MST1-rescued cells showed a slower YAP dephosphorylation than that of MAP4K4-rescued cells upon heat shock (Extended Data Fig. 2c). Moreover, the MST1-rescued cells recovered faster than the MAP4K4-rescued cells (Fig. 3h). Collectively, our data indicate that MST1 or MAP4K4 unlikely acts as a key mediator relaying heat shock signal to LATS regulation, although they may still phosphorylate LATS1 during the heat shock.
HSP90 is involved in heat shock-induced YAP dephosphorylation
HSF-1, HSP70, and HSP90 play critical roles in heat shock response 21. It has been reported that inhibition of HSP90 by 17-AAG caused YAP dephosphorylation and nuclear localization22, 23. However, both studies were solely based on pharmacological HSP90 inhibition and heat shock response was not examined. To test the roles of these genes in YAP regulation, we generated HEK293A knockout cell lines by CRISPR-Cas9 technology 24. Deletion of HSF-1 completely abolished expression of HSF-1 target genes (Extended Data Fig. 3a). However, the heat shock-induced YAP mobility shifts were not affected by HSF-1 deletion (Fig. 4a). Pretreatment of cells with chemical chaperone 4-PBA or DMSO25 did not block YAP dephosphorylation upon heat shock (Extended Data Fig. 3b).
Figure 4. HSP90 deletion compromises heat shock-induced dephosphorylation of LATS and YAP.

(a) Heat shock-induced YAP dephosphorylation is not altered in HSF-1 KO cells. HEK293A WT and HSF-1 KO cells (generated by CRISPR) were subjected to heat shock and cell lysates were analyzed by Western blot. Two independent HSF-1 KO clones are shown.
(b) HSP70-1/2 deletion moderately delays YAP dephosphorylation induced by heat shock. HEK293A WT and HSP70-1/2 DKO cells were subjected to heat shock and phos-tag gel was used to detect YAP phosphorylation. Two independent HSP70 KO clones are shown.
(c) Deletion of HSP90α/β compromises dephosphorylation of LATS1 and YAP by heat shock. HEK293A WT and HSP90α/β DKO cells were subjected to heat shock and cell lysates were analysed by Western blot. Two independent HSP90α/β DKO clones are shown. LE denotes long exposure of the Western blot.
(d) Knockdown of HSP90α/β compromises the heat shock-induced dephosphorylation of YAP and LATS1. HEK293A cells were transfected with control siRNA or siRNAs for HSP90α and HSP90β. Two independent siRNAs were used.
(e) The knockdown efficiency of HSP90α/β was confirmed by quantitative Real-Time PCR. Data are presented as mean ± s.d.; n = 3 biologically independent samples. Two-way ANOVA test.
(f) HSP90α/β knockdown delays the heat shock-induced LATS inactivation. Endogenous LATS1 was immunoprecipitated from heat shocked-high density HEK293A cells with HSP90α/β knockdown or overexpression. In vitro kinase assays were performed using recombinant GST-YAP as the substrate. Phosphorylation of GST-YAP was determined by immunoblotting with the pYAP (Ser127) antibody.
(g) Heat shock increases the interaction between HSP90α and LATS1 or MAP4K4. The plasmids for FLAG-MAP4K4, HA-LATS1, MYC-HSP90AA1 (left panel) or MYC-HSP90AB1 (right panel) were transiently co-transfected into HEK293A cells. One day after transfection, cells were subjected to heat shock for the indicated times. MYC antibodies were used for immunoprecipition and the associated FLAG-MAP4K4 and HA-LATS1 were detected by Western blot. Immunoblotting in panels a-d, f and g has been performed two times with similar results. Source data are available online.
Deletion of HSP70-1/2 had only a minor delay in YAP dephosphorylation (Fig. 4b), indicating that HSP70 is largely dispensable for YAP regulation by heat shock. HSP90 is essential for cell growth and survival and has two major isoforms HSP90α (encoded by HSP90AA1, inducible) and HSP90β (encoded by HSP90AB1, constitutive)26. We utilized two HSP90α/β partial knockout clones for our experiments. Partial deletion of HSP90α/β compromised the heat shock-induced dephosphorylation of YAP and LATS1 (Fig. 4c, Extended Data Fig. 3c, d). LATS1 degradation was also compromised in HSP90α/β DKO cells (Fig. 4c). Transient knockdown of HSP90α/β by short interfering RNA (siRNA) also largely blocked the heat shock-induced dephosphorylation of YAP and LATS1 (Fig. 4d, e, Extended Data Fig. 3e). Furthermore, HSP90 knockdown delayed LATS inactivation by heat shock whereas HSP90 overexpression had no effect (Fig. 4f).
We observed that heat shock rapidly enhanced the association of the inducible HSP90α with LATS1 (Fig. 4g), especially considering the reduction of LATS protein. In contrast, heat shock had little effect on the interaction of the constitutively expressed HSP90β with LATS1 or MAP4K4 (Fig. 4g). Taken together, HSP90 plays a critical role in heat shock-induced LATS1 degradation and dephosphorylation, the latter is possibly via the heat shock-inducible interaction of HSP90-LATS.
PP5 is involved in LATS dephosphorylation by heat shock
Our data indicate that dephosphorylation appears to be primarily responsible for rapid LATS inactivation by heat shock. Protein phosphatase 5 (PP5) is known to be activated by association with HSPs, including HSP9027, 28. We found that the dephosphorylation of LATS1 and YAP by heat shock was diminished in the PP5 KO cell pools generated by two independent CRISPR sequences (Fig. 5a). Similar results were observed in cells with PP5 knockdown by siRNA (Fig. 5b, c). Treatment with LB-100, which inhibits both PP2A and PP5, also blocked the dephosphorylation of YAP and LATS1 (Fig. 5d). These observations support a role of PP5 in heat shock-induced LATS dephosphorylation.
Figure 5. PP5 interacts with LATS1 and is involved in LATS1 dephosphorylation by heat shock.

(a) PP5 deletion diminishes the dephosphorylation of LATS1 and YAP by heat shock. HEK293A WT and PP5 deficient cell pools were subjected to heat shock. Two independent CRISPR guide sequences were used.
(b) PP5 knockdown delays the heat shock-induced dephosphorylation of LATS1 and YAP.
(c) Verification of PP5 siRNA knockdown by RT-PCR. Data are presented as mean ± s.d.; n = 3 biologically independent samples. Two-sided unpaired t-test.
(d) PP5 inhibition blocks the dephosphorylation of YAP and LATS1 induced by heat shock. High density HEK293A cells were pretreated with LB-100 (3μM or 5μM) for 3 hours and then subjected to heat shock at 43°C for indicated times.
(e) PP5 knockout preferentially affects YAP dephosphorylation by heat shock. HEK293A WT and PP5 deficient cell pool #2 were serum starved, then treated with LPA or heat shock.
(f-g) Heat shock stimulates LATS1-PP5 interaction. (f) HEK293A cells transfected with FLAG-PP5 were heat shocked for the indicated times. FLAG antibodies were used for immunoprecipition and the co-precipitated endogenous LATS1 was detected by Western blot. (g) HEK293A cells were transiently co-transfected with indicated plasmids and then subjected to heat shock one day after transfection. FLAG-PP5 (left panels) or HA-LATS1 (right panels) antibodies were used in immunoprecipitation, and the respectively co-precipiated HA-LATS1 (left panels) or FLAG-PP5 (right panels) were detected by Western blot.
(h) HSP90 deficiency reduces LATS1-PP5 interaction induced by heat shock. HEK293A cells were transfected with control siRNA or a siRNA pool targeting HSP90α and HSP90β. 48 h after siRNA transfection, cells were subjected to heat shock for the indicated times, followed by immunoprecipition with FLAG antibody. LE denotes long exposure of the Western blot.
(i) PP5 depletion delays LATS1 inactivation induced by heat shock. LATS1 was immunoprecipitated from heat shocked HEK293A cells with control or PP5 siRNA knockdown. LATS1 activity was determined using GST-YAP as a substrate. Phosphorylation of GST-YAP was determined with the pYAP (Ser127) antibody. Immunoblotting in panels a, b and d-i has been performed two times with similar results. Source data are available online.
(j) A proposed model for heat shock regulation of YAP/TAZ via HSP90 and PP5. See discussion for details.
To test whether PP5 is specifically involved in LATS regulation by heat shock, we examined the effect of LPA, which is known to induce LATS inactivation by acting upstream of MST and MAP4K29, 30, in PP5 deficient cells. YAP dephosphorylation induced by LPA was not affected by PP5 deficiency (Fig. 5e) whereas the effect of heat shock was significantly altered, suggesting a selective role of PP5 in heat shock response. We observed that heat shock increased the interaction of transfected FLAG-PP5 with endogenous LATS1 (Fig. 5f). Similarly, heat shock increased the association between HA-LATS1 and FLAG-PP5 (Fig. 5g). Interestingly, the interaction between LATS1 and PP5 was reduced in HSP90α/β knockdown cells (Fig. 5h). Moreover, PP5 deletion delayed LATS inactivation by heat shock (Fig. 5i). These data suggest a model that HSP90 acts as a scaffold to facilitate the heat shock-induced LATS1 association with and dephosphorylation by PP5 (Fig. 5j). This model explains why heat shock is epistatic to many YAP inhibitory signals (Fig. 1) as these signals act upstream of MST1/2 and MAP4Ks.
YAP/TAZ enhance heat shock transcriptome and cell survival
Heat stress induces gene expression to provide adaptation and protection from harmful environment31. We deleted YAP/TAZ in mouse melanoma B16-OVA cells (Fig. 6a, Extended Data Fig. 4a). It is noteworthy that the YAP/TAZ knockout cells grow slower. YAP/TAZ deletion was functionally confirmed by the reduced expression of target genes CTGF and CYR61 (Extended Data Fig. 4b). We found that 43°C heat shock caused a small increase of cell death while 45°C caused more dramatic cell death, thus we used 45°C in the cell survival experiments. 45°C heat shock also induced YAP dephosphorylation (Fig. 6b). We compared cell viability of B16-OVA WT, YAP/TAZ DKO#2, and LATS1/2 DKO cells after 45°C heat shock for 1 hour. LATS1/2 DKO cells showed similar viability compared with WT cells; however, YAP/TAZ deletion significantly decreased viability from 80% to 45% (Fig. 6c). YAP/TAZ DKO#2 cells displayed more early apoptosis (Annexin V positive and 7-AAD negative) and late apoptosis/necrosis (double positive in Annexin V and 7-AAD), while no difference was found between WT and LATS1/2 DKO cells (Fig. 6d). Notably, basal apoptosis was slightly elevated by YAP/TAZ deletion. Trypan blue staining also confirmed more death of YAP/TAZ DKO#2 cells (Fig. 6e, f). The above data indicate that YAP/TAZ activation plays an important protective role in cellular heat stress adaptation.
Figure 6. YAP/TAZ have a protective role in heat shock response.

(a) YAP/TAZ knockout in mouse B16-OVA melanoma cells.
(b) 45°C heat shock induces YAP dephosphorylation in B16-OVA cells. Cells under high density were treated at 45°C for the indicated times.
(c) YAP/TAZ deletion increases B16-OVA sensitivity to heat shock. B16-OVA WT, YAP/TAZ DKO#2, and LATS1/2 DKO cells were subjected to heat shock at 45°C for 1 h and placed back at 37°C. 24 h later, cell viability was determined by CCK8 assays. Data are mean ± s.d.; n = 3 biologically independent samples. One-way ANOVA test, ns, not significant.
(d) YAP/TAZ deletion enhances heat shock-induced cell death. Flow cytometry of cells stained with PE-Annexin V and 7-AAD. Representative results from two independent experiments are shown.
(e) YAP/TAZ deletion reduces cell viability upon heat shock. Experiments were similar to panel (c). Cells were stained with trypan blue for dead cells. Representative pictures from three independent samples are shown, scale bar, 50 μm.
(f) Quantification of results from panel e. DATA are mean ± s.d.; n = 3 biologically independent samples. Two-way ANOVA test, ns, not significant.
(g) YAP/TAZ are required for proper HSP gene induction by heat shock. B16-OVA WT, YAP/TAZ DKO#2, and LATS1/2 DKO cells were treated with heat shock at 45°C for 1 h and then recovered at 37°C for 6 h. HSP gene expressions were determined by RT-PCR. Data are mean ± s.d.; n = 3 biologically independent samples. Two-way ANOVA test, ns, not significant.
(h) YAP/TAZ knockout impairs HSP protein induction by heat shock. B16-OVA cells were used in the experiment.
(i) Heat shock increases YAP binding to promoters of HSPs and CTGF. HCT116 cells under high density were treated with heat shock at 43°C for 0 h or 1h, followed by ChIP with control IgG or YAP antibody. The precipitated DNA was quantified by PCR with primers specific to promoter regions of the indicated genes. CTGF was included as a positive control. Data are mean ± s.d.; n = 3 biologically independent samples. Two-way ANOVA test. Immunoblotting in panels a, b and h has been performed two times with similar results. Source data are available online.
In response to heat stress, cells upregulate HSPs for cellular homeostasis and protection 31, 32. We measured the classic HSPs, HSP25, HSP70, and HSP90 and found that their induction by heat shock was compromised in YAP/TAZ DKO#2 cells (Fig. 6g, h). Even basal HSP90α was repressed by YAP/TAZ deletion (Fig. 6h). YAP/TAZ knockdown by siRNA in B16-OVA also diminished HSP gene induction and increased apoptosis/necrosis upon heat shock (Extended Data Fig. 5a-e). Moreover, YAP/TAZ knockdown in SCC7 squamous carcinoma cells or HCT116 colon cancer cells similarly increased sensitivity to heat shock (Extended Data Fig. 5f-i). Furthermore, we conducted chromatin immunoprecipitation (ChIP) assay and observed that YAP bound to the promoters of HSPA1A, HSPB1 and DNAJB1, and this YAP interaction with HSP promoters was stimulated by heat shock (Fig. 6i). Taken together, our observations support a model that YAP/TAZ protect cells from heat stress, by directly promoting HSP expression.
We next performed RNA-SEQ of control and YAP/TAZ knockdown B16-OVA cells. In control siRNA treated cells, heat shock lead to upregulation of 418 genes and downregulation of 307 genes (Fig. 7a). Among the upregulated genes, YAP/TAZ knockdown completely abolished the induction of 348 genes and partially diminished the induction of 48 genes (Fig. 7b). Only 22 of the 418 inducible genes were not affected by YAP/TAZ knockdown. These data demonstrate a previously unrecognized profound role of YAP/TAZ in heat shock response. Gene Ontology analysis revealed that the YAP/TAZ-dependent genes are involved in phosphorylation, hemopoiesis, angiogenesis, stress response, and apoptosis (Extended Data Fig. 6a). These genes are involved in cell growth and survival control, such as MAPK and PI3K-AKT pathways (Extended Data Fig. 6b). The altered gene expressions of representative HSPs were confirmed by the FPKM (Extended Data Fig. 6c, d). Our RNA-SEQ data show that YAP/TAZ are required for proper induction of the heat shock transcriptome.
Figure 7. YAP/TAZ knockdown sensitizes B16-OVA tumor growth to hyperthermia in vivo.

(a) RNA sequencing analysis of B16-OVA control and YAP/TAZ knockdown cells. MA-plot of differentially expressed genes (DEGs) of B16-OVA siCon or siYAP/TAZ#2 treated cells (No Treatment vs. Heat Shock). Up DEGs (FPKM>1, Log2 Fold Change>1, adjusted p value<0.05) are counted and colored red; Down DEGs (FPKM>1, Log2 Fold Change<−1, adjusted p value<0.05) are counted and colored blue. n = 3 biologically independent samples were analyzed for each condition.
(b) Pie chart showing 418 up-regulated genes in siCon cells completed or partially blocked by YAP/TAZ knockdown.
(c) Animal experiment schedule. Mice were injected subcutaneously with B16-OVA cells. MP/siRNA (10 μg siRNA/mouse) were administered via intratumoral injection twice weekly starting on day 8 after cell injection. On day 11, 15 and 18, tumors were immersed in the 43°C -water bath for 30 min. The mice were sacrificed and tumors were collected on day 21.
(d) Tumor growth was monitored at the indicated times. Data are presented as mean ± s.e.m.; n = 8 tumors for each group. Two-way ANOVA test.
(e) Average tumor weights of B16-OVA on day 21 are shown (n = 8 tumors/group). mean ± s.e.m. One-way ANOVA test.
(f) Pictures showing B16-OVA tumor burden in mice.
(g) Western blot showing the knockdown of YAP/TAZ by siRNAs in B16-OVA cells on day 15 and 21 of the syngeneic. The samples were from pooled tumors (n = 3 tumors/group). Immunoblotting in panels g has been performed two times with similar results.
(h) Immunohistochemistry of cleaved caspase-3. Image is of representative tumor tissue (scale bar, 50 μm). Data are mean ± s.e.m.; n = 5 biologically independent fields. One-way ANOVA test. Source data are available online.
YAP/TAZ knockdown sensitizes B16-OVA tumor to hyperthermia
We evaluated the effect of YAP/TAZ silencing on tumor growth in murine syngeneic models in response to hyperthermia. Tumors grown from subcutaneously injected B16-OVA were treated with local hyperthermia using a 43°C water bath for 30 min on day 3 after siYAP/TAZ injection (Fig. 7c). We harvested tumors on day 21 and found that YAP/TAZ knockdown modestly reduced tumor volume and weight. Local hyperthermia also slightly decreased tumor weights in control siRNA (siCon) group. However, tumor growth of the YAP/TAZ knockdown B16-OVA cells was markedly suppressed by local hyperthermia (Fig. 7d-f). YAP/TAZ siRNA indeed reduced YAP/TAZ expression (Fig. 7g). Immunohistochemical staining showed a strong increase of cleaved caspase-3, an apoptosis marker, in the YAP/TAZ siRNA plus hyperthermia treatment group, compared to the YAP/TAZ siRNA or hyperthermia alone group (Fig. 7h). Collectively, these results show that YAP/TAZ knockdown sensitizes B16-OVA tumor cells to hyperthermia treatment.
DISCUSSION
In this study, we discovered that YAP is strongly activated by heat shock in many cell types and conditions. Heat shock-induced YAP activation is rapid, sustained, and reversible. The heat shock effect on YAP is rather unique as it is dominant over all YAP inhibitory signals tested. Interestingly, heat shock does not appear to act through MST and MAP4Ks, but rather directly impinges upon LATS inactivation. This is likely achieved by the heat shock-promoted association of LATS with PP5. Moreover, HSP90 is required for the interaction of LATS and PP5 by heat shock. We propose that heat shock inactivates LATS by promoting its association with and dephosphorylation by PP5 in a manner dependent on HSP90 (Fig. 5j). Consistent with this model, deletion of PP5 or HSP90 compromised the dephosphorylation of both LATS and its downstream substrate YAP in response to heat shock. Our model also explains why heat shock is dominant over many other YAP inhibitory signals as heat shock acts at the level of LATS while most other signals act upstream of MST and MAP4Ks. However, we cannot exclude the possibility that PP5 may participate in YAP dephosphorylation. Moreover, besides LATS inactivation, heat shock may activate phosphatase to dephosphorylate YAP.
Intriguingly, although the kinase activity of MST1 or MAP4K4 was not altered by heat shock, their interaction with LATS was increased (Fig. 3e-g), suggesting that MST1/2, as well as MAP4K4/6/7, may still phosphorylate LATS1 during heat shock. We speculate that MST1/2 is the major kinase, but can be partly compensated by MAP4K4/6/7, for LATS phosphorylation during heat shock. This may explain the delayed recovery of YAP re-phosphorylation at 37°C and the faster dephosphorylation of YAP upon heat shock in MST1/2 DKO cells (Fig. 3a, b, d). The rescue experiment in MM8KO cells further validates this hypothesis (Fig. 3h and Extended Data Fig. 2c). We speculate that the above phenomenon could be a consequence of cellular adaptation caused by a dynamic equilibrium between LATS phosphorylation and dephosphorylation. Nevertheless, our observations suggest that MST and MAP4K4 kinases are not key mediators of LATS inactivation in response to heat shock.
Although LATS dephosphorylation by heat shock was severely compromised in the HSP90 knockout or knockdown cells (Fig. 4c, d), YAP dephosphorylation was delayed but still occurred. Moreover, PP5 knockout or knockdown delayed but did not completely abolish the dephosphorylation of LATS and YAP in response to heat shock. These data indicate that additional mechanisms contribute to heat shock-induced regulation of the Hippo pathway, thus future studies are warranted.
Heat shock inhibits LATS by two mechanisms, dephosphorylation and protein degradation, but these two mechanisms appear to operate independently and with different time courses. HSP90 knockout blocks both LATS dephosphorylation and degradation, indicating the critical role of HSP90. Several E3 have been implicated in the LATS ubiquitination, including ITCH and SIAH233-35. We have made serious efforts to address the LATS E3 ubiquitin ligases. Knockdown ITCH or SIAH2 alone or in combination did not block the heat shock-induced LATS1 reduction (Extended Data Fig. 7). However, when both ITCH and SIAH2 were knocked down, basal LATS1 protein level was elevated (Extended Data Fig. 7). Further studies are needed to reveal the mechanism of LATS ubiquitination induced by heat stress.
A trivial explanation of our findings is that LATS inactivation is a nonspecific consequence of HSP limitation under heat shock if HSP is simply required for kinase folding and stability. However, our data strongly argue against this “nonspecific” model for the following reasons. First, LATS dephosphorylation occurs prior to the reduction of LATS proteins. Inhibition of LATS degradation by MG132 does not prevent LATS inactivation by heat shock. Second, the LATS phosphorylation is rapidly reversible upon temperature switch. Third, HSP90 knockout does not exacerbate LATS dephosphorylation or degradation as would be predicted by the “nonspecific” model. In contrast, HSP90 knockout suppresses LATS dephosphorylation and degradation by heat shock. Consistently, deletion of HSF-1, which is critically important for HSPs induction, has no effect on LATS regulation. Furthermore, we examined phosphorylation of many proteins, including numerous kinases, and found that heat shock did not cause a universal protein dephosphorylation and degradation (Extended Data Fig. 8). Collectively, our study shows that the Hippo pathway is actively regulated by heat shock.
Heat shock response is a ubiquitous and highly conserved protection mechanism 31. An immediate and prominent heat shock response is the rapid induction of genes, including HSP36. The fact that heat shock always activates YAP regardless of cell types or culture conditions suggests that YAP has important physiological roles in heat stress response. Notably, the C. elegans YAP-1 has been reported to be involved in thermotolerance37. Here, we show that YAP/TAZ are required for proper induction of the heat shock transcriptome, including many HSPs. 95% of heat inducible genes is blocked (83% completely and 12% partially) by YAP/TAZ deletion in the B16-OVA melanoma cells, suggesting a previously unrecognized and surprisingly prominent role of YAP/TAZ in the expression of heat inducible transcriptome. It should be noted that activation of YAP by LATS1/2 deletion was not sufficient to induce HSP expression while deletion of HSF-1 completely abolished the induction of HSPs (Fig. 6g and Extended Data Fig. 3a). We have analyzed ENCORE data (https://genome.ucsc.edu/cgi-bin/hgTrackUi?db=hg19&g=wgEncodeRegTfbsClusteredV3) for 45 heat inducible genes that are dependent on YAP/TAZ based on our RNA-SEQ data. Interestingly, genomic co-occupancy of TEAD4 and HSF-1 is found in the enhancers/promoters of more than 75% of these genes (Supplementary Table 1). These observations suggest that YAP/TAZ cooperate with HSF-1 to induce the heat shock transcriptome at least in B16 melanoma cells. Future studies to determine YAP regulation by heat shock under more physiological conditions, such as in heart and skin keratinocytes, will broaden the biological significance of our findings.
Consistent with their role in heat shock gene induction, silencing YAP/TAZ in B16-OVA cells increases apoptosis and suppresses tumor growth upon hyperthermia treatment. This study reveals an important interplay between YAP and cellular heat shock response, and suggests a possibility that targeting the Hippo pathway may enhance clinical effectiveness of hyperthermia therapy in cancer.
METHODS
Cell culture, transfection and heat shock
All the cell lines were cultured at 37°C with humidified 5% CO2. HEK293A, A549 and NIH3T3 were cultured in DMEM (Gibco) and A549, HCT116, B16-OVA and SCC7 were cultured in RPMI 1640 (Gibco) supplemented with 10% fetal bovine serum (FBS, Gibco) and 50 μg/ml penicillin/streptomycin (Gibco). The cell lines were tested to be free of mycoplasma contamination. YAP-inhibitory signals and environmental stresses included the following: high cell density (100% confluence), serum starvation (2 h), glucose starvation (2-DG, 25 mM, 1 h), PKA activation (forskolin/IBMX, 10 μM/100 μM, 1 h), disruption of F-actin (latrunculin B, 0.25 μg/ml, 1 h), inhibition of actin polymerization (Cytochalasin D, 0.5 μM, 1 h), sorbitol (0.5 M, 0.5 h), and cell detachment (1 h).
PolyJet Reagent (SignaGen Laboratories) was used for all the in vitro plasmid DNA transfection according to the manufacturer’s protocol. For all the in vitro heat shock experiments, cells were seeded in 6 well plates (2 ml medium/well) and heat shocked in a culture incubator (Thermo Scientific) with humidified 5% CO2. The temperature of the incubator was set at least one day before the heat shock experiment and was always monitored by a mercurial thermometer. A tray filled with water was always be placed in the incubator to provide a relatively high humidity which favors efficient heat shock.
Antibodies
The following antibodies were purchased from Cell Signaling and used at the indicated dilution for western blot analysis and immunohistochemistry: YAP (D8H1X) (14074, 1:1,000), LATS1 (C66B5) (3477, 1:2,000), LATS2 (D83D6) (5888, 1:1,000), pLTAS1(T1079) (D57D3) (8654, 1:1,000), pYAP (S127) (4911, 1:1,000), MST1 (3682, 1:2,000), MST2 (3952, 1:2,000), pMST1 (T183) (E7U1D) (49332, 1:500), HSF-1 (D3L8I) (12972, 1:2,000), Ubiquitin (P4D1) (3936, 1:2,000), PP5 (2289, 1:2,000), MOB1 (E1N9D) (13730, 1:1,000), pMOB1(T35) (D2F10) (8699, 1:1,000), pAKT (D9E) (S473) (4060, 1:4,000), pSRC (Y416) (2101, 1:1,000), p70 S6 Kinase (49D7) (2708, 1:4,000), p-p70 S6 Kinase (T389) (108D2) (9234, 1:1,000), Erk1/2 (137F5) (4695, 1:4,000), pErk1/2 (T202/Y204) (D13.14.4E) (4370, 1:4,000), cleaved caspase 3 (9661, 1:1,000), HA (C29F4) (3724, 1:2,000) , HA-HRP (6E2) (2999, 1:5,000) and Myc-HRP (9B11) (2040, 1:5,000). The following antibodies were purchased from Santa Cruz Biotechnology and used at the indicated dilution for western blot analysis: YAP/TAZ (63.7) (sc-101199, 1:1,000), GAPDH (sc-25778, 1:4,000) and HSP90β (sc-7947, 1:4,000). FLAG (M2) (F1804, 1:2,000), FLAG-HRP (M2) (A8592, 1:5,000) and GST (2H3-D10) (SAB4200237, 1:4,000) were purchased from Sigma, MAP4K4 (A301-502A, 1:2,000) and MAP4K7 (A310-985A, 1:1,000) were purchased from Bethyl Laboratories, HSP90α (610418, 1:2,000) was purchased from BD Biosciences, HSP70 (10995-1-AP, 1:4,000), ITCH (20920-1-AP, 1:2,000), SIAH2 (12651-1-AP, 1:2,000) and AKT (10176-2-AP, 1:4,000) was purchased from Proteintech, SRC (JF0947) (ET1702-03, 1:5,000) was purchased from HuaAn Biotechnology Co., Ltd and HSP25 (ADI-SPA-801-D, 1:4,000) was purchased from Enzo Life Sciences. Alexa Fluor 488 secondary antibodies (A-11001, 1:1000) was purchased from Life Technologies.
CRISPR-mediated gene deletion
CRISPR genomic editing technology was used to delete genes in HEK293A and B16-OVA cells. The guide RNA sequences were cloned into the px459 plasmid (Addgene 48319). The constructed plasmids were transfected into HEK293A or B16-OVA using PolyJet. 24 h after transfection, the transfected cells were enriched by 2 μg/ml puromycin selection for 2–3 days and then were sorted into 96-well plates with only one cell in each well by FACS (UCSD; Human Embryonic Stem Cell Core, BDInflux). The clones were screened by western blot analysis with gene-specific antibodies. B16-OVA/LATS1/2 DKO, HEK293A/MST1/2 DKO and MAP4K4/6/7 TKO cells were generated as previously described38,39.
The single-guide RNA (sgRNA) sequences targeting individual genes were as follows:
human HSF-1#1: 5’-CAGCTTCCACGTGTTCGACC-3’;
human PP5#1: 5’-CGCGCTGCGAGACTACGAGA-3’;
human PP5#2: 5’-ACGCGCTGGGAGACGCCACG-3’;
human HSPA1A#2: 5’-AACCGGCATGGCCAAAGCCG-3’;
human HSPA1A#3: 5’-GGTGCTGGACAAGTGTCAAG-3’;
human HSPA1B#2: 5’-CACCGGCATGGCCAAAGCCG-3’;
human HSPA1B#3: 5’-GGTTCTGGACAAGTGTCAAG-3’;
human HSP90AA1#3: 5’-TTCTCTTGCAGGTGAACCTA-3’;
human HSP90AB1#3: 5’-CATTGCTATTTATTCCTCGT-3’;
mouse YAP1#2: 5’-GCCCAAGTCCCACTCGCGAC-3’,
mouse WWTR1#2: 5’-GCAGTGTCCCAGCCGAATCT-3’.
In vitro RNA interference
Duplex siRNAs targeting human HSP90AA1, HSP90AB1, or PP5 were purchased from Integrated DNA Technologies, Inc. (Skokie, USA) and siRNAs targeting human ITCH or SIAH2, mouse YAP1 and WWTR1 were purchased from GenePharma (Shanghai, China). siRNAs were transfected into cells in vitro with Lipofectamine® RNAiMAX (Invitrogen) in accordance with the manufacturer’s instructions.
The sequences are as follows:
siCon: UUCUCCGAACGUGUCACGUTT;
human siHSP90AA1#1: GCAUGGAAGAAGUAGACUAAUCUCT;
human siHSP90AA1#2: UUGACAAUUCUGCAUGUACUAGUCC;
human siHSP90AB1#1: GGACAGUGGUAAAGAGCUGAAAATT;
human siHSP90AB1#2: AGGCAGUAAACUAAGGGUGUCAAGC;
human siPP5#1: GUGACAAUCAGUUUCAUGAAGGAGC;
mouse siYAP1#1: GGUCAAAGAUACUUCUUAATT;
mouse siYAP1#2: CCAAUAGUUCCGAUCCCUUTT;
mouse siWWTR1#1: CCAGGAAGGUGAUGAAUCATT;
mouse siWWTR1#2: GCCGAAUCUCGCAAUGAAUTT.
human siYAP1#1: GGUCAGAGAUACUUCUUAATT;
human siYAP1#2: CCGUUUCCCAGACUACCUUTT.
human siWWTR1#1: CCUGCCGGAGUCUUUCUUUTT;
human siWWTR1#2: GGUACUUCCUCAAUCACAUTT.
human siITCH#2: CGGGCGAGUUUACUAUGUATT.
human siSIAH2#2: GUUCGAUUCAUGACGGUGUTT.
Western blot and immunoprecipitation.
Immunoblotting was performed using a standard protocol. The phos-tag reagents were purchased from Wako Chemicals, and gels containing phos-tag were prepared following the manufacturer’s instructions. For immunoprecipitation, HEK293A cells were lysed with mild lysis buffer (MLB, 50 mM Tris at pH7.5, 150 mM NaCl, 0.5% Triton-100, protease & phosphatase inhibitor cocktail) and centrifuged at 13, 000 rpm for 20 min at 4°C. Then supernatants were incubated with the appropriate antibodies with rotation overnight at 4°C and 10 μl Pierce Protein A/G Magnetic Beads (Thermo Scientific) were added in for additional 2 h. Immunoprecipitates were washed three times with mild lysis buffer, and then immunoprecipitated proteins were denatured by the addition of sample buffer and boiling for 10 min, resolved by 8% SDS–PAGE, and analyzed via western blot analysis.
In vivo LATS1 ubiquitination detection
HEK293A cells were transiently transfected with plasmids expressing HA-ubiquitin and FLAG-LATS1. 36 h after transfection, cells were treated with 10 μM MG132 (Millipore, 474790) for 6 h before harvesting. Cells were lysed in 200 μl of RIPA buffer (0.5% SDS), boiled at 95°C for 5 min, diluted with MLB to make final 0.1% SDS, then sonicated and centrifuged at 4°C (13, 000 rpm for 10 min). The supernatant was incubated with specific antibody at 4°C with rotation for 6 h and then 10 ul Pierce Protein A/G Magnetic Beads (Thermo Scientific) for another 2 h at 4°C. After washing, immunoprecipitated proteins were eluted with sample buffer, boiling for 10 min and subjected to IB analysis with anti-HA antibody to detect ubiquitylation.
RNA isolation and real-time PCR.
Cells were harvested for RNA extraction using the RNeasy Plus mini kit (Qiagen). 1 μg RNA was used for reverse transcription with iScript reverse transcriptase (Bio-Rad). cDNA was then used for real-time PCR with gene-specific primers using KAPA SYBR FAST qPCR master mix (Kapa Biosystems) and the CFX Connect real-time PCR system (Bio-Rad). Since the expression of some housekeeping genes might be affected by heat shock, we confirmed that 18S rRNA could be used as reference genes in our experiments. Relative mRNA abundance of the target gene was calculated using the ΔΔCt method normalized to the expression of 18S rRNA.
The primers (forward and reverse, respectively) used were as follows:
human CTGF, 5’-CCAATGACAACGCCTCCTG-3’ and 5’-TGGTGCAGCCAGAAAGCTC-3’;
human CYR61, 5’-AGCCTCGCATCCTATACAACC-3’ and 5’-TTCTTTCACAAGGCGGCACTC-3’;
human HSPA1A, 5’-AGAGCGGAGCCGACAGAG-3’ and 5’-CACCTTGCCGTGTTGGAA-3’;
human HSP90AA1, 5’-AGGAGGTTGAGACGTTCGC-3’ and 5’-AGAGTTCGATCTTGTTTGTTCGG-3’;
human HSPA6, 5’-GATGTGTCGGTTCTCTCCATTG-3’ and 5’-CTTCCATGAAGTGGTTCACGA-3’;
human DNAJB1, 5’-CTCTGGACGGCAGGACGATA-3’ and 5’-TCTTGATGTCTGGGGAATCCTT-3’;
human PP5, 5’-AAGACTCAGGCCAATGACTACT-3’ and 5’-CGCGTAGCCATAGCACTCAG-3’;
human ITCH, 5’-CTCTCTGCCGCCGACAAATA-3’ and 5’-TGGCAAGGGAGCTTGAGTTAC-3’;
human SIAH2, 5’-CGCCAGAAGTTGAGCTGCT-3’ and 5’-TGGTGGCATACTTACAGGGAA-3’;
mouse CTGF, 5’-AGCTGACCTGGAGGAAAACA-3’ and 5’-GACAGGCTTGGCGATTTTAG-3’;
mouse CYR61, 5’-GCTCAGTCAGAAGGCAGACC-3’ and 5’-GTTCTTGGGGACACAGAGGA-3’;
mouse HSPA1A, 5’-TGGTGCAGTCCGACATGAAG-3’ and 5’-GCTGAGAGTCGTTGAAGTAGGC-3’;
mouse HSP90AA1, 5’-TGTTGCGGTACTACACATCTGC-3’ and 5’-GTCCTTGGTCTCACCTGTGATA-3’;
mouse HSPB1, 5’-GGCAGGACGAACATGGCTACA-3’ and 5’-TCGAAAGTAACCGGAATGGTGAT-3’;
mouse YAP1, 5’-TACTGATGCAGGTACTGCGG-3’ and 5’-TCAGGGATCTCAAAGGAGGAC-3’;
mouse WWTR1, 5’-GAAGGTGATGAATCAGCCTCTG-3’ and 5’-GTTCTGAGTCGGGTGGTTCTG-3’;
18S rRNA, 5’-CGCCGCTAGAGGTGAAATTCT-3’ and 5’-CGAACCTCCGACTTTCGTTCT-3’.
Annexin V apoptosis detection
B16-OVA and SCC7 cells were heat shocked at 45°C for 1 h and collected after recovery at 37°C for 24 h for the detection of apoptosis. The PE Annexin V Apoptosis Detection Kit I (BD Pharmingen™) was used following the manufacturer protocol. Samples were then processed by the NovoCyte Flow Cytometer (ACEA Biosciences, Inc.), and the results were analyzed with FlowJo V10 software. Representative gating strategy and quantitative data for flow cytometry were provided in Supplementary Figure 1.
Cell viability detection
B16-OVA and SCC7 cells were heat shocked at the indicated temperature for 1 h and collected after recovery at 37°C for 24 h for the detection of cell viability. The Cell Counting Kit-8 (MedChemExpress) was used according to the manufacturer protocol. 100 μl of CCK8 were added in per 2 ml medium and cultured for additional 1.5 h, the absorbance at 450 nm was measured by Gen5™ Microplate Reader (BioTek Instruments) using area scanning method (3×3 points for 6 well plate). For the trypan blue exclusion experiments, appropriate trypan blue was added into the cells collected by trypsinization and the stain picture and quantification were performed by the automated cell counter (Countstar, China).
In vitro kinase assay
To analyze kinase activity, HEK293A cells with or without heat shock were collected and lysed with mild lysis buffer (50 mM Tris at pH7.5, 150 mM NaCl, 0.5% Triton-100, protease & phosphatase inhibitor cocktail) and immunoprecipitated with the indicated antibodies. The immunoprecipitates were washed three times with mild lysis buffer, followed by a single wash with TBS. Immunoprecipitated kinases were subjected to kinase assay in the 1× protein kinases buffer (BioLabs) in the presence of 0.5 mM ATP and 500 ng GST-YAP or GST-LATS2. The reaction mixtures were incubated at 30°C for 30 min with a shaker at 250 rpm. The reactions were then terminated with SDS sample buffer and subjected to SDS–PAGE. Phosphorylation of YAP and LATS was determined by phospho-YAP (Ser127) antibody and phospho-LATS (Thr1079) antibody respectively.
Immunofluorescence staining.
HEK293A and A549 cells were seeded on poly-L-ornithine (Sigma)-coated coverslips to the high density in 12 well plates. After heat shock at 43°C for the indicated time, cells were successively fixed in 4% paraformaldehyde and permeabilized with 0.2% Triton X-100 in PBS for 10 min at room temperature. Cells were blocked in PBS with 2% BSA and 2% goat serum for 30 min at room temperature and incubated overnight at 4°C in primary YAP/TAZ antibodies (Santa Cruz, 1:200) diluted in 1% BSA. After three washes with PBS, Alexa Fluor 488 secondary antibodies (Life Technologies, 1:1000) were diluted in 1% BSA and incubated for 1 h in the dark at room temperature. Slides were mounted with Prolong Gold antifade reagent with DAPI (Life Technologies). Images were captured with Olympus FV1000 confocal microscopy, immunofluorescence data were collected by NIS-Elements AR 5.11.00 imaging software and the signals from channels were merged by ImageJ (1.52a) software.
Immunohistochemistry.
Primary tumor paraffin sections were deparaffinized and rehydrated with xylene and alcohol gradients, and antigen retrieval was performed in citrate buffer (pH 6.0) followed by 3% H2O2 for 15 min to inhibit endogenous peroxidase activity. Sections were then incubated overnight at 4°C with cleaved-caspase 3 antibody and detected using SPlink Detection Kit and AEC Peroxidase Substrate kit (Zhongshan Golden Bridge Biotechnology Co., China) as per the manufacturer’s protocol.
RNA sequencing and bioinformatics analysis.
Total RNAs were extracted using TRIzol (Thermo Scientific) from siCon or siYAP/TAZ#2 treated B16-OVA cells with or without heat shock at 45°C for 1 h and recovered at 37°C for 6 h. Three replicates for each sample were generated and analysed. The resulting RNA was then used to prepare libraries using NEBNext® Ultra™ RNA Library Prep Kit for Illumina® (#E7530L, NEB, USA). The clustering of the index-coded samples was performed on a cBot cluster generation system using HiSeq PE Cluster Kit v4-cBot-HS (Illumina) and the libraries were sequenced on an Illumina platform and 150 bp paired-end reads were generated. Reads were aligned to the mm10 (Mus_musculus) reference genome. R was used to identify differentially expressed genes (DEGs). Genes with FPKM > 1, adjusted P value < 0.05 and Log2 Fold > 1 or < −1 were differentially expressed. GO and KEGG enrichment analysis of DEGs was performed also using R.
Chromatin immunoprecipitation (ChIP)
ChIP assay was performed using the SimpleChIP Enzymatic Chromatin IP Kit (Cell Signaling Technology, Cat. #9003), according to the manufacturer's instructions. Briefly, HCT116 cells were cross-linked, lysed, and digested to generate DNA fragments. A total of 50 μg of digested chromatin was incubated overnight with 2 μg of YAP antibody or control IgG. Precipitated DNA was quantitated by real-time PCR analysis.
The primers (forward and reverse, respectively) used were as follows:
human HSPA1A, 5’-TGGAGAGTTCTGAGCAGGGG-3’ and 5’-GCCTTTTCCCTTCTGAGCCA-3’;
human HSPB1, 5’-CCAGCGCCCCGCACTTTTCT-3’ and 5’-AGTCGCGGAAGGGGTCCCAG-3’;
human DNAJB1, 5’-GGCAGGAGGATGGTCTTCTT-3’ and 5’-TTAGTGGTGGGCTCTTCAGC-3’;
human CTGF, 5’-CTTTGGAGAGTTTCAAGAGCC-3’ and 5’-TCTGTCCACTGACATACATCC-3’
ChIP-seq analysis and Motif discovery
The available ChIP-seq data for transcription factors TEAD4 and HSF-1indicating genome-wide chromatin occupancy were obtained from ENCODE at UCSC Genome Browser (http://www.genome.ucsc.edu/ENCODE/), and the consensus sequence of known motifs were downloaded from Homer Motif database. (http://homer.ucsd.edu/homer/motif/HomerMotifDB/homerResults.html)
Animal work
Female BALB/c nude mice were purchased from Jiangsu GemPharmatech Co., Ltd (China). For syngeneic tumor models, B16-OVA cells (5 x 105) were injected subcutaneously into the left foot of 6-8-week-old female BALB/c nude mice. Mice were maintained in a specific pathogen-free facility (temperature 20°C-26°C, humidity 55%-65%, dark/light cycle12h/12h). Eight mice were assigned to each group. GP-siRNA-Mate Plus (MP) purchased from GenePharma (Shanghai, China) was used for the in vivo siRNA transfection in accordance with the manufacturer’s instructions. MP/siRNA complex (10 μg siRNA/50 μl total volume/mouse) were administered via intratumoral injection twice weekly beginning after the volume of the tumor reached around 30 mm3. Hyperthermia was given three days after siRNA treatment by immersing the tumor-bearing foot of mice in the 43°C water bath for 30 min. For subcutaneous tumor growth, the maximum single tumor cannot exceed 1.5 cm in diameter in mice according to the guidelines provided by the animal care program and no experiments in this study generated tumor burden over this limit. The mice were sacrificed 21 days after tumor inoculation, and tumors were harvested, weighed and fixed in 4% paraformaldehyde for immunohistochemistry. All animal experiments were performed as protocols approved by the Institutional Animal Care and Use Committee at Sichuan University.
Statistics and Reproducibility
All the experiments (including western blots) were independently repeated at least twice with similar results. Data are presented as mean ± s.d. or mean ± s.e.m as mentioned in each figure legend. P values were determined using two-sided unpaired t-test or one/two-way ANOVA test by GraphPad Prism 7 software (GraphPad Prism, San Diego, CA). The P values were considered statistically significant when p < 0.05.
Data Availability
The RNA sequencing data are available in GEO Data Sets with the accession number GSE133251. The available ChIP-seq data were obtained from ENCODE at UCSC Genome Browser (http://www.genome.ucsc.edu/ENCODE/) (UCSC accession number: wgEncodeEH002333, wgEncodeEH002345 and wgEncodeEH000754), and the consensus sequence of known motifs were downloaded from Homer Motif database (http://homer.ucsd.edu/homer/motif/HomerMotifDB/homerResults.html). Source data are provided with this paper. All other data that support the findings of this study are available on request from the corresponding author.
Extended Data
Extended Data Fig. 1. Heat shock activates YAP.
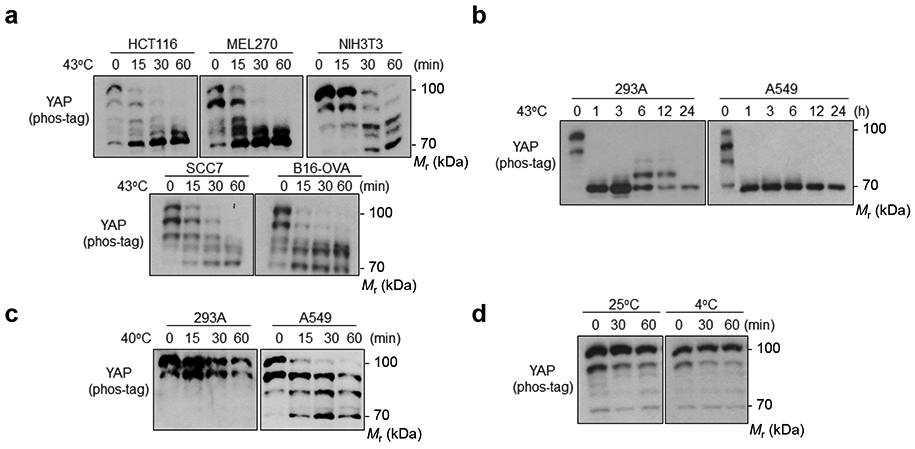
(a) Heat shock induces YAP dephosphorylation in multiple cell lines: human colon cancer HCT116; human uveal melanoma MEL270; mouse fibroblast NIH3T3; mouse squamous cell carcinoma SCC7; mouse melanoma B16-OVA. Various tumor and non-tumor cell lines were seeded onto six well plates to achieve high density (100% confluence). After 24 h, cells were heat shocked at 43°C for the indicated times and collected for YAP phos-tag gel analyses.
(b) Heat shock induced a sustained YAP dephosphorylation.
(c) Mild heat shock-induced YAP dephosphorylation is cell type-dependent. High density HEK293A and A549 cells were heat shocked at 40°C for the indicated times and collected for YAP phos-tag gel analyses.
(d) Cold stress does not affect YAP phosphorylation. High density HEK293A cells were placed at 25°C or 4°C for 30 min and 1 h; then samples were collected for YAP phos-tag gel analyses. Immunoblotting in panels a-d has been performed two times with similar results. Source data are available online.
Extended Data Fig. 2. MST1/2 and MAP4Ks are not required for heat shock-induced YAP regulation.
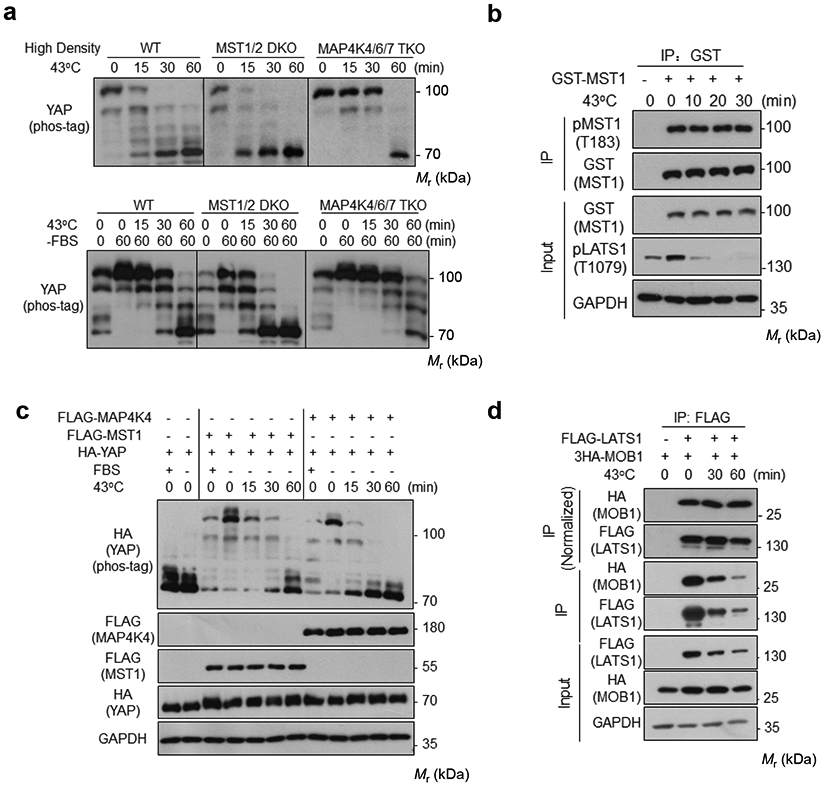
(a) HEK293A cells were cultured under high density (upper panel) or in the absence of serum (lower panel), and were subjected to heat shock for the indicated times. YAP phosphorylation was detected by the phos-tag gel.
(b) Heat shock does not affect MST1 phosphorylation. HEK293A cells were transiently transfected with GST-MST1. 24 h after transfection, cells were subjected to heat shock for the indicated times. Glutathione Sepharose 4B beads (GE Healthcare) were used to purify GTS-MST1. Phosphorylation of the purified GST-MST1 was analyzed by Western blot with pMST1 (Thr183) antibody.
(c) YAP dephosphorylation time course in MST1-rescued or MAP4K4-rescued MM8KO cells upon heat shock. Plasmids for HA-YAP and FLAG-MST1 or FLAG-MAP4K4 were transiently co-transfected into HEK293A MM8 KO cells. 24 h after transfection, cells were subcultured to new plate and reached a medium confluence the next day, treated with serum starvation for 2 h, then subjected to heat shock for indicated durations. YAP phosphorylation was detected by the phos-tag gel.
(d) Heat shock does not affect the LATS1-MOB1 interaction. HEK293A cells were transiently co-transfected with FLAG-LATS1 and 3×HA-MOB1. 24 h after transfection, cells were subjected to heat shock for the indicated times. FLAG antibodies were used for immunoprecipitation and the co-precipitated proteins were detected by Western blot. The uppermost panel were normalized against FLAG-LATS1 protein levels. Immunoblotting in panels a-d has been performed two times with similar results. Source data are available online.
Extended Data Fig. 3. Related to Figure 4.
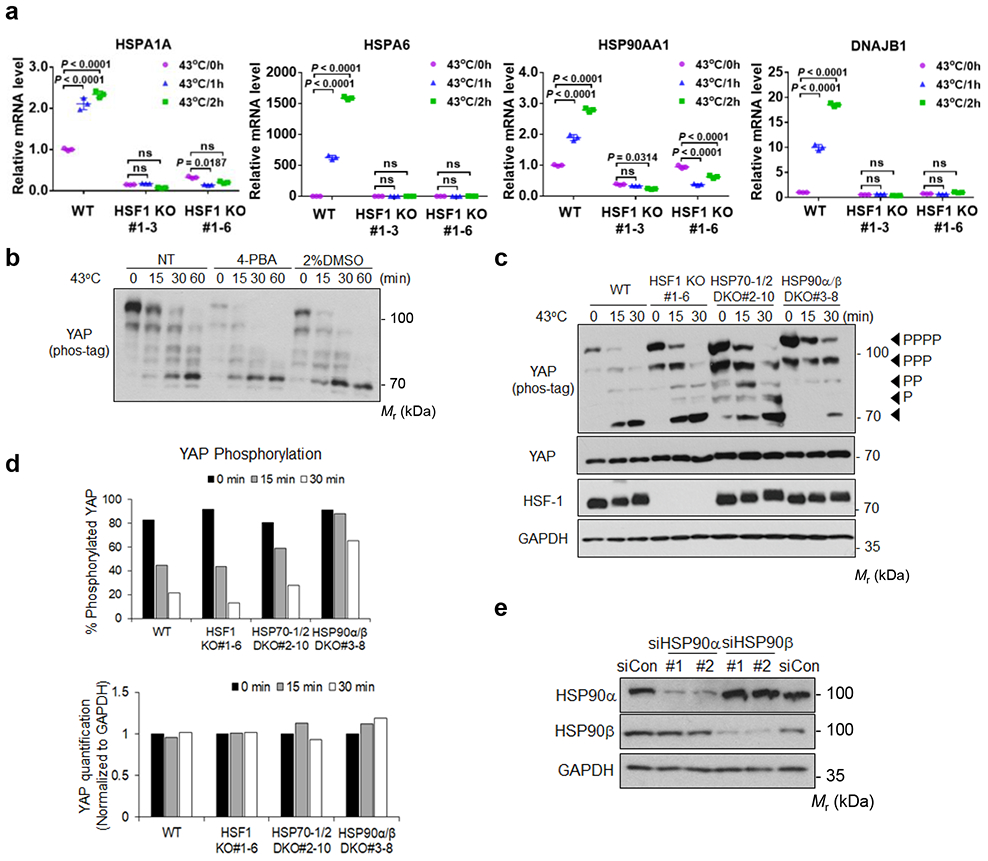
(a) Deletion of HSF-1 abolishes expression of heat shock responsive genes. High density HEK293A WT and HSF-1 KO cells were subjected to heat shock for 1 or 2 h, and mRNA expression of downstream target genes was measured by RT-PCR. Data are presented as mean ± s.d.; n = 3 biologically independent samples. Two-way ANOVA test, ns, not significant. Two independent KO clones are shown.
(b) Chemical chaperones, which can prevent protein misfolding, cannot block YAP dephosphorylation by heat shock. HEK2933A cells were pretreated with indicated chemical chaperones sodium 4-phenylbutyrate (4-PBA) or DMSO for 24 h and were then subjected to heat shock as indicated. The phosphorylation of YAP was detected by a phos-tag gel.
(c) Knockout of HSP90, but not HSF-1 or HSP70, delays YAP dephosphorylation in response to heat shock. HEK293A WT, HSF-1 KO, HSP70-1/2 DKO, and HSP90α/β DKO cells were subjected to heat shock. Samples were analyzed by western blot on phos-tag gel for YAP phosphorylation and regular gel for YAP protein levels.
(d) Quantification of the Western blots in Extended Data Figure 3c.
(e) The verification of HSP90α and HSP90β knockdown efficiency by Western blot. Two independent siRNAs were used. Immunoblotting in panels b, c and e has been performed two times with similar results. Source data are available online.
Extended Data Fig. 4. Verification of YAP/TAZ knockout in B16-OVA cells.

(a) The DNA sequence confirms inactivation of YAP1 and WWTR1 genes in the #2 clone of B16-OVA YAP/TAZ DKO cells.
(b) Functional verification of YAP/TAZ knockout. LPA induced expression of YAP/TAZ target genes is abolished in the YAP/TAZ knockout B16-OVA cells. RT-PCR results are presented as mean ± s.d.; n = 3 biologically independent samples. Two-way ANOVA test, ns, not significant. Source data are available online.
Extended Data Fig. 5. YAP/TAZ silencing sensitizes tumor cells to heat shock.

(a) Verification of YAP/TAZ siRNA knockdown in B16-OVA cells by Western blot and RT-PCR. Presented are mean ± s.d., n=3 biologically independent samples. Two-way ANOVA test.
(b) YAP/TAZ knockdown reduces B16-OVA cell viability in response to heat shock. Cells were transfected with control or two independent YAP and TAZ siRNAs, subjected to heat shock at 45°C or 46°C for 1 h, then recovered at 37°C for 24 h. Cell viability was determined by CCK8 assays. Data are mean ± s.d.; n = 3 biologically independent experiments. Two-way ANOVA test. ns, not significant; NT, no treatment.
(c) YAP/TAZ knockdown promotes B16-OVA apoptosis. Images are representative of two independent experiments with similar results.
(d) More cell debris and apoptotic bodies in YAP/TAZ knockdown cells. B16-OVA cells transfected with siRNA, heat shocked at 45°C for 1 h, and recovered at 37°C for 6 h. Cells were subjected to microscopic examination for morphology. Representative pictures from three independent samples are shown. Scale bar, 100 μm.
(e) YAP/TAZ knockdown compromises HSP induction by heat shock. Cells were similarly treated as in (d). mRNAs were quantified by RT-PCR. Data are mean ± s.d.; n = 3 biologically independent samples. Two-way ANOVA test. ns, not significant. Only siRNA#2 was chosen in this experiment.
(f, g) YAP/TAZ knockdown reduces SCC7 cell viability upon heat shock. CCK8 assay (f) and Annexin V staining (g). Data presented as mean ± s.d.; n = 3 biologically independent experiments. One-way ANOVA test.
(h) Verification of YAP/TAZ siRNA knockdown in HCT116 tumor cells. Immunoblotting has been performed two times with similar results. (i) YAP/TAZ knockdown HCT116 cells are more sensitive to heat shock stress. CCK8 assay was performed to measure cell viability. Data are presented as mean ± s.d.; n = 3 biologically independent experiments. One -way ANOVA test. Source data are available online.
Extended Data Fig. 6. YAP/TAZ contribute to the heat shock-induced transcriptome.

(a) Top 10 enriched gene ontology analysis for biological processes (BP) of 396 up-regulated genes regulated by YAP/TAZ.
(b) Top 10 enriched KEGG pathway analysis of 396 up-regulated genes regulated by YAP/TAZ.
(c) Log2 Fold Change of representative HSPs in B16-OVA siCon or siYAP/TAZ#2 treated cells (Data are from RNA-SEQ).
(d) Heat map of representative heat shock up-regulated genes regulated by YAP/TAZ (Data are from RNA-SEQ).
Extended Data Fig. 7. The ITCH and SIAH2 E3 ubiquitin ligases do not mediate heat shock-induced LATS degradation.
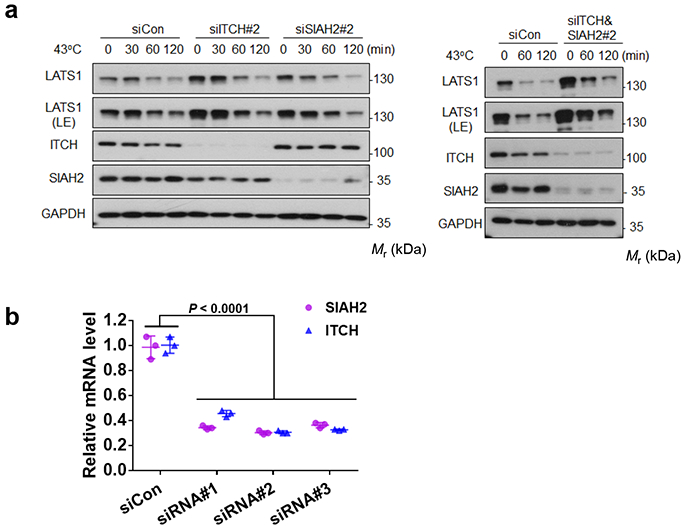
(a) Knockdown of ITCH and SIAH2 does not block the heat shock-induced LATS1 degradation. Immunoblotting has been performed two times with similar results.
(b) Verification of ITCH&SIAH2 siRNA knockdown efficiency by RT-PCR. Data are presented as mean ± s.d.; n = 3 biologically independent samples. Two-way ANOVA test. Source data are available online.
Extended Data Fig. 8. Heat shock does not induce a universal protein dephosphorylation and kinase degradation.
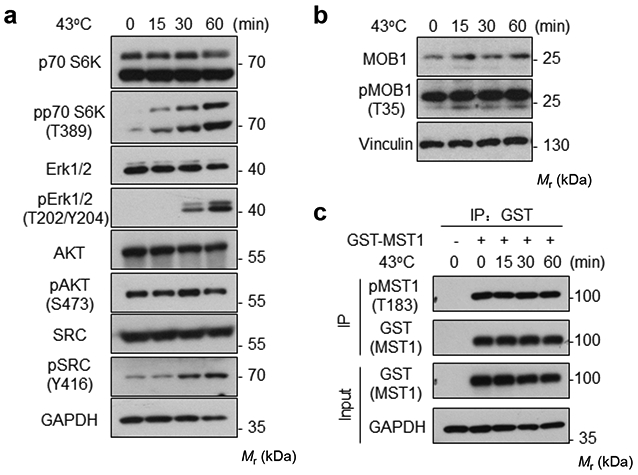
(a-b) High density HEK293A cells were heat shocked at 43°C for the indicated times and cell lysates were collected for Western blot to detect the phosphorylation and protein levels of the indicated proteins.
(c) HEK293A cells were transiently transfected with GST-MST1. 24 h after transfection, cells were subjected to heat shock for the indicated times. Glutathione Sepharose 4B beads were used to purify GST-MST1. Phosphorylation of GST-MST1 was analyzed by Western blot with pMST1 (Thr183) antibody. Immunoblotting for a-c has been performed two times with similar results. Source data are available online.
Supplementary Material
Supplementary Table Genomic co-occupancy analyses of interested genes by ChIP-seq data from ENCODE.
ACKNOWLEDGMENTS
This work was supported by grants from the National Institutes of Health (CA196878 and DE015694) to K.L.G. This study was supported by grants from National Natural Science Foundation of China (31800773), China Postdoctoral Science Foundation (2018M633369), Sichuan Science and Technology Program (2019YJ0063), The Youth Science Foundation of West China Hospital of Stomatology (WCHS-201703) to M.L. This work was supported by the National Major Scientific and Technological Special Project for “Significant New Drugs Development” (No. 2018ZX09733001, China), the Development Program of China (No. 2016YFA0201402), and by the Excellent Youth Foundation of Sichuan Scientific Committee Grant in China (No. 2019JDJQ008) to X.W.W. This research was also partly funded by the China Scholarship Council (CSC NO. 201506240035).
We thank Faxing Yu from Fudan University and Bin Zhao from Zhejiang University for reagents.
Footnotes
COMPETING FINANCIAL INTERESTS
K.-L.G. is a co-founder and has an equity interest in Vivace Therapeutics, Inc. The other authors declare no competing interests.
References
- 1.Yu F-X, Zhao B & Guan K-L Hippo pathway in organ size control, tissue homeostasis, and cancer. Cell 163, 811–828 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pan D The hippo signaling pathway in development and cancer. Developmental cell 19, 491–505 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lin KC, Park HW & Guan K-L Deregulation and Therapeutic Potential of the Hippo Pathway in Cancer. Annual Review of Cancer Biology 2, 59–79 (2018). [Google Scholar]
- 4.Harvey KF, Zhang X & Thomas DM The Hippo pathway and human cancer. Nature Reviews Cancer 13, 246 (2013). [DOI] [PubMed] [Google Scholar]
- 5.Meng Z, Moroishi T & Guan K-L Mechanisms of Hippo pathway regulation. Genes & development 30, 1–17 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dong J et al. Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell 130, 1120–1133 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhao B et al. TEAD mediates YAP-dependent gene induction and growth control. Genes & development 22, 000–000 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Åkerfelt M, Morimoto RI & Sistonen L Heat shock factors: integrators of cell stress, development and lifespan. Nature reviews Molecular cell biology 11, 545 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wust P et al. Hyperthermia in combined treatment of cancer. The lancet oncology 3, 487–497 (2002). [DOI] [PubMed] [Google Scholar]
- 10.Roti Roti JL Cellular responses to hyperthermia (40–46 C): Cell killing and molecular events. International Journal of hyperthermia 24, 3–15 (2008). [DOI] [PubMed] [Google Scholar]
- 11.Zhang H-G, Mehta K, Cohen P & Guha C Hyperthermia on immune regulation: a temperature’s story. Cancer letters 271, 191–204 (2008). [DOI] [PubMed] [Google Scholar]
- 12.Hansen CG, Moroishi T & Guan KL YAP and TAZ: a nexus for Hippo signaling and beyond. Trends Cell Biol 25, 499–513 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhao B et al. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes & Development 21, 2747–2761 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Budzyński MA, Puustinen MC, Joutsen J & Sistonen L Uncoupling Stress-Inducible Phosphorylation of Heat Shock Factor 1 from Its Activation. Molecular and Cellular Biology 35, 2530–2540 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chan EH et al. The Ste20-like kinase Mst2 activates the human large tumor suppressor kinase Lats1. Oncogene 24, 2076–2086 (2005). [DOI] [PubMed] [Google Scholar]
- 16.Hergovich A, Schmitz D & Hemmings BA The human tumour suppressor LATS1 is activated by human MOB1 at the membrane. Biochem Bioph Res Co 345, 50–58 (2006). [DOI] [PubMed] [Google Scholar]
- 17.Ni L, Zheng Y, Hara M, Pan D & Luo X Structural basis for Mob1-dependent activation of the core Mst-Lats kinase cascade in Hippo signaling. Genes Dev 29, 1416–1431 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang W et al. AMPK modulates Hippo pathway activity to regulate energy homeostasis. Nature Cell Biology 17, 490–499 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mo J-S et al. Cellular energy stress induces AMPK-mediated regulation of YAP and the Hippo pathway. Nature cell biology 17, 500–510 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hong AW et al. Osmotic stress-induced phosphorylation by NLK at Ser128 activates YAP. EMBO reports 18, 72–86 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Trinklein ND, Murray JI, Hartman SJ, Botstein D & Myers RM The role of heat shock transcription factor 1 in the genome-wide regulation of the mammalian heat shock response. Mol Biol Cell 15, 1254–1261 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huntoon CJ et al. Heat shock protein 90 inhibition depletes LATS1 and LATS2, two regulators of the mammalian hippo tumor suppressor pathway. Cancer research 70, 8642–8650 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Raghunathan VK et al. Involvement of YAP, TAZ and HSP90 in contact guidance and intercellular junction formation in corneal epithelial cells. PloS one 9, e109811 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ran FA et al. Genome engineering using the CRISPR-Cas9 system. Nature protocols 8, 2281 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Papp E & Csermely P Chemical Chaperones: Mechanisms of Action and Potential Use, in Molecular Chaperones in Health and Disease. (eds. Starke K & Gaestel M) 405–416 (Springer Berlin Heidelberg, Berlin, Heidelberg; 2006). [DOI] [PubMed] [Google Scholar]
- 26.Sreedhar AS, Kalmár É, Csermely P & Shen Y-F Hsp90 isoforms: functions, expression and clinical importance. FEBS letters 562, 11–15 (2004). [DOI] [PubMed] [Google Scholar]
- 27.Russell LC, Whitt SR, Chen M-S & Chinkers M Identification of conserved residues required for the binding of a tetratricopeptide repeat domain to heat shock protein 90. Journal of Biological Chemistry 274, 20060–20063 (1999). [DOI] [PubMed] [Google Scholar]
- 28.Das AK, Cohen PT & Barford D The structure of the tetratricopeptide repeats of protein phosphatase 5: implications for TPR-mediated protein–protein interactions. The EMBO journal 17, 1192–1199 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yu F-X et al. Regulation of the Hippo-YAP pathway by G-protein-coupled receptor signaling. Cell 150, 780–791 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen R, Xie R, Meng Z, Ma S & Guan KL STRIPAK integrates upstream signals to initiate the Hippo kinase cascade. Nat Cell Biol 21, 1565–1577 (2019). [DOI] [PubMed] [Google Scholar]
- 31.Jolly C & Morimoto RI Role of the heat shock response and molecular chaperones in oncogenesis and cell death. Journal of the National Cancer Institute 92, 1564–1572 (2000). [DOI] [PubMed] [Google Scholar]
- 32.Kregel KC Invited review: heat shock proteins: modifying factors in physiological stress responses and acquired thermotolerance. Journal of applied physiology 92, 2177–2186 (2002). [DOI] [PubMed] [Google Scholar]
- 33.Ma B et al. Hypoxia regulates Hippo signalling through the SIAH2 ubiquitin E3 ligase. Nature cell biology 17, 95 (2015). [DOI] [PubMed] [Google Scholar]
- 34.Salah Z, Melino G & Aqeilan RI Negative regulation of the Hippo pathway by E3 ubiquitin ligase ITCH is sufficient to promote tumorigenicity. Cancer research 71, 2010–2020 (2011). [DOI] [PubMed] [Google Scholar]
- 35.Ho KC et al. Itch E3 ubiquitin ligase regulates large tumor suppressor 1 stability. Proceedings of the National Academy of Sciences 108, 4870–4875 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Richter K, Haslbeck M & Buchner J The heat shock response: life on the verge of death. Mol Cell 40, 253–266 (2010). [DOI] [PubMed] [Google Scholar]
- 37.Iwasa H et al. Yes-associated protein homolog, YAP-1, is involved in the thermotolerance and aging in the nematode Caenorhabditis elegans. Experimental cell research 319, 931–945 (2013). [DOI] [PubMed] [Google Scholar]
- 38.Meng Z et al. MAP4K family kinases act in parallel to MST1/2 to activate LATS1/2 in the Hippo pathway. Nat Commun 6, 8357 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moroishi T et al. The Hippo Pathway Kinases LATS1/2 Suppress Cancer Immunity. Cell 167, 1525–1539 e1517 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table Genomic co-occupancy analyses of interested genes by ChIP-seq data from ENCODE.
Data Availability Statement
The RNA sequencing data are available in GEO Data Sets with the accession number GSE133251. The available ChIP-seq data were obtained from ENCODE at UCSC Genome Browser (http://www.genome.ucsc.edu/ENCODE/) (UCSC accession number: wgEncodeEH002333, wgEncodeEH002345 and wgEncodeEH000754), and the consensus sequence of known motifs were downloaded from Homer Motif database (http://homer.ucsd.edu/homer/motif/HomerMotifDB/homerResults.html). Source data are provided with this paper. All other data that support the findings of this study are available on request from the corresponding author.