

Summary
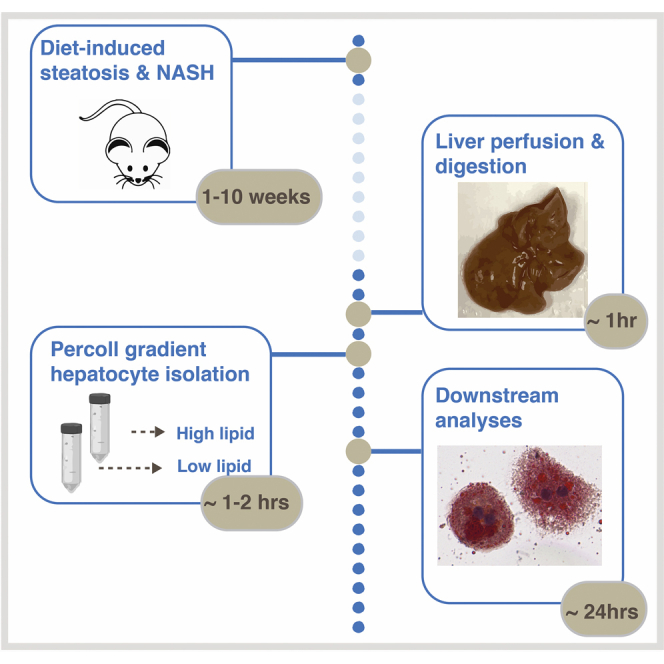
We present a protocol for isolating hepatocytes from mice with established non-alcoholic fatty liver disease. This protocol consists of liver perfusion using a peristaltic pump, followed by a modified 25% and 90% Percoll gradient centrifugation protocol to capture lipid-laden hepatocytes that are usually lost using traditional isolation protocols. This protocol enables simultaneous isolation of normal and lipid-filled hepatocytes. Lipid-filled hepatocytes can be used in cell culture systems to study drug metabolism, hepatotoxicity, or glucose and lipid metabolism.
For complete details on the use and execution of this protocol, please refer to Sharabi et al. (2017) and Kegel et al. (2016).
Subject Areas: Cell Biology, Cell culture, Cell isolation, Single Cell, Metabolism, Model Organisms
Graphical Abstract
Highlights
-
•
This protocol describes the isolation of hepatocytes from mice with fatty liver disease
-
•
Percoll gradient centrifugation enables density-based hepatocyte isolation
-
•
Examples of heterogeneity in hepatocyte lipid content are presented
We present a protocol for isolating hepatocytes from mice with established non-alcoholic fatty liver disease. This protocol consists of liver perfusion using a peristaltic pump, followed by a modified 25% and 90% Percoll gradient centrifugation protocol to capture lipid-laden hepatocytes that are usually lost using traditional isolation protocols. This protocol enables simultaneous isolation of normal and lipid-filled hepatocytes. Lipid-filled hepatocytes can be used in cell culture systems to study drug metabolism, hepatotoxicity, or glucose and lipid metabolism.
Before you begin
This protocol will isolate viable hepatocytes from healthy murine livers, as well as from mice with established non-alcoholic steatohepatitis. This protocol has been modified based on previous work to enable isolation of lipid-filled hepatocytes (Berry and Friend, 1969; Kreamer et al., 1986; Pertoft, 2000; Gonçalves et al., 2007; Li et al., 2010; Kegel et al., 2016; Sharabi et al., 2017).
Note: This protocol can be used to isolate hepatocytes from mice with different genetic backgrounds and mutations to facilitate the investigation of gene function in liver disease.
Induction of diet-induced NASH in mice
Timing: 1–10 weeks
-
1.
All animal procedures need to be approved by the Institutional Animal Care and Use Committee. All experiments in this protocol were approved by the Stanford Animal Care and Use Committee protocol #32982.
-
2.
House 6–8 week old male or female C57BL/6 mice in a cage of 4–5 mice per cage in a temperature-controlled (20°C–22°C) room on a 12-h light/dark cycle.
-
3.
Feed mice with either a chow diet or with a diet to induce non-alcoholic steatohepatitis, hereafter referred to as NASH diet (Research Diets). The NASH diet consists of 40% kcal fat (palm oil), 20% kcal fructose, and 2% cholesterol and has previously been shown to induce clinical, biochemical, and histological features of fatty liver disease in animals (Radhakrishnan et al., 2020).
-
4.
Hepatic lipid accumulation can be seen at the cellular level after a week of feeding, while whole-body changes such as body weight and blood glucose are typically seen starting at 6 weeks on this diet. Measure body weights and non-fasted glucose levels using a glucometer weekly. At 6 weeks and beyond, NASH diet-fed mice are expected to demonstrate significantly higher body weights, higher blood glucose levels, and enlarged and pale livers (Figure 1) compared to chow-fed mice.
Note: C57BL/6 mice are suitable for inducing NASH with this diet, but other strains such as 129Sv can be used as well, but the exact time of feeding might have to be optimized (Fengler et al., 2016).
Figure 1.

Induction of diet-induced non-alcoholic steatohepatitis (NASH) in mice
(A) Typical non-fasting blood glucose levels in C57BL/6 mice fed chow diet or NASH-diet for 6 weeks.
(B) C57BL/6 mice fed chow diet (left) or NASH-diet (right) for 6 weeks. Livers from NASH mice are pale and of increased size.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, peptides, and recombinant proteins | ||
| HBSS buffer | Gibco | Cat# 14175-095 |
| Potassium chloride | Sigma | Cat# P5405 |
| Sodium bicarbonate | Sigma | Cat# S6297 |
| UltraPure 0.5 M EDTA, pH 8.0 | Invitrogen | Cat# 15575-038 |
| Williams E media | Quality Biological | Cat# 112-033-101 |
| Corning regular fetal bovine serum | Corning | Cat# 35-010-CV |
| DMEM high glucose | Sigma | Cat# D6429 |
| HEPES buffered saline | Sigma-Aldrich | Cat# 51558 |
| Sodium pyruvate | Thermo Fisher | Cat# 11360070 |
| Dexamethasone | Sigma | Cat# D1756 |
| Insulin, human recombinant | Millipore Sigma | Cat# 91077C |
| Penicillin/streptomycin | Gibco | Cat# 15140-122 |
| Collagenase IV | Sigma | Cat# C5138 |
| PBS | Gibco | Cat# 10010-023 |
| 10× PBS | Gibco | Cat# 70011044 |
| Percoll | Sigma | Cat# P1644 |
| Ethanol | Fisher Scientific | Cat# 22-032-601 |
| BSA | Sigma | Cat# A7906 |
| Glutamax | Thermo Fisher | Cat# 35050061 |
| Oil red O solution | Sigma | Cat# O1391 |
| Isopropanol | Fisher Chemical | Cat# BP2632-4 |
| Formalin | SIP Brand | Cat# C4320 |
| Rat tail Collagen I | Corning | Cat# 354236 |
| Trypan blue stain (0.4%) | Invitrogen | Cat# T10282 |
| Experimental models: organisms/strains | ||
| C57BL/6 mice, 6 weeks old male or female | Jackson Laboratory | Cat #000664 |
| Other | ||
| Blood glucose meter | OneTouch UltraMini meter | N/A |
| Blood glucose strips | GenUltimate | Cat# 100-50 |
| Rodent NASH diet | Research Diets | Cat# D09100310 |
| 70 μm cell strainer | BD Falcon | Cat# 352350 |
| 25G × 7/8 needle | BD | Cat# 305124 |
| Petri dish Fisherbrand | Fisher Scientific | Cat# FB0875713 |
| Fisherbrand variable-flow peristaltic pump | Fisher Scientific | Cat# 13-876-2 |
| Perfusion tubing connectors 3/32 inch polypropylene coupler | United States Plastic Corp | Cat# 65600 |
| Perfusion tubing 1/32 inch ID × 3/32 inch OD silicone | United States Plastic Corp | Cat# 57286 |
| Benchtop centrifuge | N/A | N/A |
| Biosafety cabinet | N/A | N/A |
| Surgical scissors and tweezers | Roboz | N/A |
| Water bath | N/A | N/A |
| Corning Costar flat-bottomed cell culture plates | Fisher Scientific | Cat# 07-200-82 |
| Countess II FL automated cell counter | Life Technology | Cat# AMQAF1000 |
| Millex-GS syringe filter unit, 0.22 μm | Millipore Sigma | Cat# SLGSV255F |
| BD syringes without needle, 50 mL | Fisher Scientific | Cat# BD309653 |
Materials and equipment
Prepare all buffers and media according to the tables and keep on ice until use. Refer to the Key Resources Table and Materials and Equipment for a complete list of materials, equipment, and recipes.
CRITICAL: Prepare the digestion buffer the day of isolation. Avoid repeated freeze-thaws of the digestion buffer. Pre-warm the perfusion and digestion buffer to 42°C before starting the isolation procedure. Keep the plating media on ice.
Perfusion buffer
| Reagent | Stock concentration | Final concentration | Amount |
|---|---|---|---|
| HBSS | 1× | 1× | 500 mL |
| KCl | w/v | 0.4 g/L | 0.2 g |
| Sodium Bicarbonate | w/v | 2.1 g/L | 1.05 g |
| EDTA | n/a | 0.2 g/L | 0.1 g |
| Total | 500 mL |
Adjust pH to 7.4, sterilize using a 0.22 μm filter, store at 4°C for up to 2 months. Adjust the pH to 7.4 after the solution has reached 42°C if needed.
Digestion buffer
| Reagent | Stock concentration | Final concentration | Amount |
|---|---|---|---|
| DMEM high glucose | 1× | 1× | 89 mL |
| Penicillin/Streptomycin | 100× | 1% (v/v) | 1 mL |
| Collagenase IV | n/a | 1 mg/mL | 100 mg |
| HEPES | 10 g/L | 10 μg/mL | 100 μL |
| FBS | n/a | 10% (v/v) | 10 mL |
| Total | 100 mL |
Adjust pH to 7.4, always prepare freshly, do not store.
90% Percoll media
| Reagent | Stock concentration | Final concentration | Amount |
|---|---|---|---|
| Percoll | n/a | n/a | 9 mL |
| 10× PBS | 10× | 1× | 1 mL |
| Resuspended hepatocytes | 10 mL | ||
| Total | 20 mL |
Prepare at 20°C–24°C in a sterile environment, always prepare freshly, do not store.
25% Percoll media
| Reagent | Stock concentration | Final concentration | Amount |
|---|---|---|---|
| Percoll | n/a | n/a | 2.5 mL |
| 10× PBS | 10× | 1× | 1 mL |
| ddH2O | n/a | n/a | 6.5 mL |
| Resuspended hepatocytes | 10 mL | ||
| Total | 20 mL |
Prepare at 20°C–24°C in a sterile environment, always prepare freshly, do not store.
Plating media
| Reagent | Stock concentration | Final concentration | Amount |
|---|---|---|---|
| Williams E media | 1× | 1× | 425 mL |
| Glutamax | 200 mM | 2 mM | 5 mL |
| Penicillin/Streptomycin | 50× | 2% (v/v) | 10 mL |
| Sodium Pyruvate | 100 mM | 2 mM | 10 mL |
| Dexamethasone | 1 mM | 1 μM | 500 μL |
| Insulin | 1 mM | 0.1 μM | 50 μL |
| FBS | n/a | 10% (v/v) | 50 mL |
| Total | 500 mL |
Prepare in a sterile environment, adjust pH to 7.4, store at 4°C for up to 1 week.
Maintenance media
| Reagent | Stock concentration | Final concentration | Amount |
|---|---|---|---|
| Williams E media | 1× | 1× | 475 mL |
| Glutamax | 200 mM | 2 mM | 5 mL |
| Penicillin/Streptomycin | 50× | 2% (v/v) | 10 mL |
| Sodium Pyruvate | 100 mM | 2 mM | 10 mL |
| Dexamethasone | 1 mM | 0.1 μM | 50 μL |
| Insulin | 1 μM | 0.1 nM | 50 μL |
| BSA | n/a | 0.2% (w/v) | 1 g |
| Total | 500 mL |
Prepare in sterile environment, adjust pH to 7.4, store at 4°C for up to 1 week.
Step-by-step method details
Day of isolation: preparation of buffers and setup of perfusion pump
This step includes the preparation of buffers and preparation of the perfusion pump. The perfusion system includes a water bath and a peristaltic pump with adjustable speed. The required volumes for hepatocyte isolation from one mouse are described in the table.
One digestion reaction
| Buffers/Media | Needed for | Total amount |
|---|---|---|
| Perfusion Buffer | Liver Perfusion | ~ 40 mL |
| Digestion Buffer | Liver Perfusion | ~ 30 mL |
| Plating Media | Liver Perfusion Hepatocyte isolation Plating |
~320 mL |
| 90% Percoll Media | Hepatocyte isolation | 20 mL |
| 25% Percoll Media | Hepatocyte isolation | 20 mL |
-
1.
Prepare the perfusion buffer, digestion buffer, plating media, Percoll media, and maintenance media prior to the isolation. Place perfusion buffer and digestion buffer sufficient for one digestion reaction in two separate 50 mL tubes in a 42°C water bath for at least 30 min prior to the isolation. 40 mL perfusion buffer and 30 mL digestion buffer are sufficient for isolation of hepatocytes from one mouse.
-
2.
Set up the perfusion pump by mounting the peristaltic pump with a long 1/32” ID × 3/32” OD silicone tubing immersed in a water bath. The tubing will be connected to the pump with a 3/32” Polypropylene coupler (Figure 2A).
-
3.
Place the inlet tubing into a 50 mL tube with perfusion buffer in the 42°C water bath.
-
4.
Set the perfusion pump speed at 1–2 mL/min. Note that the settings will vary depending on the instrument used (Figure 2B).
-
5.
Place the surgical table at an angle in a waterproof container in close proximity to the perfusion pump.
-
6.
Assemble the outlet catheter by attaching a 25G 7/8 needle to the outlet tubing (Figure 2C).
-
7.
Rinse the pump with 10 mL 70% EtOH and 10 mL water.
-
8.
Run the pump with perfusion buffer for 5 min to ensure an even flow rate.
Figure 2.

Overview of the setup of the peristaltic perfusion pump, tubes, syringes, and water bath
The perfusion pump system includes a water bath (A) and a peristaltic pump with adjustable speed (B). The inlet tubing is placed into a 50 mL tube in the 42°C water bath containing the perfusion buffer (A). The silicone tubing is connected to the peristaltic pump using a polypropylene coupler. A 25G 7/8 needle is attached to the outlet tubing used to cannulate the inferior vena cava (C).
Liver perfusion and enzymatic dissociation of liver cells
This step will perfuse the liver followed by enzymatical digestion using collagenase IV to dissociate the liver cells into a single-cell suspension.
-
9.
Euthanize the mouse by CO2 asphyxiation and clean the abdominal area with 70% EtOH.
-
10.
Make an incision in the abdominal area to expose the liver, inferior vena cava, and portal vein.
-
11.
Place the catheter into inferior vena cava (Figure 3A).
-
12.
Start the perfusion pump with perfusion buffer at a rate of 2 mL/min. Ensure the correct flow rate by measuring the outlet volume as a fraction of time.
-
13.
After 2 min, cut the hepatic portal vein to clear the liver from blood.
-
14.
Continue the perfusion for an additional 10 min. This step will use 25 mL perfusion buffer in total. Successful perfusion will result in a pale, enlarged liver (Figure 3B).
-
15.
When 2–5 mL is left of the perfusion buffer, add the digestion buffer to the 50 mL tube without stopping the pump.
-
16.
Digest for 10–20 min at 2 mL/min. This will use 20–30 mL digestion buffer in total.
-
17.
When visible tissue dissociation is seen, carefully remove the liver including the gall bladder and place in a 10 cm petri dish kept on ice (Figure 3C).
-
18.
Add 10 mL cold plating medium to the dish. Without puncturing the gall bladder, dissociate the liver cells by gently swirling the liver in cold plating media. Use a pipette with a 1,000 μL tip to generate a homogenous cell suspension (Figure 3D).
-
19.
Transfer the cell suspension to a 70 μm strainer placed on a 50 mL tube on ice (Figure 3E). Wash the plate with an additional 10 mL plating medium and transfer to the 50 mL tube.
Figure 3.

Step-by-step illustration of isolation of viable hepatocytes from diet-induced NASH mice
(A) Placement of the catheter into inferior vena cava (step 11).
(B) Liver perfusion resulting in a pale, enlarged liver (step 14).
(C) Result after digestion using collagenase solution (step 17).
(D) Addition of plating medium and dislodging of the cells from the liver (step 18).
(E) Passing the cell suspension through a 70 μm strainer (step 19).
(F) Schematic illustration of centrifugation steps to isolate hepatocytes. Cell pellets will be collected after 100 × g and 1,000 × g for 10 min using 25% and 90% Percoll gradients (steps 20–25).
(G) Plated hepatocytes 24 h after isolation under a phase-contrast microscope. Scale bar denotes 100 μm.
Single-cell hepatocyte isolation using Percoll gradient centrifugation
This step will separate viable hepatocytes with different sizes of lipids from dead hepatocytes and other non-parenchymal cell types in the cell suspension using Percoll gradient centrifugation. The Percoll media is composed of colloidal silica and has been extensively used for the isolation of cells and organelles (Kreamer et al., 1986; Pertoft, 2000). The density of the Percoll solution is dependent on its concentration, the total g-force, and the centrifugation time, which will affect the gradient formation. Depending on the requirements, conditions such as centrifugal force, centrifugation time, and Percoll density gradient composition can be modified.
-
20.
Centrifuge the strained cells at 50 × g for 5 min at 20°C–24°C to separate hepatocytes from non-parenchymal cells. Hepatocytes will pellet at 50 × g.
-
21.
Discard the supernatant.
-
22.
Resuspend the pellet in 20 mL plating media and wash the cells twice at 50 × g for 5 min.
-
23.
Resuspend the pellet in 40 mL plating media. Divide the solution into four tubes with 10 mL solution in each tube.
-
24.
Prepare two sets of Percoll/hepatocyte mix for a total of 4 tubes with either 25% or 90% Percoll by adding 10 mL of 20°C–24°C 90% Percoll media to two of the tubes and 25% Percoll media to the remaining tubes. The final volume of the Percoll/hepatocyte mix will be 20 mL.
-
25.
Centrifuge one set of tubes (25% and 90% Percoll/hepatocytes) at 100 × g and the other set of tubes (25% and 90% Percoll/hepatocytes) at 1,000 × g. Centrifuging cells at 1,000 × g is predicted to sediment cells with higher lipid content compared with 100 × g. This step will isolate viable non-lipid-filled and lipid-filled hepatocytes (Figure 3F).
-
26.
Resuspend the pellets in 20 mL plating media and spin at 50 × g for 4 min twice to remove the Percoll media.
-
27.
Resuspend the final hepatocyte pellets in 1 mL media and measure cell number and viability using trypan blue and a manual or automatic hemocytometer.
-
28.
Seed cells into collagen coated plates. Plate 4 ×105 viable cells in 6-well plates or 2 × 105 cells in 12-well plates and place plates in a 37°C incubator with 5% CO2 for 3–4 h.
-
29.
After 3–4 h, wash once with PBS to remove dead cells, replace with maintenance medium (Figure 3G). Experiments can start the same or next day.
Note: Keep the Percoll medium at 20°C–24°C to maintain the pH between 8.5–9.5.
Note: Depending on the degree of hepatic steatosis, differences in cell numbers from the different Percoll gradient fractions can vary between isolations. When isolating cells from NASH mice, the largest number of hepatocytes will be collected from the 25% Percoll density gradient, while the lowest number of hepatocytes will be obtained from the 90% Percoll density gradient.
Optional: If complete removal of erythrocytes is desired, spin the hepatocyte pellet at 20 × g for 3 min after step 20 and remove the supernatant.
Optional: If non-parenchymal cells are desired, collect the supernatant from step 21 and spin the supernatant solution at 650 × g to pellet non-parenchymal cells such as stellate cells, endothelial cells, and Kupffer cells.
Optional: If the cells will be analyzed with single-cell RNA sequencing or RNA sequencing, continue directly from step 27 without plating the cells.
Pause Point: It is necessary to complete the whole procedure without pause to maintain cell viability.
Functional characterization of hepatocytes from NASH mice
This step will functionally characterize the hepatocytes using oil red O staining. Alternative analyses include drug testing, lipogenesis, and gene expression analyses. The protocol here uses hepatocytes plated in a 12-well plate.
Oil red O working solution (sufficient for one 12 well plate)
| Reagent | Stock concentration | Final concentration | Amount |
|---|---|---|---|
| Oil red O | n/a | n/a | 4.2 mL |
| ddH2O | n/a | n/a | 2.8 mL |
| Total | 7 mL |
Prepare the solution 10 min before use.
-
30.
Prepare 7 mL oil red O Solution by mixing oil red O with H2O at a 3:2 ratio. 7 mL is sufficient for one 12-well plate when using 500 μL per well.
-
31.
Gently wash the cells twice with 1 mL PBS.
-
32.
Add 0.5 mL per well of 3% Formalin diluted in PBS and incubate for 0.5–1 h at 20°C–24°C.
-
33.
Discard the formalin and wash the cells twice with 1 mL ddH2O.
-
34.
Add 0.5 mL 60% isopropanol and incubate for 5 min.
-
35.
Discard the isopropanol and apply the oil red O Working Solution. Rotate the plate to ensure even distribution and incubate for 10–20 min at 20°C–24°C.
-
36.
Discard the oil red O solution and wash the cells 2–5 times with ddH2O until clear.
-
37.
Add 1 mL ddH2O and view red lipid droplets under a microscope using a 40× objective.
Optional: For nuclei staining, add Hematoxylin after step 36 and incubate for 1 minute. After discarding Hematoxylin, wash the cells 2–5 times with ddH2O until clear. Nuclei should appear blue under the microscope.
Note: Stained cells can be stored in 4°C for up to 1 week. Keep the cells immersed in water to avoid drying.
Expected outcomes
The estimated yield is ∼ 1–2 million hepatocytes from one liver isolated from 6–9 weeks diet-induced NASH with a viability ranging from 50%–90% (Figure 4A). This can be compared to the yield from one mouse liver isolated from chow-fed mice which is ∼ 5–20 million cells. Typically, shorter time on NASH diet will result in a higher cell number and viability. Furthermore, with less severe liver steatosis, a higher number of hepatocytes will be obtained from the 90% Percoll gradient, while more severe steatosis will result in a larger fraction of hepatocytes isolated from the 25% Percoll gradient.
Figure 4.

A frequently observed outcome: number of hepatocytes isolated from different Percoll fractions, and the corresponding lipid content as measured by oil red O staining
(A) Viable hepatocyte populations from a representative isolation. The cell numbers from each Percoll gradient were measured using trypan blue and a manual hemocytometer. Values are expressed as a percentage of viable cells.
(B) Hepatocyte cell numbers from a representative isolation. The cell numbers and viability from each Percoll gradient was measured using trypan blue and a manual hemocytometer.
(C) Lipid staining using oil red O in hepatocytes from 90% and 25% Percoll gradients at 100 × g or 1,000 × g. Microscope images show results from a representative isolation using a 60× objective. Mayer’s hematoxylin solution is used as counterstain. Scale bar denotes 100 μm in the original image and 35 μm in the magnified image.
A frequently observed outcome from a hepatocyte isolation from different Percoll fractions is shown in the graph expressed as viable/dead cells from each Percoll fraction (Figure 4B). Accordingly, measuring the lipid content by oil red O staining shows increased lipid staining in the 25% Percoll gradient compared to 90%, demonstrating successful isolation of lipid-deficient and lipid-filled hepatocytes (Figure 4C).
Limitations
One distinct disadvantage of this method is the yield of viable lipid-filled hepatocytes that can be obtained in comparison to a typical hepatocyte isolation from healthy livers. Thus, the degree of NASH-induction might have to be optimized to achieve the highest yield while inducing a sufficient degree of steatosis. Additionally, cells from NASH mice are more sensitive to overdigestion. Therefore, careful observation of the timing of digestion is essential, as this otherwise can result in large variability between preparations.
Another disadvantage of culturing primary hepatocytes is the short time frame by which experiments have to be concluded (Shulman and Nahmias, 2013). Typically, primary hepatocytes will lose their functional features within 24–48 h after plating, leading to a dramatic drop in typical hepatocyte genes such as albumin, while fibroblast markers such as afp are increased, indicating a loss of cell characteristics. This limits the number and types of downstream functional assays that can be performed. However, multiple types of analyses can be performed within this time frame which might have useful applications, such as drug metabolism assays, hepatotoxicity assays, or glucose and lipid metabolic assays. Alternatively, for long-term functional analyses, our isolation method could be used in conjunction with the 5C small molecule inhibitor cocktail for the preservation of hepatocyte-specific functions and differentiation (Xiang et al., 2019).
Troubleshooting
Problem 1
Failure to cannulate inferior vena cava (step 11).
Potential solution
If the catheter insertion into inferior vena cava is unsuccessful, the catheter can be inserted into the hepatic portal vein. Proceed with the perfusion according to the protocol, but instead cut the inferior vena cava to release excess blood.
Problem 2
Low cell viability or small cell pellets (step 27).
Potential solution
Low cell viability or small cell pellets can be due to either incomplete digestion or overdigestion.
Poor perfusion can result in incomplete digestion and low recovery of cells. This will lead to the inability to mechanically dissociate the liver into a single-cell suspension. To solve this problem, ensure that the perfusion using perfusion buffer results in a pale, slightly swollen liver before introducing the digestion buffer. Excessive perfusion and digestion will lead to overdigestion and should be avoided to prevent cell death. If overdigestion is seen, lower the speed of perfusion and digestion from 2 mL/min to 1 mL/min.
The time of digestion will depend on the specific collagenase activity in the digestion buffer. Due to lot-to-lot variability in collagenase activity, optimization is warranted when using a new lot.
Problem 3
Low cell recovery during Percoll gradient cell isolation even after successful perfusion (step 25).
Potential solution
This problem might occur when the temperature of the Percoll solution is incorrect or when too many cells are added to the gradient. Keep the Percoll medium at 20°C–24°C to maintain accurate density and to keep the pH between 8.5–9.5. The volume of the density gradient is optimized for one murine liver per gradient. If more cells are needed, several livers can be pooled together. If pooling livers from several mice, increase the volume of media and gradient media accordingly.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dr. Katrin J. Svensson (katrinjs@stanford.edu).
Materials availability
This study did not generate any unique materials or reagents.
Data and code availability
This study did not generate any unique datasets or code.
Acknowledgments
This work was supported by the NIH grants DK125260 and DK111916, the Jacob Churg Foundation, and the McCormick and Gabilan Award (K.J.S.). This work was supported by the Stanford Diabetes Research Center (NIH grant P30DK116074).
Author contributions
Conceptualization, Y.J. and K.J.S.; Investigation, Y.J., M.Z., and K.J.S.; Writing – Original Draft, Y.J. and K.J.S.; Writing – Review & Editing, Y.J., M.Z., and K.J.S.; Funding Acquisition, K.J.S.; Supervision, K.J.S.
Declaration of interests
The authors declare no competing interests.
References
- Berry M.N., Friend D.S. High-yield preparation of isolated rat liver parenchymal cells: a biochemical and fine structural study. J. Cell Biol. 1969;43:506–520. doi: 10.1083/jcb.43.3.506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fengler V.H.I., Macheiner T., Kessler S.M., Czepukojc B., Gemperlein K., Müller R., Kiemer A.K., Magnes C., Haybaeck J., Lackner C. Susceptibility of different mouse wild type strains to develop diet-induced NAFLD/AFLD-associated liver disease. PLoS One. 2016;11:e0155163. doi: 10.1371/journal.pone.0155163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonçalves L.A., Vigário A.M., Penha-Gonçalves C. Improved isolation of murine hepatocytes for in vitro malaria liver stage studies. Malar. J. 2007;6:169. doi: 10.1186/1475-2875-6-169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kegel V., Deharde D., Pfeiffer E., Zeilinger K., Seehofer D., Damm G. Protocol for isolation of primary human hepatocytes and corresponding major populations of non-parenchymal liver cells. J. Vis. Exp. 2016;109:e53069. doi: 10.3791/53069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreamer B.L., Staecker J.L., Sawada N., Sattler G.L., Hsia M.T., Pitot H.C. Use of a low-speed, iso-density Percoll centrifugation method to increase the viability of isolated rat hepatocyte preparations. In Vitro Cell Dev. Biol. 1986;22:201–211. doi: 10.1007/BF02623304. [DOI] [PubMed] [Google Scholar]
- Li W.-C., Ralphs K.L., Tosh D. Isolation and culture of adult mouse hepatocytes. Methods Mol. Biol. 2010;633:185–196. doi: 10.1007/978-1-59745-019-5_13. [DOI] [PubMed] [Google Scholar]
- Pertoft H. Fractionation of cells and subcellular particles with Percoll. J. Biochem. Biophys. Methods. 2000;44:1–30. doi: 10.1016/s0165-022x(00)00066-x. [DOI] [PubMed] [Google Scholar]
- Radhakrishnan S., Ke J.-Y., Pellizzon M.A. Targeted nutrient modifications in purified diets differentially affect nonalcoholic fatty liver disease and metabolic disease development in rodent models. Curr. Dev. Nutr. 2020;4:nzaa078. doi: 10.1093/cdn/nzaa078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharabi K., Lin H., Tavares C.D.J., Dominy J.E., Camporez J.P., Perry R.J., Schilling R., Rines A.K., Lee J., Hickey M. Selective chemical inhibition of PGC-1α gluconeogenic activity ameliorates type 2 diabetes. Cell. 2017;169:148–160.e15. doi: 10.1016/j.cell.2017.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shulman M., Nahmias Y. Long-term culture and coculture of primary rat and human hepatocytes. Methods Mol. Biol. 2013;945:287–302. doi: 10.1007/978-1-62703-125-7_17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang C., Du Y., Meng G., Soon Yi L., Sun S., Song N., Zhang X., Xiao Y., Wang J., Yi Z. Long-term functional maintenance of primary human hepatocytes in vitro. Science. 2019;364:399–402. doi: 10.1126/science.aau7307. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This study did not generate any unique datasets or code.