

Summary
Mosaic analysis with double markers (MADM) technology enables concomitant fluorescent cell labeling and induction of uniparental chromosome disomy (UPD) with single-cell resolution. In UPD, imprinted genes are either overexpressed 2-fold or are not expressed. Here, the MADM platform is utilized to probe imprinting phenotypes at the transcriptional level. This protocol highlights major steps for the generation and isolation of projection neurons and astrocytes with MADM-induced UPD from mouse cerebral cortex for downstream single-cell and low-input sample RNA-sequencing experiments.
For complete details on the use and execution of this protocol, please refer to Laukoter et al. (2020b).
Subject Areas: Single Cell, Flow Cytometry/Mass Cytometry, Genetics, Neuroscience
Graphical Abstract
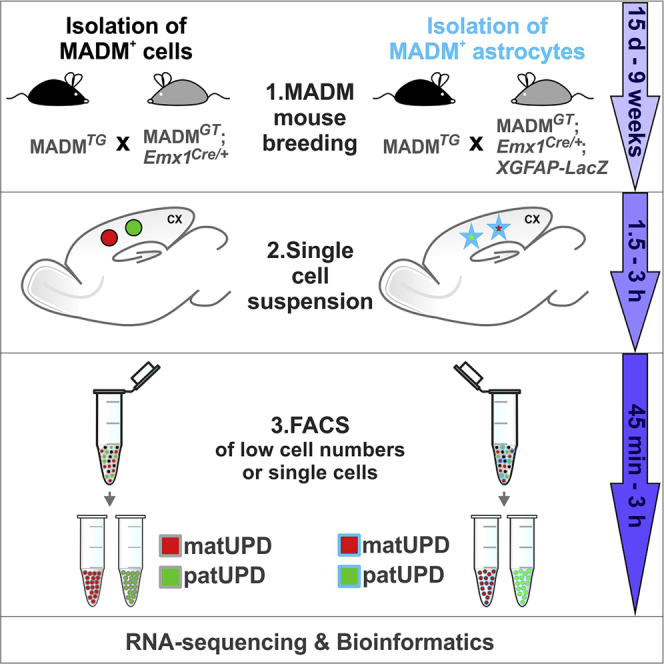
Highlights
-
•
Generation of cells with uniparental chromosome disomy (UPD) using MADM technology
-
•
Isolation of cells with MADM-induced UPD for single-cell downstream applications
-
•
Analysis of cortical cells with MADM-induced UPD using scRNA-seq
-
•
Analysis of neurons and astrocytes with MADM-induced UPD to probe genomic imprinting
Mosaic analysis with double markers (MADM) technology enables concomitant fluorescent cell labeling and induction of uniparental chromosome disomy (UPD) with single-cell resolution. In UPD, imprinted genes are either overexpressed 2-fold or are not expressed. Here, the MADM platform is utilized to probe imprinting phenotypes at the transcriptional level. This protocol highlights major steps for the generation and isolation of projection neurons and astrocytes with MADM-induced UPD from mouse cerebral cortex for downstream single-cell and low-input sample RNA-sequencing experiments.
Before you begin
Background
Genomic imprinting is an epigenetic phenomenon in mammalian genomes that results in monoallelic parent-of-origin dependent gene expression. Therefore, certain genes are only expressed from the paternally inherited allele and others only from the maternally inherited allele. A key feature of imprinted genes is their imperative dosage-sensitivity. Uniparental chromosome disomy (UPD, somatic cells containing either two maternal or two paternal chromosomes) has previously been used to probe genomic imprinting mainly in whole animal, and revealed major principles of genomic imprinting (Tucci et al., 2019).
MADM is a genetic technology that allows concomitant fluorescent labeling and induction of UPD with unprecedented single-cell resolution in a defined genetic lineage (Contreras et al., 2020, Hippenmeyer et al., 2013, Laukoter et al., 2020a, Laukoter et al., 2020b). MADM is based on the use of split marker genes that consist of partial coding sequences for green fluorescent protein (GFP) and tandem dimer Tomato (TdT), interspersed by LoxP sites. MADM requires crossing of two different mouse lines, the GT (N-GFP/C-TdT) line and the TG (N-TdT/C-GFP) line, one of which also carries a Cre driver of interest, for the generation of experimental animals (Figures 1A and 1B).
Figure 1.

Experimental MADM paradigm for generating UPD with single-cell resolution
MADM utilizes Cre/LoxP-mediated interchromosomal recombination to reconstitute two reciprocally chimeric marker genes (GT/TG). The two reciprocally chimeric marker genes are targeted separately to identical loci on homologous chromosomes. The marker genes consist of partial coding sequences for green (GFP, G) and red (TdT, T) fluorescent proteins separated by an intron containing a LoxP site. Recombination in G2 phase of the cell cycle followed by X-Segregation (the two recombinant chromosomes are transmitted to distinct daughter cells) leads to individual green (GFP+) and red (TdT+) labeled cells, respectively, with near complete uniparental chromosome disomy (UPD). Recombination in G2 phase of the cell cycle with segregation of both recombinant chromosomes (including fully reconstituted TdT and GFP marker genes) to the same daughter cell (Z-Segregation) does not alter the genotype and results in one yellow cell which serves as control. Z-Segregation at the same time also gives rise to an unlabeled cell (unchanged genotype).
(A) MADM paradigm to generate UPD using forward crossing scheme: If the TG MADM cassette is inherited from the father and the GT MADM cassette from the mother as indicated, cells labeled in red show unimaternal chromosome disomy (MM; matUPD) and green cells unipaternal chromosome disomy (PP, patUPD). Consequently, maternally expressed genes (MAT, shown in red) are expressed at the double dose and paternally expressed genes are not expressed in red matUPD cells. In contrast, green cells with patUPD express paternally expressed genes (PAT, shown in blue) at twice the normal dose and maternally expressed genes are not expressed. Thus, imprinted genes are differentially expressed depending on the UPD. Biallelically expressed genes (BAE, non-imprinted genes, shown in black) are not affected.
(B) MADM paradigm to generate UPD using reverse crossing scheme. Here, the TG MADM cassette is inherited from the mother and the GT MADM cassette from the father. Thus, matUPD (2x maternally expressed genes, 0x paternally expressed genes) are labeled in green (GFP+) and patUPD (2x paternally expressed genes, 0x maternally expressed genes) are labeled in red (TdT+). Symbols are detailed in the key.
Parts of the figure and its legend are reused but adapted from previous publications (Hippenmeyer et al., 2013, Hippenmeyer et al., 2010, Laukoter et al., 2020b) with permission.
Cells with MADM-induced UPD are generated by Cre/LoxP-dependent mitotic recombination during the G2 phase in dividing stem cells followed by X-segregation. G2-X events produce near complete UPD of a particular chromosome carrying the MADM cassettes (Figures 1A and 1B). Here, we established an experimental pipeline for the isolation of MADM-labeled cells carrying UPD for downstream applications, including small sample and single-cell RNA-sequencing (scRNAseq). Our protocol is based on the use of Emx1-Cre driver in combination with MADM cassettes targeted to chr. 7, which harbors many imprinted genes, but can be applied to any chromosome with MADM cassettes (Contreras et al., 2020).
In cells with MADM-induced UPD, imprinted genes located on the chromosome carrying the MADM cassettes are either homozygosed to be expressed in double dose or are not expressed. MADM-induced UPD thus provide a functional approach to simultaneously test loss-of-function and gain-of-function of imprinted gene dosage. Cells with MADM-induced UPD are visualized with distinct fluorescent colors, e.g., patUPD cells labeled in green (GFP+) and matUPD cells labeled in red (tdT+) (Figure 1A) (or vice versa (Figure 1B), also outlined in breeding strategy in Figures 2A and 2B). Importantly, the dosage of imprinted gene expression is predictable in MADM-induced UPD. Thus, ensuing cellular and transcriptional phenotypes, originating from imbalanced imprinted gene expression, can be analyzed in a cell-type-specific manner. MADM-induced UPD thus provide a unique functional assay to probe genomic imprinting with single-cell resolution.
Figure 2.

Breeding strategy for experimental mice
(A) Breeding strategy A illustrates the generation of experimental MADM mice for sorting of cells originating from a genetically defined lineage. Experimental mice contain red (TdT+) cells with unimaternal disomy (matUPD: 2x maternally expressed genes, 0x paternally expressed genes). Green (GFP+) cells carry unipaternal disomy (patUPD: 2x paternally expressed genes, 0x maternally expressed genes).
(B) Breeding strategy B illustrates reversal of fluorescent colors in cells with MADM-induced UPD. In this setup green (GFP+) cells contain matUPD and red (TdT+) cells carry patUPD.
(C) Breeding strategy C shows the generation of experimental MADM-LacZ mice for astrocyte sorting. Astrocytes are identified based on LacZ expression under the control of the human GFAP promoter. GFP+, LacZ+ astrocytes harbor matUPD, while TdT+, LacZ+ positive cells carry patUPD.
(D) Breeding strategy D indicates reversal of fluorescent colors in cells with MADM-induced UPD in experimental animals for MADM+LacZ astrocyte sorting. TdT+, LacZ+ cells carry matUPD and GFP+, LacZ+ cells harbor patUPD.
Parts of the figure and its legend are reused but adapted from previous publications (Hippenmeyer et al., 2013, Hippenmeyer et al., 2010, Laukoter et al., 2020b) with permission.
For more details on the MADM technology, please refer to (Beattie et al., 2017, Beattie et al., 2020, Contreras et al., 2020, Hippenmeyer et al., 2013, Hippenmeyer et al., 2010, Laukoter et al., 2020a, Laukoter et al., 2020b, Zong et al., 2005).
Generation of experimental MADM mice for FACS isolation of cells with MADM-induced UPD
Timing: 15 days and up to 9 weeks
-
1.Cross MADM-7TG/TG animals with MADM-7 GT/GT;Emx1Cre/+ animals (Figures 2A and 2B).Note: Breeding strategy A: cross MADM-7TG/TG males (homozygous for the MADM cassette that contains the N-terminal domain of TdT and C-terminal domain of GFP) with MADM-7GT/GT;Emx1Cre/+ females (homozygous for the MADM cassette that contains the N-terminal domain of GFP and C-terminal domain of TdT). In the experimental animals (F1 generation), red cells (TdT+) will carry unimaternal disomy (matUPD; double dose of maternally expressed genes, no expression of paternally expressed genes) and green cells (GFP+) will contain unipaternal disomy (patUPD; 2x expression dose of paternally expressed genes and no expression of maternally expressed genes).Breeding strategy B: Fluorescent colors of UPD cells are swapped. Reversing fluorescent colors in cells with UPD serves as control for potential effects resulting from the expression of the fluorescent marker genes themselves. Cross MADMTG/TG females with MADMGT/GT;Emx1Cre/+ males. In the F1 experimental generation red cells contain patUPD (2x expression dose of paternally expressed genes and no expression of maternally expressed genes) and green cells carry matUPD (2x expression dose of maternally expressed genes and no expression of paternally expressed genes).
-
a.Check for vaginal plug to monitor successful mating and to facilitate the experimental planning.
-
b.Males and females of the F1 generation can be used as experimental animals (genotype is MADM-7GT/TG, Emx1Cre/+).
-
c.Perform experiment at developmental stage(s) of choice.Note: Experimental animals can be used at embryonic stages, P0, early postnatal stages and up to adulthood (P21, P42). Here we made use of Emx1-Cre that primarily targets radial glial progenitors in the dorsal forebrain, which give rise to excitatory projection neurons and glial cells in cerebral cortex (Gorski et al., 2002). Distinct proportions of these cortical cell types are present at different developmental time points. For details refer to Table 1.Note: Here we used MADM cassettes targeted to chr. 7. In principle the protocol for isolation of cells with MADM-induced UPD can be applied to any MADM chromosome (Contreras et al., 2020). Besides Emx1-Cre, any other Cre driver can be used, provided Cre is expressed in dividing stem and progenitor cells, to target cell types and lineages of interest. Depending on the use of specific chr. with targeted MADM cassettes and the particular Cre driver, interchromosomal recombination and MADM-labeling efficiencies in experimental animals might vary.
-
a.
Table 1.
Developmental time point and corresponding cell types present in Emx1-lineage
| Time point | Radial glial progenitor cells | Astrocyte intermediate progenitors | Excitatory projection neurons | Astrocytes | Oligodendrocytes | Olfactory bulb neuroblasts |
|---|---|---|---|---|---|---|
| E15 | fraction | – | majority | – | – | fraction |
| P0 | fraction | fraction | majority | fraction | fraction | fraction |
| P7 | – | – | majority | fraction | fraction | fraction |
| P14 | – | – | majority | fraction | fraction | fraction |
| P21 | – | – | majority | fraction | fraction | fraction |
Generation of experimental MADM mice for FACS isolation of astrocytes with MADM-induced UPD
Note: Astrocytes are specifically targeted using a LacZ transgene expressed under the control of the human GFAP promoter (Brenner et al., 1994). The transgene is located on the X-chromosome. Due to random X-inactivation, labeling frequency varies in females. For consistency (i.e., maximal LacZ expression) it is therefore recommended to primarily use males as experimental animals.
-
2.Cross MADM-7GT/GT;Emx1Cre/+ males with MADM-7TG/TG;XGFAP-LacZ+/− females (UPDs are colored as follows: matUPD in green; patUPD in red) (as shown in breeding strategy C in Figure 2C).
 CRITICAL: Make sure that the XGFAP-LacZ transgene is transmitted from females in order to obtain XGFAP, LacZ positive male experimental animals.Note: In order to reverse the fluorescent colors in cells with UPD (matUPD in red and patUPD in green) use MADM-7TG/TG males and MADMGT/GT;Emx1Cre/+;XGFAP-LacZ+/− females (Figure 2D).
CRITICAL: Make sure that the XGFAP-LacZ transgene is transmitted from females in order to obtain XGFAP, LacZ positive male experimental animals.Note: In order to reverse the fluorescent colors in cells with UPD (matUPD in red and patUPD in green) use MADM-7TG/TG males and MADMGT/GT;Emx1Cre/+;XGFAP-LacZ+/− females (Figure 2D).-
a.Check for vaginal plug to monitor successful mating and to facilitate the experimental planning.
-
b.Males of the F1 generation can be used as experimental animals (genotype is MADM-7GT/TG;Emx1Cre/+;XGFAP-LacZ+).
-
c.Include a negative control by also preparing single-cell suspension of one MADM-7GT/TG;Emx1Cre/+ (no XGFAP-LacZ expression) animal. The negative control will allow proper set up of the FACS gating strategy (see below).
-
d.Perform experiment at developmental stage of choice (in our lab we have successfully applied the protocol at P0, P4, and up to P14).
-
a.
Prepare equipment (on the day of experiment)
-
3.
Clean all dissection tools (2 curved forceps and scissors) with 70% ethanol and RNase Away, a decontamination reagent that removes RNase from equipment and surfaces.
-
4.
Pre-warm shaking water bath (37°C) for single-cell suspension of Emx1-lineage or Thermoshaker (37°C) for single-cell suspension of astrocytes.
-
5.Prepare PCR tube holders for sorting:
-
a.Take 1.5 mL tubes and cut off the lid.
-
b.Fill the 1.5 mL tubes with paper tissue or Parafilm (Figure 3).
-
c.Insert a PCR tube in the middle of the stuffed 1.5 mL tube.
-
d.After sorting, PCR tubes can be taken out of the 1.5 mL tubes with forceps and PCR tube holders can be reused by inserting a new PCR tube.
-
a.
Figure 3.

Self-made PCR tube holders for collecting sorted cells at FACS Aria
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, peptides, and recombinant proteins | ||
| Papain vial source | Worthington | Cat#PAP2 |
| DNase vial source | Worthington | Cat#D2 |
| Inhibitor vial source | Worthington | Cat#OI-BSA |
| EBSS | Thermo Fisher Scientific | Cat#24010043 |
| DMEM/F12 | Thermo Fisher Scientific | Cat#21041025 |
| Heat-inactivated fetal bovine serum (FBS) | Thermo Fisher Scientific | Cat#10082147 |
| Heat-inactivated horse serum (HS) | Thermo Fisher Scientific | Cat#26050088 |
| Triton X 100 | Sigma-Aldrich | Cat#T878 |
| RNase inhibitor | Takara | Cat#2313A |
| RNase away | Thermo Fisher Scientific | Cat#10328011 |
| RNase-free water | Thermo Fisher Scientific | Cat#10977035 |
| Critical commercial assays | ||
| FACS Blue LacZ beta galactosidase detection kit | Abcam | Cat#ab189815 |
| Experimental models: organisms/strains | ||
| Mouse: MADM-7-GT | The Jackson Laboratory | RRID:IMSR_JAX:021457 |
| Mouse: MADM-7-TG | The Jackson Laboratory | RRID:IMSR_JAX:021458 |
| Mouse: Emx1-Cre | The Jackson Laboratory | RRID:IMSR_JAX:005628 |
| Mouse: XGFAP-LacZ | The Jackson Laboratory | RRID: IMSR JAX: 003487 |
| Software and algorithms | ||
| FACS Diva | BD Biosciences | https://www.bdbiosciences.com/en-us/instruments/research-instruments/research-software/flow-cytometry-acquisition/facsdiva-software |
| R statistical environment | https://www.r-project.org/ | |
| DESeq2 | Love et al., 2014 | http://www.bioconductor.org/ |
| Seurat | Butler et al., 2018 | http://www.bioconductor.org/ |
| Other | ||
| FACS Aria III | BD Biosciences | N/A |
| AluminaSeal | Merck | Cat#Z740251-100EA |
| Hard-Shell PCR Plates (96-well) | Bio Rad | Cat#HSP9631 |
| PCR tubes and caps, RNase-free, 0.2 mL (8-strip format) | Thermo Fisher Scientific | Cat#AM12230 |
| Falcon 5 mL polystyrene round-bottom tube with cell strainer cap (Falcon 352235) (40 μm cell strainer) | Fisher Scientific | Cat#10585801 |
| Microfuge tubes 1.5 mL | Thermo Fisher Scientific | Cat#AM12450 |
| TPP TubeSpin bioreactor tubes | Merck | Cat#Z761028-180EA |
Materials and equipment
Solutions from Worthington kit
Resuspend Papain containing L-cysteine and EDTA (in 5 mL EBSS), DNase (in 0.5 mL EBSS) and Ovomucoid protease inhibitor (in 32 mL EBSS) according to manufacturer’s instructions.
Papain and DNase should be prepared freshly on the day of the experiment.
Resuspended Ovomucoid protease inhibitor can be stored at 4°C and used for several months.
Papain-DNase solution
| Reagent | Final concentration (mM or μM) | Amount |
|---|---|---|
| Papain | 20 units/mL | 5 mL |
| DNase | 1,000 units/mL | 250 μL |
Solution 2
| Reagent | Final concentration (mM or μM) | Amount |
|---|---|---|
| EBSS | n/a | 5,350 μL |
| DNase | 166.7 units/mL | 250 μL |
| Ovomucoid protease inhibitor | 0.67 mg | 400 μL |
| Total | n/a | 6,000 μL |
Media
| Reagent | Final concentration (mM or μM) | Amount |
|---|---|---|
| DMEM/F12 | n/a | 14.4 mL |
| FBS | 10% | 1,800 μL |
| HS | 10% | 1,800 μL |
| Total | n/a | 18 mL |
Solutions of FACS blue LacZ beta galactosidase detection kit
Resuspend and aliquot reagents according to manufacturer’s instructions.
Lysis buffer
| Reagent | Final concentration (mM or μM) | Amount |
|---|---|---|
| Triton X-100 | 0.2% | 0.08 μL |
| RNase Inhibitor | 2 units/μL | 0.2 μL |
| RNase-free water | n/a | 3.72 μL |
| Total/well | n/a | 4 μL |
Note: This lysis buffer can be used to collect single cells for scRNAseq or low inputs of cells (up to 400 cells). RNA-sequencing with these amounts of cells was successfully performed using the Smart-seq V2 protocol (Picelli et al., 2014).
Step-by-step method details
Preparation of single-cell suspension for sorting cells with MADM-induced UPD of the Emx1 lineage in developing cerebral cortex
This section of the protocol documents the dissection and preparation of single-cell suspension from mouse cerebral cortex for sorting cells with MADM-induced UPD and fluorescent labeling derived from Emx1 lineage.
-
1.Prepare all solutions required for the tissue dissociation (details are listed above)
-
a.Papain-DNase solution
-
b.Solution 2
-
c.DMEM/F12
-
d.Media
-
a.
Volumes of reagents are adjusted according to the amount of used tissue/sample
| Tissue | Papain-DNase solution | Solution 2 (step 5) | Solution 2 (step 7) | DMEM/F12 |
|---|---|---|---|---|
| Single cortex (embryonic brain) | 650 μL | 500 μL | 250 μL | 1,000 μL |
| One hemisphere (P0 animal) | 650 μL | 500 μL | 250 μL | 1,000 μL |
| Multiple embryonic cortices (up to 3 cortices) | 1,300 μL | 1,200 μL | 300 μL | 1,000 μL |
| Cortex of single individual (P0, adult animals) | 1,300 μL | 1,200 μL | 300 μL | 1,000 μL |
| Multiple cortices (up to 3 cortices at any postnatal age) | 2,600 μL | 2,400 μL | 500 μL | 2,000 μL |
-
2.Sacrifice experimental animal
-
a.Decapitate the animal.
-
b.Remove the skin overlaying the skull. Then perform a long cut of the skull along the midline, a short cut at the naso-frontal suture (in front of the olfactory bulbs) and two short cuts along the parietal-interparietal sutures. By flipping the bone parts to the side, the brain is easily accessible. Place curved forceps underneath the entire brain, and thereby lift up the entire brain to remove it from the skull (Figure 4).
-
c.Remove the hippocampus to avoid contamination from other cell types.
-
d.Dissect the cortex. Here we specifically focus on the isolation of cortical cell types. In our hands, careful and precise dissection of the area of interest is most efficient to avoid contamination from cell types originating from other brain areas (Figure 4).
-
a.
-
3.
Transfer cortex to Papain-DNase solution in a 50 mL bioreactor tube harboring a filter-cap and incubate for 30 min at 37°C in a shaking water bath.
-
4.
During the incubation time, prepare lysis buffer and the respective collection tubes (PCR tubes) or 96-well plate.
-
5.
After 30 min, remove the bioreactor tube containing the dissociated cortical tissue from the water bath and add Solution 2. Mix thoroughly by pipetting up and down to dissolve remaining tissue parts.
-
6.
Centrifuge for 5 min with 100 × g at 20°C–22°C.
-
7.
Remove supernatant and resuspend the pellet in Solution 2. Add DMEM/F12 as a wash buffer. Mix well.
-
8.
Centrifuge for 5 min with 200 × g at 20°C–22°C.
-
9.
Remove supernatant and resuspend pellet in 300 – 500 μL media. Single-cell suspension is now ready for FACS. Keep suspension on ice until sorting can be started.
-
10.
For collection of a low number of cells, proceed with step 11. For sorting single cells, proceed with step 20.
Figure 4.

Dissection of the cortex from postnatal brain
(A) After removing the skull-overlaying skin (not shown), cut the bone tissue of the skull along the midline.
(B) Cut along the parietal-interparietal sutures.
(C) After removing the bone parts the brain is easily accessible.
(D) Carefully place the curved forceps underneath the brain in order to release it from the skull.
(E) Whole brain after removal from the skull.
(F) The whole hemisphere can be flipped to the side. Hippocampus is visible and accessible now.
(G) Removal of hippocampus.
(H) Hemisphere after hippocampus was removed.
(I) Dissection of the cortex.
(J) Dissected cortex which is used as starting material for preparation of single-cell suspension.
Isolation of cells with MADM-induced UPD using FACS (collection of low cell numbers)
In this section of the protocol MADM+ cells are sorted according to their fluorescent colors and collected for RNA-sequencing.
-
11.Ensure that the cytometer (here: FACS Aria III) is set up for your sorting experiment.
-
a.Use the 100 μm nozzle.
-
b.Set the sample temperature and the temperature for the collection tubes to 4°C.
-
c.Flush the system with RNase Away for 1–2 min.
-
d.Flush the system with RNase-free water for 1–2 min.
-
e.Adjust the side streams so that they hit the middle of the collection tubes.
-
a.
-
12.
Filter your entire sample through a 40 μm cell strainer. Dilute your sample with 200–300 μL media, which is additionally added through the cell strainer.
-
13.
Prepare 0.2 mL PCR tubes containing 4 μL lysis buffer as collection tubes for your sorted cells.
-
14.
Place the collection tubes into 1.5 mL tubes, which are then placed into the appropriate FACS Aria tube holder.
-
15.
Load your sample and record 30,000 - 50,000 events. Adjust the gates to select the surviving cell population (see Troubleshooting Problem 1), for removing doublets and for sorting GFP+ and TdT+ (optional: GFP+ TdT+ double positive) cells (Figure 5A) (see Troubleshooting Problem 2).
-
16.
Open the sort layout and select 4-way purity as the sorting mode to sort your sample.
-
17.
Start sorting. Depending on your experimental needs, either set a maximum number of labeled cells that shall be collected or sort the entire volume of sample.
Note: Lysis buffer was used and optimized for single cells and low amounts of cells (max. 400 cells). If a greater number of cells will be collected with FACS, a different lysis buffer needs to be used (Laukoter et al., 2020b).
-
18.
Once sorting is completed, transfer the entire volume of the collected sample into an empty well in a 96-well plate, which should remain on dry ice throughout the procedure. Be fast with pipetting since samples freeze in the pipette tip if this step is executed too slowly.
-
19.
Collect all replicates for your RNA-sequencing experiment in this 96-well plate. Either fill the entire plate on a single day, or add few samples on a day-to-day basis. When adding few samples over a time course of several days or weeks, store the plate at −80°C until further processing or additional samples can be added.
Note: This setup with collecting samples on a 96-well plate proved to be very useful for preparing samples for RNA-sequencing. However, sample collection and handling can be adapted to individual use and respective purposes.
Note: Since sorting can be done only for one sample at the time, it is important to decide on the number of cells you want to collect for your sequencing experiment. The longer a sample is kept on ice before it is processed by FACS, the higher the number of dead cells (this is of critical importance when cells from postnatal brains are sorted).
Figure 5.

Gating strategy for FACS experiments
(A) Gating steps for sorting MADM-labeled cells.
(B) Gating for red LacZ− population is set up based on a negative control (no XGFAP-LacZ expression).
(C) Gating for red astrocytes (TdT+, LacZ+).
(D) Gating for green LacZ− population is set up based on a negative control (no XGFAP-LacZ expression).
(E) Gating for green astrocytes (GFP+, LacZ+).
Isolation of cells with MADM-induced UPD using FACS for single-cell RNA-sequencing
This section of the protocol lists details on sorting MADM+ cells for single-cell RNA-sequencing.
-
20.Ensure that the cytometer (here: FACS Aria III) is set up for your sorting experiment.
-
a.Use the 100 μm nozzle.
-
b.Set the sample temperature and the plate holder temperature to 4°C.
-
c.Flush the system with RNase Away for 1–2 min.
-
d.Flush the system with RNase-free water for 1–2 min.
-
e.Make sure that the stream is hitting the wells of the 96-well plate properly in the middle (use “test sort” of unlabeled cells on the cover of the plate).
-
a.
-
21.
Filter your entire sample through a 40 μm cell strainer. Dilute your sample with 200–300 μL media, which is additionally added through the cell strainer.
-
22.
Place 96-well plate in the plate holder. All wells need to be filled with 4 μL lysis buffer.
-
23.
Load your sample and record 30,000 – 50,000 events. Adjust the gates to select the live cell population (see Troubleshooting Problem 1), for removing doublets and for sorting GFP+ and TdT+ (optional: GFP+ TdT+ double positive) cells (see Figure 5A) (same comments as above) (see Troubleshooting Problem 2).
-
24.
Open the sort layout and set the purity to single cells.
-
25.
Select the collection of a single cell per well.
Note: Design a layout for collecting your single cells. Keep in mind that you may want to sort a higher number of cells (up to 400 cells) as a positive control for your sequencing experiment in one of the wells. For this purpose, you can also make use of the MADM-unlabeled cells as there is no limitation with cell numbers.
Consult with your sequencing facility regarding the plate layout since empty wells and a positive control might be needed at specific positions. An example for a plate layout is provided in Table 2.
-
26.
Start sorting.
-
27.
After sorting is completed, seal the plate with AluminaSeal, quick freeze immediately on dry ice and store at −80°C until further processing. In order to ensure maximum sequencing efficiency plates should be processed within three months after collection.
Table 2.
Potential plate layout for collection of single cells which are later processed for RNA-sequencing
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A | 1 TdT+ cell | 1 GFP+ cell | 1 TdT+ cell | 1 GFP+ cell | 1 TdT+ cell | 1 GFP+ cell | 1 TdT+ cell | 1 GFP+ cell | 1 TdT+ cell | 1 GFP+ cell | 1 TdT+ cell | 1 GFP+ cell |
| B | 1 TdT+ cell | 1 GFP+ cell | 1 TdT+ cell | 1 GFP+ cell | 1 TdT+ cell | 1 GFP+ cell | 1 TdT+ cell | 1 GFP+ cell | 1 TdT+ cell | 1 GFP+ cell | 1 TdT+ cell | 1 GFP+ cell |
| C | 1 TdT+ cell | 1 GFP+ cell | 1 TdT+ cell | 1 GFP+ cell | 1 TdT+ cell | 1 GFP+ cell | 1 TdT+ cell | 1 GFP+ cell | 1 TdT+ cell | 1 GFP+ cell | 1 TdT+ cell | 1 GFP+ cell |
| D | 1 TdT+ cell | 1 GFP+ cell | 1 TdT+ cell | 1 GFP+ cell | 1 TdT+ cell | 1 GFP+ cell | 1 TdT+ cell | 1 GFP+ cell | 1 TdT+ cell | 1 GFP+ cell | 1 TdT+ cell | 1 GFP+ cell |
| E | 1 TdT+ cell | 1 GFP+ cell | 1 TdT+ cell | 1 GFP+ cell | 1 TdT+ cell | 1 GFP+ cell | 1 TdT+ cell | 1 GFP+ cell | 1 TdT+ cell | 1 GFP+ cell | 1 TdT+ cell | 1 GFP+ cell |
| F | 1 TdT+ cell | 1 GFP+ cell | 1 TdT+ cell | 1 GFP+ cell | 1 TdT+ cell | 1 GFP+ cell | 1 TdT+ cell | 1 GFP+ cell | 1 TdT+ cell | 1 GFP+ cell | 1 TdT+ cell | 1 GFP+ cell |
| G | 1 TdT+ cell | 1 GFP+ cell | 1 TdT+ cell | 1 GFP+ cell | 1 TdT+ cell | 1 GFP+ cell | 1 TdT+ cell | 1 GFP+ cell | 1 TdT+ cell | 1 GFP+ cell | 1 TdT+ cell | 1 GFP+ cell |
| H | 1 TdT+ cell | 1 GFP+ cell | 1 TdT+ cell | 1 GFP+ cell | 1 TdT+ cell | 1 GFP+ cell | 1 TdT+ cell | 1 GFP+ cell | 1 TdT+ cell | empty | empty | 50 cells |
Preparation of single-cell suspension for sorting astrocytes with MADM-induced UPD
This section of the protocol explains the dissection and preparation of single-cell suspension for sorting MADM+ astrocytes with MADM-induced UPD.
-
1.Prepare all solutions required for the tissue dissociation (details are listed above)
-
a.Papain-DNase solution
-
b.Solution 2
-
c.DMEM/F12
-
d.Media
-
e.Substrate Reagent of FACS Blue LacZ beta galactosidase detection kit
-
a.
Volumes of reagents for one replicate are adjusted depending on the age of the experimental animal
| Age | Papain-DNase solution | Solution 2 (step 5) | Solution 2 (step 7) | DMEM/F12 |
|---|---|---|---|---|
| P0/P4/P7 | 400 μL | 400 μL | 200 μL | 800 μL |
| P10/P14 | 500 μL | 500 μL | 250 μL | 1,000 μL |
-
2.Sacrifice experimental animal. Individual animals were used as biological replicates for downstream bioinformatics analyses upon RNA-seq.
-
a.Decapitate the animal.
-
b.Remove the skin overlaying the skull. Then perform a long cut of the skull along the midline, a short cut at the naso-frontal suture (in front of the olfactory bulbs) and two short cuts along the parietal-interparietal sutures. By flipping the bone parts to the side, the brain is easily accessible. Place curved forceps underneath the entire brain, and thereby lift up the entire brain to remove it from the skull (Figure 4).
-
c.Remove the hippocampus to avoid contamination from other cell types.
-
d.Dissect the cortex. In this protocol, we specifically focus on the isolation of cortical cell types. Thus, careful and precise dissection of the area of interest is most efficient to avoid contamination from cell types originating from other brain areas (Figure 4).
-
a.
-
3.
Transfer cortex into Papain-DNase solution in a 1.5 mL tube and incubate for 30 min at 37°C in a Thermoshaker.
-
4.
During the enzymatic digestion, prepare LacZ staining solution by diluting LacZ substrate 1:50 in DMEM/F12. Pre-activate the staining solution by incubating at 37°C until step 10.
-
5.
Also prepare lysis buffer and the respective collection tubes (PCR tubes).
-
6.
Add Solution 2 to the Tissue-Papain-DNase mix. Mix well by pipetting up and down and dissociate remaining pieces of tissue.
-
7.
Centrifuge for 8 min at 100 × g at 20°C–22°C.
-
8.
Remove the supernatant with a pipette and resuspend the pellet in Solution 2. Add DMEM/F12 as wash buffer.
-
9.
Centrifuge for 8 min at 200 × g at 20°C–22°C.
-
10.
Remove supernatant and resuspend pellet in 100 μL pre-activated LacZ staining solution. Incubate for 25 min at 37°C in a Thermoshaker.
-
11.
Stop the reaction by adding 100 μL media. Mix well.
-
12.
Centrifuge for 8 min at 200 × g at 20°C–22°C.
-
13.
Resuspend the cell pellet in 200 μL media. Keep samples at 20°C–22°C and in the dark until sorting is started.
Note: LacZ staining is temperature sensitive. Staining is most stable at RT.
Isolation of astrocytes with MADM-induced UPD using FACS (collection of low cell numbers)
In this section of the protocol, astrocytes are sorted according to MADM-labeling and LacZ substrate fluorescent signal, and are then collected for bulk RNA-sequencing.
-
14.Ensure that the cytometer (here: FACS Aria III) is set up for your sorting experiment.
-
a.Use the 100 μm nozzle.
-
b.Set the sample temperature to 20°C–22°C and the temperature for the collection tubes to 4°C.
-
c.Flush the system with RNase Away for 1–2 min.
-
d.Flush the system with RNase-free water for 1–2 min.
-
e.Adjust the side streams so that they hit the middle of the collection tubes.
-
a.
-
15.
Filter your entire sample through a 40 μm cell strainer. Dilute your sample with 200 μL media, which is additionally added through the cell strainer.
-
16.
First load the LacZ negative, MADM positive sample and record 100,000 cells. Use the negative control to adjust the gates for the live cell population (see Troubleshooting Problem 1) as well as for the MADM+ cells (see Troubleshooting Problem 2), and also adjust the LacZ gate (Figures 5A–5E).
-
17.
Prepare 0.2 mL PCR tubes containing 4 μL lysis buffer as collection tubes for your sorted cells.
-
18.
Place the collection tubes into 1.5 mL tubes, which are then placed in the appropriate FACS Aria tube holder.
-
19.
Load your LacZ positive sample and start sorting. Astrocytes are either GFP+ LacZ+ or TdT+ LacZ+ double positive. LacZ− but GFP+ and TdT+ cells are enriched for neurons of the Emx1+ lineage, but also contain oligodendrocytes and olfactory bulb neuroblasts. Either set a maximum number of labeled cells that shall be collected, or sort the entire volume of sample (max. 400 cells).
-
20.
Once sorting is completed, transfer the entire volume of the collected sample into an empty well in a 96-well plate, which should remain on dry ice throughout the whole procedure. Be fast with pipetting since samples freeze in the pipette tip in case this step is executed too slowly.
-
21.
Collect all replicates for your RNA-sequencing experiment in this 96-well plate. Either fill the entire plate on a single day, or add few samples on a day-to-day basis. When adding few samples over a time course of several days or weeks, store the plate at −80°C until further processing or additional samples can be added.
Note: The setup with collecting samples on a 96-well plate proved very useful for preparing samples for RNA-sequencing. However, this can be adapted to individual use and respective purposes.
Expected outcomes
In our experience, the fraction of living cells in suspension is higher in younger animals. However, sorting of MADM+ cells using Emx1-Cre for single-cell RNA-sequencing was always successful, even at adult stages. Collection of 400 TdT+ cells and 400 GFP+ cells was easily feasible from individual animals dissected at P0, P7, and up to P21. The time required for sorting increased with the age of the experimental animal due to an increase of myelin-containing debris in the single-cell suspension.
Sorting MADM+ astrocytes from a single animal usually yields relatively low cell numbers. We typically aim for 200 cells at P0, 50–150 cells at P7 and 10–50 cells at P14 per MADM fluorescent color from a single cortex. In order to reach these cell numbers, usually the entire volume of single-cell suspension was sorted.
RNA-sequencing and statistical analysis
Preparation of cDNA libraries from sorted cells was performed as described in (Picelli et al., 2014).
Standard bioinformatics quality control and statistical analysis are used to analyze the RNA-seq data. We used R statistical environment for all RNA-seq analyses (https://www.r-project.org/). In our hands DESeq2 and Seurat packages proved to be especially suitable to analyze bulk and single-cell data, respectively. For detailed description on data analysis with these packages, we refer the interested reader to the publications, which link to documentations and online tutorials (Butler et al., 2018, Love et al., 2014). Since cells with MADM-induced UPD contain predictable imbalances of imprinted gene dosage, transcriptional analysis provided a powerful approach to study genomic imprinting (Laukoter et al., 2020b).
Besides the probing of genomic imprinting using UPD, MADM technology can be utilized to perform functional analysis of candidate genes (Beattie et al., 2017, Beattie et al., 2020, Hippenmeyer et al., 2010, Laukoter et al., 2020a). In MADM-based functional genetic analysis, fluorescent labeling of individual cells corresponds to mutant and wild-type genotypes, respectively (e.g., green is mutant and red is wild type). Application of the above protocol in MADM-based mutant analysis allows investigation of mutant-specific transcriptional responses (Laukoter et al., 2020a). In such cases, we recommend to always verify that the mutation is linked to one particular UPD in order to gain reproducible results between different replicates, experimental batches, or genotypes. Importantly, reverse breeding schemes (resulting in color swap of mutant and wild-type MADM cells) permit the separation of UPD phenotypes from genetic loss of gene function phenotypes.
Limitations
Both protocols for preparing single-cell suspensions to isolate neurons and glial cells, or specifically astrocytes, have been optimized for early postnatal stages. Application of the protocol for late adult stages might require additional optimization due to increased cell death and increased myelin-containing debris.
MADM is a technology that can be used to sparsely induce UPD and fluorescently label cells (Contreras et al., 2020). Thus, collection of more than 20–30K cells is not easily possible. For instance, if using MADM-7 to isolate cells from the Emx1 lineage at P0 multiple cortices were pooled to increase cell numbers in the samples. Consequently, MADM is most suitable for downstream applications that require limited biological material, up to a few thousand cells and/or for single-cell applications.
Troubleshooting
Problem 1
High amount of dead cells
Potential solution 1
-
1.
Make sure to not deprive cells of air during single-cell suspension preparation (use ventilated cape tubes or leave 1.5 mL tubes partly open).
-
2.
Limit the time between sample preparation and sorting. When many samples need to be processed at the same time, it is preferable to work in a team so that certain protocol steps can be done in parallel. Alternatively, split sample preparation into batches of maximum 3–5 samples, and sort these before moving on to the next batch.
Problem 2
Low number of MADM-labeled cells
Potential solution 2
-
1.
If individual animals were used for the single-cell suspension, pool multiple cortical samples to prepare the single-cell suspension for sorting.
-
2.
Cover your sample tubes with aluminum foil to protect the samples from light.
-
3.
MADM-labeled cells might have died during the preparation. Please refer to section above “Problem: High amount of cell death.”
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact Simon Hippenmeyer (simon.hippenmeyer@ist.ac.at).
Materials availability
This study did not generate new unique reagents.
Data and code availability
No custom code is necessary to perform this protocol. All datasets mentioned in this protocol have been described elsewhere (Laukoter et al., 2020b).
Acknowledgments
This research was supported by the Scientific Service Units (SSU) at IST Austria through resources provided by the Bioimaging (BIF) and Preclinical Facilities (PCF). N.A received support from the FWF Firnberg-Programm (T 1031). This work was also supported by IST Austria institutional funds; FWF SFB F78 to S.H.; NÖ Forschung und Bildung n[f+b] life science call grant (C13-002) to S.H.; the People Programme (Marie Curie Actions) of the European Union’s Seventh Framework Programme (FP7/2007-2013) under REA grant agreement no. 618444 to S.H.; and the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (grant agreement no. 725780 LinPro) to S.H.
Author contributions
All authors conceived the project. S.L. established and optimized the protocol for single-cell suspension of MADM+ cells with input from F.M.P. and S.H. N.A. optimized isolation and FACS of astrocytes. F.M.P. established bioinformatics analysis. All authors edited and proofread the manuscript.
Declaration of interests
The authors declare no competing interests.
References
- Beattie R., Postiglione M.P., Burnett L.E., Laukoter S., Streicher C., Pauler F.M., Xiao G., Klezovitch O., Vasioukhin V., Ghashghaei T.H., Hippenmeyer S. Mosaic analysis with double markers reveals distinct sequential functions of Lgl1 in neural stem cells. Neuron. 2017;94:517–533.e3. doi: 10.1016/j.neuron.2017.04.012. [DOI] [PubMed] [Google Scholar]
- Beattie R., Streicher C., Amberg N., Cheung G., Contreras X., Hansen A.H., Hippenmeyer S. Lineage tracing and clonal analysis in developing cerebral cortex using mosaic analysis with double markers (MADM) J. Vis. Exp. 2020 doi: 10.3791/61147. [DOI] [PubMed] [Google Scholar]
- Brenner M., Kisseberth W.C., Su Y., Besnard F., Messing A. GFAP promoter directs astrocyte-specific expression in transgenic mice. J. Neurosci. 1994;14:1030–1037. doi: 10.1523/JNEUROSCI.14-03-01030.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler A., Hoffman P., Smibert P., Papalexi E., Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018;36:411–420. doi: 10.1038/nbt.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contreras X., Davaatseren A., Amberg N., Hansen A.H., Sonntag J., Andersen L., Bernthaler T., Heger A., Johnson R., Schwarz L.A., Luo L., Rülicke T., Hippenmeyer S. A genome-wide library of MADM mice for single-cell genetic mosaic analysis. bioRxiv. 2020 doi: 10.1101/2020.06.05.136192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorski J.A., Talley T., Qiu M., Puelles L., Rubenstein J.L., Jones K.R. Cortical excitatory neurons and glia, but not GABAergic neurons, are produced in the Emx1-expressing lineage. J. Neurosci. 2002;22:6309–6314. doi: 10.1523/JNEUROSCI.22-15-06309.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hippenmeyer S., Johnson R.L., Luo L. Mosaic analysis with double markers reveals cell-type-specific paternal growth dominance. Cell Rep. 2013;3:960–967. doi: 10.1016/j.celrep.2013.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hippenmeyer S., Youn Y.H., Moon H.M., Miyamichi K., Zong H., Wynshaw-Boris A., Luo L. Genetic mosaic dissection of Lis1 and Ndel1 in neuronal migration. Neuron. 2010;68:695–709. doi: 10.1016/j.neuron.2010.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laukoter S., Beattie R., Pauler F.M., Amberg N., Nakayama K.I., Hippenmeyer S. Imprinted Cdkn1c genomic locus cell-autonomously promotes cell survival in cerebral cortex development. Nat. Commun. 2020;11:195. doi: 10.1038/s41467-019-14077-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laukoter S., Pauler F.M., Beattie R., Amberg N., Hansen A.H., Streicher C., Penz T., Bock C., Hippenmeyer S. Cell-type specificity of genomic imprinting in cerebral cortex. Neuron. 2020;107:1160–1179.e9. doi: 10.1016/j.neuron.2020.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picelli S., Faridani O.R., Bjorklund A.K., Winberg G., Sagasser S., Sandberg R. Full-length RNA-seq from single cells using Smart-seq2. Nat. Protoc. 2014;9:171–181. doi: 10.1038/nprot.2014.006. [DOI] [PubMed] [Google Scholar]
- Tucci V., Isles A.R., Kelsey G., Ferguson-Smith A.C., Erice Imprinting G. Genomic imprinting and physiological processes in mammals. Cell. 2019;176:952–965. doi: 10.1016/j.cell.2019.01.043. [DOI] [PubMed] [Google Scholar]
- Zong H., Espinosa J.S., Su H.H., Muzumdar M.D., Luo L. Mosaic analysis with double markers in mice. Cell. 2005;121:479–492. doi: 10.1016/j.cell.2005.02.012. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No custom code is necessary to perform this protocol. All datasets mentioned in this protocol have been described elsewhere (Laukoter et al., 2020b).