

Abstract
The ability to alter genomes specifically by CRISPR-Cas gene editing has revolutionized biological research, biotechnology, and medicine. Broad therapeutic application of this technology, however, will require thorough preclinical assessment of off-target editing by homology-based prediction coupled with reliable methods for detecting off-target editing. Several off-target site nomination assays exist, but careful comparison is needed to ascertain their relative strengths and weaknesses. In this study, HEK293T cells were treated with Streptococcus pyogenes Cas9 and eight guide RNAs with varying levels of predicted promiscuity in order to compare the performance of three homology-independent off-target nomination methods: the cell-based assay, GUIDE-seq, and the biochemical assays CIRCLE-seq and SITE-seq. The three methods were benchmarked by sequencing 75,000 homology-nominated sites using hybrid capture followed by high-throughput sequencing, providing the most comprehensive assessment of such methods to date. The three methods performed similarly in nominating sequence-confirmed off-target sites, but with large differences in the total number of sites nominated. When combined with homology-dependent nomination methods and confirmation by sequencing, all three off-target nomination methods provide a comprehensive assessment of off-target activity. GUIDE-seq's low false-positive rate and the high correlation of its signal with observed editing highlight its suitability for nominating off-target sites for ex vivo CRISPR-Cas therapies.
Introduction
The development of engineered nucleases that target specific sites in the genome has driven advances in basic and applied research at a rapid pace.1,2 The more recent advent of facile CRISPR-Cas methods for programmable RNA-guided genome editing has further quickened the pace of both discovery and clinical application.3,4 The Cas9 nuclease, in combination with a chimeric guide RNA (gRNA), cleaves genomic sites in a sequence-specific manner. The gRNA comprises a spacer (typically 17–22 bases long) that hybridizes to the target genomic DNA strand and a trans-activating crRNA (tracrRNA) that interacts with Cas9.1 Cleavage of the target site by Cas9 also requires a short DNA sequence adjacent to the complementary sequence, known as the protospacer adjacent motif (PAM). Genomic sites with a PAM and perfect complementarity to the gRNA spacer sequence are referred to as on-target sites. Cas9 may also cleave DNA at off-target sites, which are genomic sites with mismatches (MM) and/or gaps with respect to the gRNA spacer sequence.5 Once DNA is cleaved, cellular repair machinery usually repairs the double-strand breaks (DSBs), resulting in either a perfect repair that is available for recutting or insertions and deletions (indels) at the cut site, resulting in disruption of the native genomic sequence at on- and off-target sites alike.
For clinical application of genome editing, CRISPR-Cas systems must achieve therapeutically relevant levels of editing at the on-target site with minimal editing at any off-target sites. Typically, two orthogonal approaches are used to identify potential off-target sites. In the first approach, termed “homology-dependent,” computational tools are used to nominate potential off-target sites based on the presence of a PAM sequence and the degree of homology between the adjacent target DNA sequence and the gRNA spacer sequence. Homology-dependent approaches are based on the principle that sequences that closely match the gRNA have a greater probability of being cleaved than sites with a greater number of MM and gaps do.5–8 In the second approach, termed “homology-independent,” genome-wide assays combined with next-generation sequencing (NGS) are used to nominate sites empirically that could potentially be cleaved by the Cas9-gRNA ribonucleoprotein (RNP).9–13 Potential off-target sites nominated by homology-dependent and homology-independent approaches are then deep sequenced after CRISPR-Cas editing to detect editing activity.
Cell-based and in vitro biochemical genome-wide assays have been developed for homology-independent nomination of off-target sites. Cell-based assays, such as IDLV,14 GUIDE-seq,10 and DISCOVER-seq,13 exploit endogenous cellular repair machinery to mark the location of DSBs that occur during editing. Genomic DNA fragments bearing these marks are enriched during Illumina library preparation to a degree that renders them detectable by NGS. Biochemical assays, such as CIRCLE-seq11 and SITE-seq,12 which use purified genomic DNA as the substrate, also enrich sites cleaved by the RNP. The major difference between the two assay types is that cell-based assays nominate sites in accessible regions of the genome in a cell type-specific manner, whereas biochemical assays potentially nominate cleavable sites irrespective of chromatin accessibility. GUIDE-seq utilizes incorporation of a short double-stranded oligodeoxynucleotide (dsODN) at DSBs in cells. Anchoring primer binding sites in the dsODN sequence enables identification of dsODN insertion sites and nomination of off-target sites. CIRCLE-seq uses purified, circularized genomic DNA as a substrate for a CRISPR-Cas cleavage reaction. Circles linearized through cleavage are then sequenced to identify break points and nominate off-target sites. In SITE-seq, DSBs are enriched using biotinylated adaptors that are ligated to adenylated, cleaved, purified genomic DNA. Sequence read pileups that terminate at cut sites are detected computationally to nominate off-target sites.
While both cell-based and biochemical assays have been used to nominate off-target sites,15–18 the relative abilities of these methods to identify true sequence-confirmed off-targets have not been systematically investigated. It is also unclear how quantitative the readouts from these nomination methods are and whether these assays predict true editing in a cellular context. An understanding of the relative performance of these methods is critical for deciding which genome-wide assay is best suited to nominate off-target sites when developing CRISPR-Cas-based therapies.
Here, we evaluated and compared the performance of the cell-based assay GUIDE-seq and the biochemical assays CIRCLE-seq and SITE-seq in nominating true off-target sites for eight gRNAs. We benchmarked the three methods against homology-nominated sites that were sequence confirmed using hybrid capture followed by sequencing. Based on our findings, we suggest how these genome-wide assays may be best used to nominate potential off-target sites for CRISPR-Cas editing therapies.
Methods
Cell culture
Cellular and molecular experiments were carried out using HEK293T/Cas9, a HEK293T cell line engineered with a cassette comprising an Streptococcus pyogenes Cas9 (SpCas9) cDNA under control of a strong constitutive CBh promoter19 and a puromycin N-acetyltransferase gene stably integrated into the AAVS1 locus (GeneCopoeia, Rockville, MD). Cells were cultured in Dulbecco's modified Eagle's medium (Thermo Fisher Scientific, Carlsbad, CA) supplemented with 10% fetal bovine serum (Sigma–Aldrich, St. Louis, MO), 1 × Pen-Strep (50 IU/mL each of penicillin G and streptomycin sulfate; Thermo Fisher Scientific) and 1 mg/mL puromycin (InvivoGen, San Diego, CA) in a humidified 5% CO2 atmosphere at 37°C. Cells were passaged at a ratio of 1:10 upon treatment with standard trypsin-EDTA solution.
GUIDE-seq
GUIDE-seq was performed in HEK293T/Cas9 cells according to a published protocol.10 To prepare the dsODN, a 50 μL solution consisting of 100 mM Tris-HCl (pH 7.5), 1 mM NaCl, 10 mM EDTA, and 40 μM of each modified oligonucleotide (Supplementary Table S1) was prepared. Using a thermocycler block, the dsODN was generated by first heating the solution at 95°C for 4 min and then cooling the solution at a rate of 0.1°C/s to a final temperature of 23°C.
A Lonza 4D nucleofector was used with the manufacturer's program (HEK293) to electroporate 2 μg gRNA (Supplementary Table S2) and 500 nM dsODN into 2 × 105 cells that were re-suspended in 20 μL SF Nucleofector solution (Lonza, Basel, Switzerland). For control reactions, cells were only electroporated with dsODN. Following electroporation, cells were re-suspended in culture medium and allowed to recover for 48 h. Genomic DNA was then extracted from all cells using a DNeasy blood and tissue kit (Qiagen, Germantown, MD). Integration of the dsODN was verified through Sanger sequencing and indel analysis using the Tracking of Indels by Decomposition method.20
A Covaris LE220 ultrasonicator (Covaris, Woburn, MA) was used to shear 400 ng genomic DNA to an average size of 200 bp. The sheared DNA was then end repaired, A-tailed, and adapter ligated according to the published protocol.10 The discovery protocol was followed, in which both the sense and antisense library strands were amplified through two rounds of nested anchored polymerase chain reaction to create Illumina libraries. The GUIDE-seq libraries were sequenced on a NextSeq 500 sequencer (Illumina, San Diego, CA) on a high-output flow cell with the following cycle parameters: 66 × 16 × 16 × 70. Three replicates of GUIDE-seq were carried out for each gRNA and for the control.
CIRCLE-seq
Genomic DNA was purified from 109 HEK293T/Cas9 cells using the DNeasy blood and tissue kit (Qiagen) and then sheared to an average length of 300 bp using the Covaris LE220 ultrasonicator. Sheared DNA was then circularized through a series of reactions, as previously detailed.11 Circularized DNA preparations were treated with plasmid-Safe ATP-dependent DNase (Epicentre) to eliminate linear DNA molecules remaining after the circularization reaction. The resulting circular DNA was then purified with paramagnetic SPRI beads (Beckman Coulter, Indianapolis, IN) and eluted off beads into 10 mM TRIS-HCl, pH 8.0.
To prepare the SpCas9 RNP used in the CIRCLE-seq assay, synthesized gRNAs (Supplementary Table S2) were first melted at 95°C for 2 min and then allowed to cool slowly at room temperature for 5 min before placing on ice. A 50 μL solution containing 2 × NEBuffer™ 3.1 (New England Biolabs, Ipswich, MA), 600 nM renatured gRNA, and 180 nM Engen® Spy Cas9 (New England Biolabs) was constructed, mixed, and then incubated at 37°C for 5 min to form SpCas9 RNP. Following this incubation, the solution containing SpCas9 RNP was placed on ice. SpCas9 cleavage reactions were prepared by adding a 50 μL solution containing 180 ng circularized genomic DNA to freshly prepared SpCas9 RNP (final concentration of SpCas9 RNP was 90 nM). SpCas9 RNP cleavage reactions were then incubated for 60 min at 37°C, as previously described.11 For control reactions, SpCas9 without gRNA was incubated with 180 ng circular DNA for 60 min at 37°C. DNA from SpCas9 RNP digestions and control reactions was then purified by using paramagnetic SPRI beads and used to prepare CIRCLE-seq libraries, as previously detailed.11 Three replicates of CIRCLE-seq were carried out for each gRNA and for the SpCas9-only control. CIRCLE-seq libraries were sequenced on the NextSeq 500 sequencer on a mid-output flow cell with the following cycle parameters: 75 × 6 × 6 × 75.
SITE-seq
SITE-seq was performed according to a published protocol.12 Briefly, synthesized gRNAs (Supplementary Table S2) were melted at 95°C for 2 min and then allowed to cool slowly at room temperature for 5 min before placing on ice. Next, SpCas9 RNP digestions of high molecular weight genomic DNA (10 mg) from HEK293T/Cas9 cells were set up in triplicate as described12 and then incubated at 37°C for 4 h. The final concentration of SpCas9 RNP in digestions was 1 μM. For control reactions, 10 mg high molecular weight genomic DNA from HEK293T/Cas9 cells was incubated with SpCas9 (without gRNA) at 37°C for 4 h in triplicate. SpCas9 RNP digestions and control reactions were terminated by adding proteinase K and RNase A and incubating reactions 37°C for 20 s and then 55°C for 20 s. The DNA in reactions was purified with paramagnetic SPRI beads (1 × v/v; Beckman Coulter) and eluted off beads into 10 mM TRIS-HCl, pH 8.0. Purified gDNA was processed through a series of reactions previously detailed12 that generate SITE-seq libraries enriched for DSBs in DNA following the digestion or control reactions. SITE-seq libraries were sequenced on the NextSeq 500 sequencer on a mid-output flow cell with the following cycle parameters: 75 × 8 × 8 × 75.
Hybrid capture
The CCTop algorithm21 adapted for gapped searches was used to complete a homology search for up to 5 MM with no gaps or up to 3 MM with 1 gap (5 MM 0 gap, 3 MM 1 gap) with the PAM sequences specified as NNN for each gRNA. A filtering step was applied to permit specific PAMs, the canonical NGG PAM as well as the following non-canonical PAMs: NAG, NGA, NAA, NCG, NGC, NTG, and NGT. For sites nominated by the homology-independent methods outside the 5 MM 0 gap, 3 MM 1 gap homology space, cut sites and PAM were determined by aligning the gRNA to the DNA target. For each nominated site, hybrid capture probes were generated by encompassing the candidate site with 100 bp 5′- and 3′-flanking regions and then tiling 120 bp probes across the region in 1 bp increments. A separate SureSelect custom DNA probe set was ordered for each gRNA (Agilent Technologies, Santa Clara, CA).
Treated samples were generated by electroporating 2 × 105 HEK293T/Cas9 cells re-suspended in 20 μL SF Nucleofector solution (Lonza) with 2 μg synthesized gRNAs (Supplementary Table S2) using a Lonza 4D nucleofector with the manufacturer's program HEK293. After electroporation, cells were re-suspended in culture media and allowed to recover for 48 h. Untreated controls were not subjected to electroporation. Genomic DNA was then extracted using the DNeasy blood and tissue kit (Qiagen).
A Covaris LE220 ultrasonicator was used to shear 200 ng genomic DNA to 150–200 bp. Fragmented DNA was end repaired, A-tailed, adapter ligated, and amplified with Agilent's SureSelect XT HS Target Enrichment System for Illumina Paired-End Multiplexed Sequencing Library kit according to the manufacturer's protocol. Prepared libraries (1 μg) were hybridized to probes (Agilent) and captured on streptavidin beads after a series of washes. Captured libraries were amplified and purified. Libraries were sequenced on 28 lanes of Illumina's HiSeq sequencing platform with dual index, 2 × 150 bp configuration (Genewiz, South Plainfield, NJ). For all eight gRNA, replicates 1 and 2 were generated, processed, and sequenced together. Replicate 3 was generated, processed, and sequenced independently of replicates 1 and 2.
Hybrid capture libraries of sites that fell outside the 5 MM 0 gap, 3 MM 1 gap homology space were sequenced on a NextSeq 500 sequencer on a high-throughput flow cell with the following cycle parameters: 150 × 8 × 8 × 150. Three replicates of treated and untreated control samples were performed for all eight gRNAs.
Multiplexed amplicon sequencing
Multiplexed amplicon sequencing (rhAmpSeq) primers were ordered for 209 sites from Integrated DNA Technologies (IDT, Coralville, IA). Primers were successfully designed for 207 sites (206 as a pool and one single assay). Following the manufacturer's protocol, rhAmpSeq libraries were generated from an input of 25 ng DNA from the same samples analyzed with hybrid capture deep sequencing. Resulting rhAmpSeq libraries were then purified, quantified, pooled, and sequenced on a NextSeq 500 sequencer on a high-output flow cell with the following cycle parameters: 149 × 10 × 10 × 149.
Computational Methods
All analysis was performed on an Amazon EC2 instance with Ubuntu 16.04.2 LTS. Version numbers of important packages are listed in Supplementary File S6. Human genome build hg38 was used as the reference genome for all analyses. Reads were mapped using BWA22 v0.7.15, which performs alt-aware alignment to align reads preferentially to standard chromosomes. Reads mapping to alternative chromosomes were ignored for this study.
GUIDE-seq analysis
Analysis of GUIDE-seq reads was performed with the published pipeline (https://github.com/aryeelab/guideseq)10 with a few modifications, as described below. Sequencing runs downloaded from the Illumina BaseSpace platform were converted to FASTQ format using bcl2fastq conversation software (Illumina). For each sample, the sense and antisense orientation FASTQ files were concatenated together. The concatenated files were processed with fastQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/) to check overall data quality. The molecular barcodes at the beginning of R1 and R2 read sequences were concatenated and subject to UMI tag deduplication as described in https://github.com/aryeelab/umi. A slight modification was made in the code for the tag deduplication step to account for the location of the UMI tag in NextSeq sequencing reads. The UMI reads were consolidated as specified in the published pipeline and then aligned to the human genome build hg38 with BWA. Off-targets sites were identified with the “identify” step in the published pipeline. Identified sites were realigned to gRNA sequence using the Needleman–Wunsch algorithm from EMBOSS.23 Sites with up to seven MM and five gaps were defined as nominated. After processing, an average UMI-corrected sequencing coverage of 3,795 (median: 223) was obtained per nominated site for GUIDE-seq.
CIRCLE-seq analysis
Our off-target site nomination workflow was modified to process CIRCLE-seq reads using functions from the published CIRCLE-seq pipeline (https://github.com/tsailabSJ/circleseq)11 with a few modifications, as described below. Sequencing runs downloaded from the Illumina BaseSpace platform were converted to FASTQ format using bcl2fastq. The FASTQ files were processed with fastQC to check overall data quality. Reads were then merged by concatenating the reverse complement of the R1 read with the R2 read as specified in the “mergeReads.py” file of the published pipeline. Merged reads were aligned to the human genome build hg38 with BWA. Off-target sites were identified using the “findCleavageSites.py” file of the published pipeline with modifications in the “tabulate_merged_start_positions” function to analyze 2 × 75 bp sequencing reads with no read thresholding applied. Identified sites with up to 10 MM and 5 gaps were realigned to the gRNA sequence using the Needleman–Wunsch algorithm from EMBOSS.23 Identified sites with identical start and end coordinates but different alignments were then combined into one nominated site by summing up read counts. Finally, to assess noise in the assay, an empirical distribution was constructed from average read counts across three replicates of control samples, as recommended in the CIRCLE-seq pipeline, using the empirical cumulative distribution function (ecdf, R stats v3.6.3). The read count threshold was defined as the minimum read count for which the empirically determined p-value in control samples was <0.01 and was calculated to be six reads for this study. Sites with up to 7 MM and 5 gaps and average read counts across three replicates of treated samples above the read count threshold were defined as nominated. After processing, an average sequencing coverage of 89.9 reads (median: 12.3) was obtained per nominated site for CIRCLE-seq.
SITE-seq analysis
Our off-target site nomination workflow was modified to process SITE-seq reads using the code provided in the publication.12 Sequencing runs were downloaded from the Illumina BaseSpace platform and converted to FASTQ format using bcl2fastq. The FASTQ files were processed with fastQC to check overall data quality. R1 reads were aligned to human genome build hg38 with BWA. Initial read pileups in aligned R1 reads were calculated with the “find_initial_read_pileups” function available in the SITE-seq publication using the default depth threshold of five reads. Off-targets sites were identified using the default parameters with the “call_site_seq_features” function from the SITE-seq publication with a modification to provide a longer flanking sequence length for nominated sites from 20 to 35 bp. Identified sites with up to 10 MM and 5 gaps were realigned to the relevant gRNA sequence using the Needleman–Wunsch algorithm from EMBOSS.23 Identified sites with identical start and end coordinates but different alignments were combined into one nominated site by summing up read counts. Sites with up to 7 MM and 5 gaps were defined as nominated. After processing, an average sequencing coverage of 36.6 reads (median: 13) was obtained per nominated site for SITE-seq.
Hybrid capture analysis
For hybrid capture sequencing, Illumina base calls were converted to FASTQ format and de-multiplexed using bcl2fastq. Reads were aligned to the human genome build hg38 using the BWA MEM24 algorithm with default parameters. Aligned reads were then sorted with “samtools sort”25 with maximum memory set to 1 Gb and indexed using “samtools index.” The aligned reads were de-duplicated using “samtools rmdup” and then re-indexed with “samtools index.”
For each nominated site, a SpCas9 cut site was predicted based on the alignment of the off-target site to the gRNA and a cleavage offset of 3 bp upstream of the PAM position. Reads mapping in a 1,000 bp region on either side of each cut site were analyzed individually using the “AlignedSegment class” obtained from the “AlignmentFile.fetch()” method in the python package pysam (https://github.com/pysam-developers/pysam). Soft-clipped and supplementary alignments were not considered for indel quantification. If the read contained the cleavage position plus 20 bp on either side of the cleavage position, then it was considered for indel quantification. All reads containing indels overlapping a region spanning ±3 bp on either side of the predicted cleavage position were counted, and were output along with the total number of reads passing the aforementioned filtering criteria. For every cut site, the indel frequency was calculated by dividing the number of reads containing indels by the total number of reads.
Sites sequenced at an average read depth of more than 500 across the three treated and three untreated samples, and with <30% indel frequency in the untreated samples, were subject to statistical testing. These minimums exclude sites that would have a low read count that is typical of difficult to characterize genomic sites or those with a small difference in indels (0.2% of 500 = 1 read, 0.2% of 3,000 = 6 reads). p-Values for significant difference in indel frequency between treated and untreated samples were obtained using the Cochran–Mantel–Haenszel test stratified by replicates26 (mantelhaen.test from R stats v3.6.3). Multiple hypothesis correction was applied27 to the p-values obtained for sites nominated for each gRNA using the p.adjust (method = “BH”) from R stats v3.6.3. Sites were annotated as sequence confirmed if they had an adjusted CMH p-value of <0.05, >0.2% indel frequency difference between at least one treated and the untreated samples, and a positive average indel frequency difference between treated and untreated samples (Δ indel frequency). Median sequencing coverage of 2,313 de-duplicated reads was obtained per site for hybrid capture libraires for homology sites. Average sequencing complexity for hybrid capture libraries for homology sites was 70 million reads with a wide range (45–150 million) based on probe set sizes.
rhAmpSeq analysis
Illumina base calls were converted to FASTQ format and de-multiplexed using bcl2fastq. The FASTQ files were processed with fastQC to check for overall data quality. Adapters were trimmed for R1 and R2 reads with Trimmomatic28 without any quality trimming. Trimmed reads were merged with the PEAR algorithm with default parameters. Reads that had a minimum base phred quality score of <10 were removed. Merged, trimmed, and quality filtered reads were aligned to the human genome hg38 build with run-bwamem tool from bwa.kit, followed by sorting and indexing of aligned files. Alignments were evaluated using the “AlignmentFile.fetch()” method in the python package pysam. As described for hybrid capture analysis above, if the read contained the cleavage position and 20 bp on either side of the cleavage position, then it was considered for indel quantification. All reads containing indels overlapping a region spanning ±3 bp on either side of the predicted cleavage position were counted, and were output along with the total number of reads passing the aforementioned filtering criteria. The Δ indel frequency was calculated for sites sequenced at an average read depth of more than 500 across the three treated and three untreated samples, and with <30% indel frequency in the untreated samples. Median sequence coverage of 35,378 reads was obtained per site for rhAmpSeq.
Results
Study design
To evaluate the performance of GUIDE-seq, CIRCLE-seq, and SITE-seq comparatively, we used eight previously published gRNAs (Table 1). The gRNAs cover a wide range of specificities, as demonstrated by the number of genomic sites bioinformatically nominated21 in a homology space covering sites with up to 5 MM with no gaps, and up to 3 MM with 1 gap (Supplementary Fig. S1). Some of these gRNAs have multiple sequence-confirmed off-target sites in human cell lines.9,10 We intentionally selected non-therapeutic gRNAs with multiple known off-target sites to maximize the number of data points available for evaluating the three off-target nomination methods.
Table 1.
List of gRNAs Used in this Study
| gRNAReference | Position in hg38 genome | Sequence | Sites predicted in 5 MM 0 gap, 3 MM 1 gap space |
|---|---|---|---|
| HEK111 | Intergenic | GGGAAAGACCCAGCATCCGT | 4,030 |
| HEK311 | lncRNA AC114971.1 | GGCCCAGACTGAGCACGTGA | 5,336 |
| RNF29, 10 | RNF gene, Exon 2 | GTCATCTTAGTCATTACCTG | 6,264 |
| FANCF9, 10 | FANCF gene, Exon 1 | GGAATCCCTTCTGCAGCACC | 6,678 |
| VEGFA19, 10 | Intergenic | GGGTGGGGGGAGTTTGCTCC | 7,957 |
| IL2RG28 | IL2RG, Exon1 | TGGTAATGATGGCTTCAACA | 9,412 |
| HEK211 | lncRNA AC114971.1 | GAACACAAAGCATAGACTGC | 13,563 |
| CCR56 | CCR2, Exon2/CCR5, Exon 3 | GTGTTCATCTTTGGTTTTGT | 22,312 |
gRNA, guide RNA; MM, mismatch
We performed GUIDE-seq in triplicate on HEK293T cells that constitutively express SpCas9. We performed CIRCLE-seq and SITE-seq assays in triplicate on genomic DNA purified from the same cell line used for GUIDE-seq assays. For each gRNA, we performed confirmatory sequencing in edited HEK293T/Cas9 cells. We used hybrid capture probes to enrich and sequence all genomic sites that were computationally determined to have a homology distance up to 5 MM with 0 gap or up to 3 MM with 1 gap (5 MM 0 gap, 3 MM 1 gap) from the gRNA (Table 1). We also sequenced a subset of sites nominated by homology-independent methods that fell outside of the 5 MM 0 gap, 3 MM 1 gap homology space. All experiments included negative controls from which gRNAs had been omitted to control for fragile sites and random DSBs, as well as artifacts from library preparation or sequencing errors.
Below, we compare the three off-target nomination methods to each other and to hybrid capture followed by sequencing. First, we assess these methods for reproducibility and enrichment of signal over noise. Second, we assess how well these methods capture sequence-confirmed edited sites and how many unedited sites these methods nominate.
Number of sites nominated by genome-wide assays
For this study, we filtered GUIDE-seq, CIRCLE-seq, and SITE-seq data to remove nominated sites that did not align to the gRNA sequence. While the standard practice is to filter sites with more than six edit distance (defined here as the total number of MM and gaps in the DNA target),10,11 we included all sites with up to 7 MM and 5 gaps to be comprehensive (Supplementary File S1). The cell-based GUIDE-seq assay nominated an average of 40 sites per gRNA, the lowest average among the three assays (Supplementary Table S3). This result is expected for GUIDE-seq because unlike purified chromatin-free DNA used in biochemical assays, the chromatin state of a site in a cell can affect its accessibility to CRISPR-Cas-mediated cleavage and dsODN insertion. CIRCLE-seq and SITE-seq, which are biochemical assays performed on purified DNA, nominated an average of 5,244 and 2,093 sites per gRNA, respectively (Supplementary Tables S4 and S5).
Comparing reproducibility between replicates of genome-wide assays
The three assays were assessed for reproducibility in nominating sites across replicates, as well for reproducibility in the number of reads obtained per nominated site. GUIDE-seq was the least reproducible with respect to the identity of the sites nominated across three replicates. Across the eight gRNAs, 30% of GUIDE-seq nominated sites were reproducibly nominated by all three replicates. For CIRCLE-seq and SITE-seq, 72% and 60.4% of all nominated sites were reproducibly nominated by all three replicates, respectively (Fig. 1A). The percentage of total sites nominated by all three replicates was consistent across different gRNAs for GUIDE-seq and SITE-seq (Supplementary Tables S3 and S5). In contrast, for CIRCLE-seq, we observed gRNA-dependent reproducibility of sites across replicates (Supplementary Table S4).
FIG. 1.

(A) Overlap among three replicates for sites nominated by GUIDE-seq, CIRCLE-seq, and SITE-seq across all eight gRNAs. (B) Correlation of read counts for nominated sites between two replicates for GUIDE-seq (R2 = 0.54–0.67), CIRCLE-seq (R2 = 0.80–0.83), and SITE-seq (R2 = 0.90–0.91) read counts. The plots present data for replicates 1 and 2, and the R2 range is calculated from all three comparisons (1 vs. 2, 2 vs. 3, 1 vs. 3). (C) Distributions of read counts for nominated sites obtained by for GUIDE-seq, CIRCLE-seq, and SITE-seq grouped by the number of mismatches and gaps in the site with respect to gRNA sequence. gRNA, guide RNA.
For sites nominated by multiple replicates, all three homology-independent methods were highly reproducible in terms of the number of reads obtained (Fig. 1B and Supplementary Fig. S2). The range of Pearson R2 values for the three replicate-to-replicate correlations was 0.54–0.67 for GUIDE-seq, 0.80–0.83 for CIRCLE-seq, and 0.90–0.91 for SITE-seq. We observed that sites nominated by multiple replicates of a single assay had lower edit distances than those nominated by only one replicate (Supplementary Fig. S3).
Enrichment of on-target sites
CRISPR-Cas9 generally cleaves on-target sites with higher efficiency than off-target sites, with the reduction in cutting activity influenced by the number and positions of mismatches and gaps.6–8,29 Nomination assays can provide insights into gRNA specificity and mismatch/gap tolerance if the nomination signal quantitively reflects Cas9 activity in cells. Our analysis across eight gRNAs demonstrates that while read counts for all three assays decrease with increasing number of mismatches and gaps between the nominated off-target sites and the gRNA sequence, GUIDE-seq and CIRCLE-seq read counts for on-target sites are significantly higher than read counts for off-target sites (Fig. 1C and Supplementary Fig. S4). For GUIDE-seq and CIRCLE-seq, respectively, only 1.29% (4/311) and 0.07% (29/41,949) of off-target sites had read counts that fell within the on-target site read count distribution consistent with read signal correlating with expected SpCas9 activity in cells. In contrast, for SITE-seq 8.7% (1,454/16,735) of off-target sites had as many reads as the on-target sites, suggesting a potential lack of correlation in SITE-seq signal and SpCas9 activity in cells.
Overlap among assays
Given that the three assays nominate widely different numbers of off-target sites, a key question is the degree to which the same off-target sites are nominated by each method. CIRCLE-seq and SITE-seq both nominated ∼86% of the sites nominated by GUIDE-seq (Fig. 2A and Supplementary Tables S6 and S7). The CIRCLE-seq assay nominated 78% of the sites nominated by SITE-seq (Fig. 2A and Supplementary Table S8). Sites nominated by any two assays had lower edit distance than sites that were nominated by only one assay (Fig. 2B; p < 0.05), suggesting that sites nominated by multiple assays are more likely to be true gRNA-dependent sites, rather than the result of random DSBs.
FIG. 2.

(A) Overlap among three genome-wide assays for sites nominated across eight gRNAs. (B) Edit distances of nominated sites across eight gRNAs shared among and unique to different homology-independent nomination methods. The x-axis represents overlap with the three genome-wide assays. The y-axis shows the sum of mismatches and gaps for a site when aligned to the gRNA sequence. All three distributions were significantly different from each other (Wilcoxon p-value <2e-16).
Benchmarking with targeted deep sequencing
We further evaluated the performance of GUIDE-seq, SITE-seq, and CIRCLE-seq by measuring the level of editing at homology-nominated sites through targeted deep sequencing of gRNA-edited HEK293T/Cas9 cells. For each gRNA, we determined how many sites with NGS-confirmed editing were nominated by each assay (sequence-confirmed sites, true positives), how many unedited sites were nominated by each assay (false-positives of the nomination step), and whether there was editing at off-target sites not nominated by one or more of the genome-wide assays (false-negatives of the nomination step).
For nomination of homology-dependent sites, we searched the human genome for sites similar to the spacer sequences of the eight gRNAs using the CCTop bioinformatic program21 in the 5 MM 0 gap, 3 MM 1 gap (Table 1) homology space and designed hybrid capture probes for each of the eight gRNAs to enrich the predicted homology sites. Hybrid capture-based Illumina sequencing on gRNA-treated and control samples was performed in triplicate in HEK293T/Cas9 cells, with ∼2,300 × median sequencing coverage (Supplementary Fig. S5A). Sites with at least 500 average read depth across all samples, a statistically significant difference in indel frequency between treated and control samples, an indel frequency difference of at least 0.2% in at least one pair of treated and untreated samples, and <30% indel frequency in control samples (to filter germline indels) were considered to be sequence-confirmed sites (Supplementary File S2). Out of 75,552 predicted sites, 66,165 sites had more than 500 average read depth across the treated and control samples and <30% indel frequency in control samples. Of these sites, 51 were annotated as sequence-confirmed edited sites or true positives (Supplementary Fig. S5B and Supplementary File S3). Sites that did not pass the statistical test for indel frequency difference or which had <0.2% indel frequency difference between treated and control samples were annotated as unedited sites. Sequence-confirmation calling of edited or unedited sites was robust to choice of read coverage threshold (100, 200, 300, 400, or 500 reads) and false-discovery rates (Benjamini–Hochberg adjusted CMH p-value thresholds of 0.05 and 0.1; Supplementary Table S9).
Sequence-confirmed edited sites
We compared the list of sites nominated by GUIDE-seq, CIRCLE-seq, and SITE-seq with the list of sequence-confirmed edited sites and unedited sites from hybrid capture sequencing in cells. Of the 51 sequence-confirmed edited sites, 50 were nominated by at least one genome-wide assay (Table 2 and Fig. 3), and 45 of these sites were nominated by all three replicates of all three genome-wide off-target nomination assays, suggesting an appreciable overlap among the methods (Supplementary Table S10). The five sites not nominated by all three replicates of all three assays were all edited at low frequencies <0.5% (Supplementary Fig. S6). Out of the 45 sites, nine were on-target sites with perfect complementarity to the gRNA sequence (CCR5 gRNA targets to two genomic sites with perfect complementarity).
Table 2.
Intersection of Sequence-Confirmed or Unedited Homology-Nominated Sites with Sites Nominated by Homology-Independent Methods
| Hybrid capture on 5 MM, 0 gap and 3 MM, 1 gap sites | Nominated by at least one replicate of |
Shared by three genome-wide assays | |||
|---|---|---|---|---|---|
| GUIDE-seq | CIRCLE-seq | SITE-seq | |||
| Edited sites | 51 | 50 | 49 | 50 | 49 |
| Unedited sites | 66,114 | 172 | 6,602 | 3,585 | 158 |
| Precision | 0.225 | 0.007 | 0.014 | ||
FIG. 3.

Overlap between the number of sites nominated by the three genome-wide assays and sites sequence-confirmed as edited in HEK293T/Cas9 cells following hybrid capture sequencing in the 5 MM 0 gap, 3 MM 1 gap homology space. Sites sequenced with an average depth of more than 500 reads and with <30% indel frequency in the untreated samples are included. MM, mismatch; indel: insertions and deletions.
The one site that was sequence confirmed as edited in hybrid capture analysis but not nominated by any replicate of SITE-seq, CIRCLE-seq, or GUIDE-seq can be classified as a false-negative of the nomination step. However, upon closer scrutiny, this site is more likely to be a false-positive of the sequence-confirmation step. It has a homopolymeric stretch around the SpCas9 cut site, and sequencing through homopolymers can be error prone (Supplementary Fig. S7). It is likely that these sequencing errors produced high variability in indel frequencies observed across the hybrid capture samples (Fig. 4). Also, one site not nominated by CIRCLE-seq but nominated by GUIDE-seq and SITE-seq was just below the CIRCLE-seq mean read count threshold that we applied (seven, six, and five read counts in the three replicates).
FIG. 4.
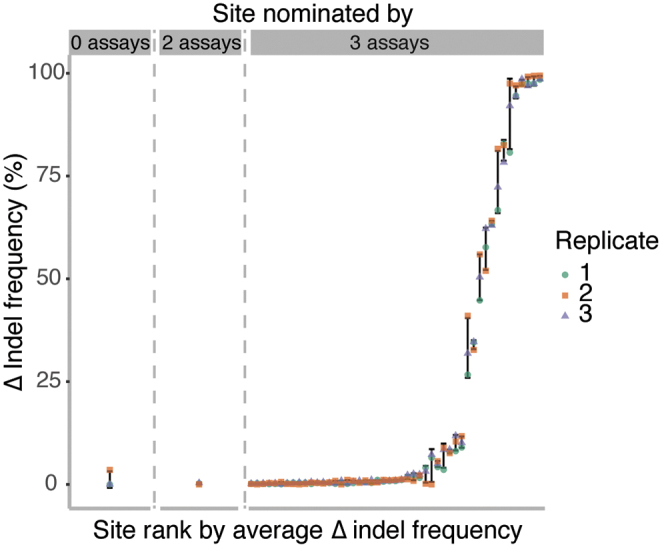
Indel frequencies (%) differences between treated and control samples from three replicates of hybrid capture followed by sequencing in HEK293T/Cas9 cells are shown for all 51 edited sites, ranked by average indel frequency difference. Each color represents one replicate. Vertical bars represent standard deviation in indel frequency difference across replicates. Panels depict overlap with genome-wide assays. The first panel shows one site not nominated by any genome-wide assay. The second panel shows one site nominated by two genome-wide assays. The third panel shows 49 sites nominated by all three genome-wide assays.
False-positives of off-target nominations assays
While all three genome-wide methods nominated almost all of the sequence-confirmed edited sites, they differed vastly in the number of false-positives nominated (sites found to be unedited by hybrid capture sequencing; Table 2 and Fig. 3). In this sense, the cellular assay GUIDE-seq has the highest precision and CIRCLE-seq has the lowest. The biochemical assays CIRCLE-seq and SITE-seq nominated many false-positive sites within the 5 MM 0 gap, 3 MM 1 gap space, presumably as a result of the entire genome being open and accessible to CRISPR-Cas9 gRNA complex. Reaction kinetics may also be different between biochemical and cellular assays due to rather different concentrations of nuclease and gRNA.
Among those considered false-positives of off-target nomination assays, 158 sites were nominated by all three genome-wide methods (Table 2). Given that these sites were nominated by a cellular-assay and two biochemical assays, we wondered if these sites were truly unedited or false-negatives of the sequence-confirmation step. False-positives and false-negatives can arise during sequence confirmation if the sites are not sequenced to a sufficient depth. However, we note that these 158 sites were sequenced at a median read depth of 3,000 reads (Supplementary Fig. S8A). At 154 of these sites, the average indel frequency differences between treated and control samples were within 0.2% (Supplementary Fig. S8B). We evaluated edit distances of these sites to assess their likelihood of being edited in cells, and found that the 158 sites have higher edit distances compared to sites sequence confirmed as edited (Supplementary Fig. S8C). While these sites were found to be unedited by targeted sequencing at the depth explored in this study, it remains possible that editing occurs at these sites at very low frequencies <0.2%.
The initial round of confirmatory targeted sequencing of 75,552 homology-nominated sites was intended to evaluate the reliability of genome-wide methods to nominate sequence-confirmed sites within the 5 MM 0 gap, 3 MM 1 gap homology space. We subsequently expanded the scope of this analysis to the sites nominated by the three homology-independent nomination methods outside of the 5 MM 0 gap, 3 MM 1 gap homology space (Supplementary Table S11). Because exhaustive sequence confirmation of these 38,265 sites would be prohibitive, we performed a second round of hybrid capture sequencing in HEK293T/Cas9 cells focused on a subset of more than 2,800 sites that had been nominated for CCR5, RNF2, HEK1, and HEK3 gRNAs within the broader homology space of up to 6 MM and three gaps (Supplementary Fig. S9A and B and Supplementary Table S12). A total of 2,328 of the selected sites were nominated outside of the 5 MM 0 gap, 3 MM 1 gap space and were nominated by one or more homology-independent assays. Apart from on-target sites, no sites nominated within this broader homology space were sequence confirmed as edited with a statistically significant indel frequency difference of at least 0.2% between treated samples and untreated controls (Supplementary Fig. S9C and Supplementary File S4).
Validation of hybrid capture results with rhAmpSeq
We interrogated sites of most interest with rhAmpSeq to quantify indel frequencies using a different experimental method and at a higher depth. All nine on-target sites, 40 sequence-confirmed off-target sites, and 152 potential false-negatives were sequenced with more than 500 average read depth (Supplementary File S5). We found that indel frequencies measured from hybrid capture sequencing and rhAmpSeq were highly correlated (R2 = 0.97; Supplementary Fig. S10). Out of 152 potential false-negative sites (sites nominated by all three genome-wide assays but not sequence confirmed by hybrid capture), only six sites had indel frequency differences >0.2% in rhAmpSeq, and ranged between 0.22% and 0.36%. Additionally, the one site that was sequence confirmed as edited in hybrid capture analysis but not nominated by any replicate of SITE-seq, CIRCLE-seq, or GUIDE-seq was found to have a very low indel frequency difference between treated samples and untreated controls (0.00015%), providing evidence that this site was likely a false-positive of the sequence-confirmation step.
Correlation of nomination signal with editing rate in cells
Nomination assays would have greater utility beyond off-target site nomination if the read counts they produced were to reflect editing in cells quantitatively. This would enable prioritization of gRNAs based on the inferred frequency of editing at off-target sites. Since we observed that GUIDE-seq and CIRCLE-seq assays enrich for on-target sites and potentially highly cleaved sites, we expected their signal to correlate with indel frequency difference between treated and control samples measured by hybrid capture sequencing in cells. We found that GUIDE-seq read counts correlated highly with the indel frequency difference empirically determined by sequencing (R2 = 0.7), with on-target sites having the highest GUIDE-seq read counts (Fig. 5A). In the case of CIRCLE-seq, on-target sites had the highest read counts, but the correlation of read counts to observed indel frequency difference was lower than for GUIDE-seq (Fig. 5B; R2 = 0.36). For SITE-seq, on-target sites generated the same number of reads as off-target sites and false-positives, and there was no correlation observed between read counts and observed indel frequency difference between treated and control samples (Fig. 5C; R2 = 0.01).
FIG. 5.

Correlation of read counts for nominated sites obtained in (A) GUIDE-seq, (B) CIRCLE-seq, and (C) SITE-seq with an average indel frequency (%) difference between treated and control samples from three replicates of hybrid capture followed by sequencing in HEK293T/Cas9 cells. Color images are available online.
Discussion
Robust off-target assessment methods are essential for the safe therapeutic application of CRISPR-Cas9 and related genome editing approaches. This study provides the most comprehensive assessment to date of homology-independent off-target nomination methods by benchmarking to sequencing data from more than 66,000 off-target sites. The sequencing of a large number of computationally nominated (homology-dependent) sites for eight gRNAs allowed us to determine if the genome-wide assays have an appreciable incidence of false-negatives, that is, true off-target sites that are not nominated by these methods. We used hybrid capture enrichment as our sequence-confirmation method in this study because of the ease of multiplexing thousands of sites. It has been shown previously that hybrid capture sequencing can accurately quantify species with large indels, especially with long probes.30,31 We also found a high correlation between indel frequency differences measured by hybrid capture and multiplexed amplicon sequencing.
The three genome-wide off-target site nomination assays—GUIDE-seq, CIRCLE-seq, and SITE-seq—performed similarly in nominating true off-target sites that were sequence confirmed to have indels. Of 51 sites sequence confirmed as edited across eight gRNAs, 50 were nominated by GUIDE-seq and SITE-seq, whereas CIRCLE-seq nominated 49 of the 51 edited sites. These results indicate that the three homology-independent methods do not have obvious blind spots for the detection of true off-target editing events.
The sequence-confirmed off-target site for CCR5 gRNA that had not been nominated by any of the three genome-wide assays was a false-positive of the sequence-confirmation step as confirmed by rhAmpSeq. This site highlights that while targeted deep sequencing is used as the gold standard for off-target analysis, it is not without limitations. Many regions of the genome are challenging to sequence and are susceptible to sequencing errors and false editing calls, emphasizing the need to compare the sequencing reads carefully between the treated and untreated samples. CRISPR-Cas-mediated cleavage results in characteristic patterns of indel around the cleavage site32 that could be used in future research to train methods to differentiate low-frequency CRISPR indels from sequencing artifacts.
A sequence-confirmed site that was nominated by GUIDE-seq and SITE-seq but not by CIRCLE-seq had a very low average indel frequency difference between treated and untreated samples (<0.2%). This site had 2 MM and 1 gap with respect to the gRNA sequence and therefore would have been nominated bioinformatically, even with a small homology search space of up to 2 MM and 1 gap. Overall, we would have successfully nominated all sequence-confirmed edited sites for all eight gRNAs in this study with a combination of any one of the homology-independent genome-wide assays and homology-dependent nomination in the homology space up to 2 MM and 1 gap. As these criteria are comprehensive for these eight gRNAs of varying promiscuity, we expect that off-target sites for therapeutic gRNAs, which are generally selected based on their predicted off-target profile, will be comprehensively nominated using a combination of any genome-wide assay and a homology-dependent search in 2 MM, 1 gap space.
Most sequence-confirmed sites (45/51) were nominated by all three replicates of all three methods. The five sites that were nominated by genome-wide assays, but not by every replicate, had very low indel frequencies (<0.5%). Certain replicates of nomination methods might miss very low frequency sites, even when sequenced at high depth, but combining hits from multiple replicates improves the reliability of these assays.
In this study, the genome-wide assays nominated a higher number of homology-independent sites with more read counts per site than in previous publications,11,12,33 likely due to our use of larger homology space and increased sequencing depth. We also chose experimental conditions that would maximize off-target cleavage; GUIDE-seq was performed in HEK293T cells that constitutively express SpCas9, whereas CIRCLE-seq and SITE-seq were performed with high RNP concentration. The number of off-target sites and their indel frequencies would potentially be reduced with a reduced exposure to RNP as in ex vivo therapies.
Our data support the use of GUIDE-seq as an efficient and comprehensive homology-independent off-target nomination method. GUIDE-seq is particularly well suited for situations in which cell type specific chromatin structure is an important determinant of editing efficiency or when the edited cell type is readily available, as is the case with therapeutic ex vivo CRISPR-Cas editing. Given that CIRCLE-seq nominated all but one sequence-confirmed site in this study, it should be considered a valuable homology-independent method for contexts in which experimental limitations preclude the use of GUIDE-seq, including therapeutic in vivo CRISPR-Cas editing. Although SITE-seq performed similarly with respect to capturing true off-target sites, the lack of correlation between SITE-seq read counts and indel frequencies determined by sequencing is a drawback of this method. In this study, we used a saturating concentration of SpCas9 RNP for SITE-seq to maximize the number of nominated sites. The lack of correlation between indel frequencies and SITE-seq read counts was also observed in the study by Cameron et al.12 (Supplementary Fig. S11), wherein they examined the relationship between several concentrations of SpCas9 and the number of sites generated by the SITE-seq assay.
Having analyzed the data in this work and other off-target studies, we suggest the following practices to guide assessment of off-target editing with CRISPR-Cas systems.
-
(1)
The use of computational off-target and on-target site prediction methods to prioritize candidate gRNAs.
-
(2)
The use of both a homology-independent (empirical) method and a homology-dependent (computational) method used to nominate potential off-target sites. While this study has demonstrated that homology-independent methods are comprehensive and have low false-negative rates, a conservative approach should include both nomination methods. For ex vivo therapeutics, GUIDE-seq will provide the lowest false-positive rate of the methods evaluated. For in vivo therapeutics, CIRCLE-seq may be able to provide the broadest nomination strategy.
-
(3)
Sequencing at sufficient depth and the use of multiple replicates for genome-wide assays and sequence confirmation. Sequencing at a read depth of more than 1,000 at on- and off-target sites will generally allow for the detection of <1% editing, while a read depth of more than 5,000 will increase sensitivity to <0.2%. The use of replicates enables comprehensive nomination of sites edited at low frequencies and provides essential statistical power.
-
(4)
The use of rational selection criteria to nominate off-target sites for further evaluation. For example, one may choose targets within an edit distance of three from the on-target site or choose targets with more than 10 CIRCLE-seq reads. This contrasts with the common practice of assaying an arbitrary number of top sites.
-
(5)
The use of appropriate statistical methods for calling off-target edits as sequence confirmed.
-
(6)
Consideration of potential sequence artifacts from low complexity regions, such as homopolymeric stretches.
-
(7)
Analysis of indel patterns at statistically significant sites to confirm that they resemble typical CRISPR-Cas-mediated edits.
-
(8)
Consideration of the genomic context of the off-target site on the expression, regulation, and function of genes. Does editing occur in a promoter or coding region of a gene? Does editing occur in or near a tumor suppressor or oncogene? Or does editing occur in an intergenic region, far from any gene?
Conclusions
The goal of this study was to characterize and thoroughly compare the homology-independent laboratory methods that are commonly used to evaluate gRNAs for therapeutic application. Our primary concern was that one or more of the methods would fail to detect a particular class of off-target sites. While we cannot completely rule out this possibility, the data presented here indicate that these methods reliably detect off-target sites for a range of gRNAs and potential off-target sites. Nevertheless, we believe that it remains prudent to perform assessment of sites nominated by homology-dependent as well as homology-independent methods, and that adoption of practices discussed here will enable thorough evaluation of gRNAs for preclinical assessment and clinical application.
Supplementary Material
Acknowledgments
We thank many employees of CRISPR Therapeutics as well as several external colleagues for very helpful discussions and feedback on the manuscript. Raw FASTQ files associated with CIRCLE-seq, SITE-seq, GUIDE-seq, hybrid capture sequencing, and rhAmpSeq can be accessed at BioProject PRJNA670368 in the Sequence Read Archive (SRA), NCBI.
Author Disclosure Statements
H.G.C., J.P., S.J.S., N.F., M.C.L.Z., E.H., A.S.K., J.M.T., C.A.S., A.W.N., T.W.H., J.D.K., and A.K. are current employees and hold stock and/or stock options in CRISPR Therapeutics AG. H.J.W. and T.J.C. are former employees and shareholders of CRISPR Therapeutics AG.
Funding Information
This work was fully funded by CRISPR Therapeutics.
Supplementary Material
References
- 1. Jinek M, Chylinski K, Fonfara I, et al. . A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012;337:816–821. DOI: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Carlson DF, Fahrenkrug SC, Hackett PB. Targeting DNA with fingers and TALENs. Mol Ther Nucleic Acids 2012;1:e3 DOI: 10.1038/mtna.2011.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Porteus M. Genome editing: a new approach to human therapeutics. Annu Rev Pharmacol Toxicol 2016;56:163–190. DOI: 10.1146/annurev-pharmtox-010814-124454. [DOI] [PubMed] [Google Scholar]
- 4. You L, Tong R, Li M, et al. . Advancements and obstacles of CRISPR-Cas9 technology in translational research. Mol Ther Methods Clin Dev 2019;13:359–370. DOI: 10.1016/j.omtm.2019.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pattanayak V, Lin S, Guilinger JP, et al. . High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nat Biotechnol 2013;31:839–843. DOI: 10.1038/nbt.2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cradick TJ, Fine EJ, Antico CJ, et al. . CRISPR/Cas9 systems targeting beta-globin and CCR5 genes have substantial off-target activity. Nucleic Acids Res 2013;41:9584–9592. DOI: 10.1093/nar/gkt714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fu Y, Foden JA, Khayter C, et al. . High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat Biotechnol 2013;31:822–826. DOI: 10.1038/nbt.2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hsu PD, Scott DA, Weinstein JA, et al. . DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol 2013;31:827–832. DOI: 10.1038/nbt.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kim D, Bae S, Park J, et al. . Digenome-seq: genome-wide profiling of CRISPR-Cas9 off-target effects in human cells. Nat Methods 2015;12:237–243, 231 p following 243. DOI: 10.1038/nmeth.3284. [DOI] [PubMed] [Google Scholar]
- 10. Tsai SQ, Zheng Z, Nguyen NT, et al. . GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat Biotechnol 2015;33:187–197. DOI: 10.1038/nbt.3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tsai SQ, Nguyen NT, Malagon-Lopez J, et al. . CIRCLE-seq: a highly sensitive in vitro screen for genome-wide CRISPR-Cas9 nuclease off-targets. Nat Methods 2017;14:607–614. DOI: 10.1038/nmeth.4278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cameron P, Fuller CK, Donohoue PD, et al. . Mapping the genomic landscape of CRISPR-Cas9 cleavage. Nat Methods 2017;14:600–606. DOI: 10.1038/nmeth.4284. [DOI] [PubMed] [Google Scholar]
- 13. Wienert B, Wyman SK, Richardson CD, et al. . Unbiased detection of CRISPR off-targets in vivo using DISCOVER-Seq. Science 2019;364:286–289. DOI: 10.1126/science.aav9023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gabriel R, Lombardo A, Arens A, et al. . An unbiased genome-wide analysis of zinc-finger nuclease specificity. Nat Biotechnol 2011;29:816–823. DOI: 10.1038/nbt.1948. [DOI] [PubMed] [Google Scholar]
- 15. Gyorgy B, Nist-Lund C, Pan B, et al. . Allele-specific gene editing prevents deafness in a model of dominant progressive hearing loss. Nat Med 2019;25:1123–1130. DOI: 10.1038/s41591-019-0500-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kim MY, Yu KR, Kenderian SS, et al. . Genetic inactivation of CD33 in hematopoietic stem cells to enable CAR T cell immunotherapy for acute myeloid leukemia. Cell 2018;173:1439–1453.e19. DOI: 10.1016/j.cell.2018.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Maeder ML, Stefanidakis M, Wilson CJ, et al. . Development of a gene-editing approach to restore vision loss in Leber congenital amaurosis type 10. Nat Med 2019;25:229–233. DOI: 10.1038/s41591-018-0327-9. [DOI] [PubMed] [Google Scholar]
- 18. Wu Y, Zeng J, Roscoe BP, et al. . Highly efficient therapeutic gene editing of human hematopoietic stem cells. Nat Med 2019;25:776–783. DOI: 10.1038/s41591-019-0401-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gray SJ, Foti SB, Schwartz JW, et al. . Optimizing promoters for recombinant adeno-associated virus-mediated gene expression in the peripheral and central nervous system using self-complementary vectors. Hum Gene Ther 2011;22:1143–1153. DOI: 10.1089/hum.2010.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Brinkman EK, Chen T, Amendola M, et al. . Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res 2014;42:e168 DOI: 10.1093/nar/gku936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Stemmer M, Thumberger T, Del Sol Keyer M, et al. . CCTop: an intuitive, flexible and reliable CRISPR/Cas9 target prediction tool. PLoS One 2015;10:e0124633 DOI: 10.1371/journal.pone.0124633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Li H, Durbin R. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 2010;26:589–595. DOI: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rice P, Longden I, Bleasby A. EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet 2000;16:276–277. DOI: 10.1016/s0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- 24. Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013 [Google Scholar]
- 25. Li H, Handsaker B, Wysoker A, et al. . The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009;25:2078–2079. DOI: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gastwirth J. Statistical methods for analyzing claims of employment discrimination. Indust Labor Relat Rev 1984;38 DOI: 10.2307/2523801 [DOI] [Google Scholar]
- 27. Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B 1995;57:289–300 [Google Scholar]
- 28. Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 2014;30:2114–2120. DOI: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lin Y, Cradick TJ, Brown MT, et al. . CRISPR/Cas9 systems have off-target activity with insertions or deletions between target DNA and guide RNA sequences. Nucleic Acids Res 2014;42:7473-7485. DOI: 10.1093/nar/gku402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Walsh T, Lee MK, Casadei S, et al. . Detection of inherited mutations for breast and ovarian cancer using genomic capture and massively parallel sequencing. Proc Natl Acad Sci U S A 2010;107:12629-12633. DOI: 10.1073/pnas.1007983107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ueno T, Yamashita Y, Soda M, et al. . High-throughput resequencing of target-captured cDNA in cancer cells. Cancer Sci. 2012;103:131-135. DOI: 10.1111/j.1349-7006.2011.02105.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chen W, McKenna A, Schreiber J, et al. . Massively parallel profiling and predictive modeling of the outcomes of CRISPR/Cas9-mediated double-strand break repair. Nucleic Acids Res 2019;47:7989–8003. DOI: 10.1093/nar/gkz487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kim D, Kim JS. DIG-seq: a genome-wide CRISPR off-target profiling method using chromatin DNA. Genome Res 2018;28:1894–1900. DOI: 10.1101/gr.236620.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.