

Abstract
CRISPR-Cas systems typically consist of a CRISPR array and cas genes that are organized in one or more operons. However, a substantial fraction of CRISPR arrays are not adjacent to cas genes. Definitive identification of such isolated CRISPR arrays runs into the problem of false-positives, with unrelated types of repetitive sequences mimicking CRISPR. We developed a computational pipeline to eliminate false CRISPR predictions and found that up to 25% of the CRISPR arrays in complete bacterial and archaeal genomes are located away from cas genes. Most of the repeats in these isolated arrays are identical to repeats in cas-adjacent CRISPR arrays in the same or closely related genomes, indicating an evolutionary relationship between isolated arrays and arrays in typical CRISPR-cas loci. The spacers in isolated CRISPR arrays show nearly as many matches to viral genomes as spacers from complete CRISPR-cas loci, suggesting that the isolated arrays were either functionally active recently or continue to function. Reconstruction of evolutionary events in closely related bacterial genomes suggests three routes of evolution of isolated CRISPR arrays: (1) loss of cas genes in a CRISPR-cas locus, (2) de novo generation of arrays from off-target spacer integration into sequences resembling the corresponding repeats, and (3) transfer by mobile genetic elements. Both combination of de novo emerging arrays with cas genes and regain of cas genes by isolated arrays via recombination likely contribute to functional diversification in CRISPR-Cas evolution.
Introduction
CRISPR-Cas are adaptive immunity systems, the principal function of which is to protect bacteria and archaea from viruses and other mobile genetic elements (MGE).1–4 A typical CRISPR-cas locus consists of a CRISPR array and adjacent cas genes that form one or more operons. The CRISPR arrays consist of two to several hundred direct repeats (typically 25–36 bp in size) separated by spacers, some of which are homologous to segments of virus or plasmid genomes. The Cas proteins mediate the three stages of the CRISPR immune response: (1) adaptation (incorporation of new spacers into CRISPR arrays), (2) processing of pre-CRISPR RNAs (pre-crRNAs) into mature crRNAs, and (3) interference when the crRNAs serve as guides to bind and cleave complementary target DNA or RNA specifically.5,6 Additionally, many CRISPR-Cas systems encompass various accessory genes that modulate the basic immune functions.6,7 The CRISPR-Cas systems show striking diversity of cas gene composition and genomic loci architecture. Based on the presence of signature genes, cas gene content, and locus architecture, CRISPR-Cas systems have been classified into two classes, six types, and more than 30 subtypes.6
Although the majority of CRISPR-Cas systems contain both CRISPR arrays and cas genes, there are many derived variants that lack major components, in particular CRISPR arrays.7–11 Some of the type III CRISPR-Cas systems that lack CRISPR arrays have been shown to utilize, in trans, crRNA produced by arrays adjacent to type I loci.12–14 Conversely, bacterial and archaeal genomes encompass numerous isolated CRISPR arrays that are not adjacent to cas genes.6,15 In general, it remains unclear which of the isolated arrays are functionally active and, in particular, whether they can complement suits of cas genes that lack arrays. However, multiple lines of evidence indicate that at least some, and probably many, isolated CRISPR arrays are functional. In particular, many isolated arrays are preceded by a promoter-containing leader sequence that is identical to the leaders of CRISPR arrays adjacent to cas genes in the same genome, and some isolated arrays have been shown to capture new spacers.16,17 Furthermore, production of mature crRNAs from isolated arrays has been observed in several experimental systems.18–21 In some bacteria, isolated arrays are conserved across the genomes of numerous strains, which is suggestive of conserved functionality. For example, somewhat paradoxically, in Enterococcus faecalis, an isolated array shows a broader conservation than any of the cas-adjacent arrays.22
In certain cases, isolated arrays might perform specialized roles. In particular, it has been demonstrated that isolated arrays in some Escherichia coli strains contain repeats identical to those of subtype I-F CRISPR-Cas systems that are located in an equivalent genomic position in other E. coli strains and spacers against subtype I-F cas genes,23 and indeed prevent the uptake of complete I-F loci on plasmids.24 Furthermore, functionality of isolated arrays is suggested by the discovery of CRISPR mini-arrays (i.e., arrays that typically contain a single spacer flanked by repeats) that have been detected in the genomes of some bacterial and archaeal viruses.25,26 These viral mini-arrays typically contain spacers homologous to genomic sequences from related viruses, and indeed have been shown to contribute to inter-virus competition by recruiting host Cas proteins and inhibiting the reproduction of the target viruses.
We were interested in the diversity, evolution, and potential functions of CRISPR arrays that are located away from cas genes. Here, we report a comprehensive census of such isolated arrays from complete bacterial and archaeal genomes, show that these arrays contain many spacers matching virus genomes, and investigate possible evolutionary scenarios of their origin. Although many of the isolated arrays probably evolved through the most obvious scenario, namely, as a result of the loss of the accompanying cas genes, some seem to have emerged via other routes, namely, de novo origin resulting from off-target spacer incorporation and dissemination by MGE.
Methods
Genomic database and CRISPR arrays data set
The previously assembled data set of complete prokaryotic genomes was used for all searches.6 This database contains genomes that were available in GenBank27 in March 2019. For the data set of 13,116 genomes, 14,585 potential CRISPR arrays were found using the minCED tool with default parameters (https://github.com/ctSkennerton/minced). Altogether, 7,916 CRISPR-Cas systems were identified and analyzed in the genomes contained in the screened database using the previously compiled collection of profiles for Cas protein families.6 As a result of the profile search, CRISPR-Cas subtypes were assigned for 9,732 CRISPR arrays located in the previously described CRISPR-cas loci6 (1,816 CRISPR-cas loci contained two or three arrays), and the remaining 4,853 arrays were isolated.
All protein sequences annotated in these genomes were clustered using MMSEQ228 with a similarity threshold of 0.75. The fraction of proteins in shared clusters was obtained for all genome pairs. Genome-to genome distances were calculated as –ln[max(f1,f2)], where f1 and f2 are the fractions for each genome in the pair. An UPGMA tree was constructed from the genome distance matrix using the hclust() function of the R package (R Foundation for Statistical Computing, Vienna, Austria). For a given subset of genomes, weights were calculated from the corresponding subtree, as previously described29 (briefly, the total weight allocated to a [sub]tree is distributed between all its subtrees proportionally to the sum of branch lengths).
CRISPR leader prediction
The CRISPRleader prediction tool30 was used to search for potential leader sequences in the vicinity of identified CRISPR arrays. Arrays and 600 bp flanking regions were used as input sequences for CRISPRleader with the default parameters, except that partial genomes was turned on. Two types of outputs were parsed from the results: potential leader sequences, that is, sequences with similarity to the repeats,30 and predicted leaders, that is, potential leaders with similarity to any previously identified leader. These leader sequences were used as BLASTN queries to search the genomic database, with the word size cutoff set to 50. Coordinates for BLASTN hits were added to the leader set for arrays without potential leader predicted by CRISPRleader if hit was inside of 600 bp flanks of the array.
Virus database
Accession numbers were downloaded from the nuccore NCBI database27 for sequences that are annotated as viral and have bacterial or archaeal hosts on 16 December 2019. Non-prokaryotic viruses and nonviral sequences were manually filtered out. A nucleotide sequence database containing 21,285 sequences and 843,956,625 bp was assembled using sequences left after the filtration.
Protospacer search
From the 274,663 spacers present in all acquired CRISPR arrays, the set of 191,790 unique spacer sequences was used as a query for BLASTN search against the assembled viral database, using a word size of eight and with low complexity filtering turned off.31 The results were filtered for 95% identity and 95% coverage against the query spacers set. Altogether 29,938 protospacers were found for 4,289 spacers.
Validation of CRISPR arrays
All 9,732 CRISPR arrays adjacent to cas genes (separated by no more than five open reading frames [ORFs]) were considered true positives. The remaining 4,853 isolated arrays were processed with a filtering pipeline (Fig. 1). Among the isolated arrays, 574 contained at least one spacer with a possible protospacer in the viral database. DNA sequences of detected possible CRISPR arrays were translated in six frames to find possible protein coding sequences inside. ORF coverage for CRISPR arrays (as length of largest ORF divided by length of the CRISPR array) was calculated to compare CRISPR arrays with known types and isolated arrays (Fig. 2A). Isolated arrays >400 bp and ORF overlap >0.95 were filtered out. All ORFs detected in six frames translation of CRISPR arrays were annotated with Conserved Domain Database (CDD)32 profiles using PSI-BLAST33 with a 10−4 e-value cutoff (Fig. 2B). Isolated arrays with <850 bp that had BLAST hits were filtered out. For each isolated array, pairwise distances between spacers were calculated as the number of matches in the longest BLASTN34 hit (word size of six, e-value threshold 100, no filtering) divided by the length of the smaller spacer in the pair. Spacers within a CRISPR array were clustered using single linkage clustering with a cutoff of 0.3. A spacers similarity index was calculated for each CRISPR array as the number of clusters divided by number of spacers in the array (index value of 1, meaning that all spacers are different; Fig. 2C). Altogether, 1,735 isolated arrays were filtered out as false-positives (Fig. 1).
FIG. 1.

Computational pipeline for analysis of CRISPR arrays. The filtering pipeline was designed to eliminate spurious arrays. The rectangular shapes show input and output, and the diamond shapes show filtering criteria. The numbers at the bold vertical lines show the number of arrays processed at each step.
FIG. 2.

Characteristics of isolated CRISPR arrays and arrays adjacent to cas genes before and after filtering. (A) Coverage of CRISPR arrays by ORFs. The 2D density plots of the fraction of an array covered by an ORF versus the array length (in bp). The density scale shows the 2D kernel smoothed number of arrays. The red lines show the filtering cutoffs (isolated arrays >400 bp and covered by an ORF >0.95 of their length were removed). The left panel shows cas-associated arrays; the middle panel shows the unfiltered isolated arrays; the right panel shows the isolated arrays after filtering. (B) Presence of PSI-BLAST hits from the CDD database profiles into translated arrays. The normalized counts of arrays with CDD (hits per bp) versus the array length (in bp). The red line shows the filtering cutoff (isolated arrays <850 bp with CDD hits were removed). The left panel shows cas-associated arrays; the middle panel shows the unfiltered isolated arrays; the right panel shows the isolated arrays after filtering. (C) Fraction of unique spacers in arrays. The 2D density plots of the ratio of the number of unique spacers to the total number of spacers in an array versus the number of spacers in the array. Arrays where all spacers are different (the fraction of unique spacers is equal to 1) are not shown. Density scale shows the 2D kernel smoothed number of arrays. The red line shows the filtering cutoff (isolated arrays with the fraction of unique spacers <0.85 were removed). The left panel shows cas-associated arrays; the middle panel shows the unfiltered isolated arrays; the right panel shows the isolated arrays after filtering. ORF, open reading frame; CDD, Conserved Domain Database.
Spacers clustering
A total of 191,790 unique spacer sequences (274,663 spacers) were used to calculate pairwise similarities between each pair of spacers as the number of matches in the longest BLASTN hit (word size of six, e-value threshold 100, no low complexity filtering) divided by the length of the smaller spacer in the pair. Similarities were converted to distances (d = -ln s); spacer clusters were constructed from the distance matrix using the single linkage clustering method (Supplementary Fig. S1) with a clustering threshold of 0.1. As a result, 187,863 spacer clusters were formed.
Arrays clustering by spacer content
CRISPR arrays were clustered based on their spacer content with single linkage clustering and zero clustering thresholds. Distances between CRISPR arrays were calculated as minus log of Jaccard similarity for the set of spacer clusters (as described above) present in each pair of arrays. In cases where all spacers for one of CRISPR arrays were a subset of another, the distances for these clusters were set to zero (identical arrays). As result 9,338 clusters were formed, with 2,333 of them containing more than one isolated CRISPR array. For each content cluster, initial weight (value 1) was evenly distributed between all CRISPR arrays present in the cluster.
The array weights are essential for obtaining meaningful statistics on features of arrays because arrays with highly similar spacers are clearly non-independent and should not be counted separately. The clustering scheme replaces array counts with sums of weights; the sum of weights across the whole data set is equal to the number of clusters (effectively, the number of independent arrays). Unlike reducing the complete set of arrays to a set of cluster representatives, the weighting scheme provides for members of the same cluster to contribute individually to the calculated characteristics, such as, for example, distribution of isolated arrays across the taxa or CRISPR-Cas types.
Repeats clustering
A total of 5,766 unique repeat sequences (obtained from filtered CRISPR arrays) were used to calculate pairwise distances. Distance was calculated as the number of matches divided by the minimal length of repeat in the pair for the longest BLASTN hit (word size of six, e-value threshold 100, no low complexity filtering) for hits where the length of aligned sequences was >15 and the number of mismatches in the aligned sequences was <3. To obtain the random expectation, the same procedure was applied to the set of 36,360 repeat sequences retrieved from our previous study,35 where nucleotides were shuffled inside of each repeat sequence (Supplementary Fig. S2).
Entropy for each repeat cluster was calculated as for each CRISPR-Cas type present in the cluster, where wt is spacer content weight of the CRISPR arrays of the CRISPR-Cas type t, and W is the total weight of the arrays represented by the repeat cluster. Entropy for all clusters was calculated as for all repeat clusters, where Wgs the total weight of all CRISPR arrays (Supplementary Fig. S3A). Entropy per CRISPR-Cas subtype was calculated for all well-represented CRISPR-Cas subtypes (weight of the arrays >25) using the same formula, however only for the clusters where selected CRISPR-Cas subtype was assigned at least to one CRISPR array (Supplementary Fig. S3B).
Single linkage clustering with clustering thresholds of 0.1 and 0.16 was applied to find repeat clusters and permissive repeat clusters, respectively (Supplementary Table S1).
Analysis of unique orphan arrays
A total of 303 repeat sequences from unique orphan arrays were used as BLASTN queries against the nucleotide collection (nt) database36 using a word size of 15. Blast hits were filtered by size (>19 nt) and by co-location (minimum distance >50 bp for start positions of the hits). Repeat hits were aggregated into pseudo-arrays if they were within a 500 bp vicinity of each other. Arrays with fewer than four repeats as well as arrays that occurred in more than 10 instances (arrays with same repeat sequence) in a genome were discarded. For the final set of 10,136 pseudo arrays, 5 kb upstream and downstream regions were annotated with CRISPRCasTyper (https://github.com/Russel88/CRISPRCasTyper) to fetch coding sequences and annotation for potential cas genes. Coding sequences were also annotated with the CDD32 using PSI-BLAST.33
Calculation of clusters of orthologous gene categories distribution for genes flanking isolated arrays
All of the genomic database was annotated separately with clusters of orthologous genes (COGs)37 and all CDD profiles using PSIBLAST with an e-value cutoff of 10−4. For each isolated array in the genome, one gene was randomly selected from the self-genome for the following gene sets: housekeeping genes (Supplementary Data S1), non-housekeeping genes, and non-cas defense genes. Additionally, for each isolated array, a random position in the self-genome was selected. For each coordinate retrieved, plus coordinates for isolated arrays and coordinates of cas loci,6 five flanking genes from each side were retrieved. For each flanking gene (excluding the initial gene that was used to identify loci), the COG category was identified by COG annotation and summed using the weight of the genome (Supplementary Table S2). Genome weights were calculated using the tree of genomes hosting all Cas loci and CRISPR arrays.
Guilt by association analysis
Analysis was performed using the previously developed pipeline38 with the following parameters: five flanking genes from the baits (isolated arrays), 0.3 sequence similarity for permissive protein clustering, and ab e-value cutoff of 10−4 for PSIBLAST search. Base on these data, an effective number of proteins in the vicinity of the isolated arrays and entire genomic database was calculated for each permissive protein cluster, as well as effective median distance and level of association (Supplementary Table S3).
Alignable Tight Genome Clusters analysis
The genomes from the Alignable Tight Genome Clusters (ATGC) collection39 that were present in assembled genomic database were surveyed for the presence of isolated arrays. The top 3 ATGCs with the highest fraction of genomes containing isolated arrays were studied in detail. DNA sequences of the isolated arrays and 3 kb flanking regions were used as a BLASTN query to the genomic database (with dust no parameter). BLAST hits with a coverage length of <3 kb were filtered out. For each hit, 20 ORFs upstream and downstream were retrieved and annotated using CDD profiles and custom Cas protein profiles.6 Phylogenetic trees were downloaded from the ATGC Web site (http://dmk-brain.ecn.uiowa.edu/ATGC/atgc.html). All loci retrieved with the BLAST search were connected to tree leaves and visualized using ETE3 python.40
Sequence alignments of the protein sequences encoded by the selected genes were constructed using MAFFT41 with the adjustdirectionaccurately parameter turned on.
Results
Detection and classification of isolated CRISPR arrays
The database of completely sequenced prokaryotic genomes available as of March 1, 2019, was searched for CRISPR arrays. Of the 13,116 genomes in the database, CRISPR-like arrays were detected in 6,001. Previously, cas genes were identified in 5,689 genomes from the same data set.6 Cas genes were identified in the vicinity (15 genes in each direction) of 9,732 of the 14,585 detected CRISPR arrays. The remaining 4,853 arrays were located in gene neighborhoods with no identifiable cas genes and thus were classified as putative isolated arrays. In order to filter out false-positive CRISPR-like sequences (Methods) that could come from proteins with repetitive structures or various intergenic regions and might contaminate the set of isolated arrays (Supplementary Data S2), a dedicated computational pipeline was constructed (Fig. 1), where filtering parameters were obtained from bona fide CRISPR arrays (Fig. 2). After running this pipeline, 1,269 arrays that overlapped protein-coding genes and thus likely corresponded to protein repeats erroneously recognized as CRISPR were discarded, and 466 more were discarded because they contained multiple identical spacers. The remaining 3,118 arrays were considered to be bona fide isolated CRISPR arrays.
All 12,850 detected CRISPR arrays (after eliminating false-positives), including both isolated arrays and arrays adjacent to cas genes, were analyzed in detail (Supplementary Table S1). Using sequence identity to define the similarity between repeats, two clustering thresholds (0.1 and 0.16; see Methods) were employed to identify strict and permissive clusters of CRISPR. For the strict clustering threshold of 0.1, clusters were found to represent CRISPR arrays of distinct CRISPR-Cas subtypes (Supplementary Fig. S3). The permissive threshold of 0.16 was found to be the upper bound value at which randomized CRISPR sequences started to form clusters (Supplementary Fig. S2). Repeats from Class 1 CRISPR-Cas systems typically clustered at higher thresholds (greater sequence similarity) than those from Class 2 systems, especially of subtypes II-A, B, and V-A. CRISPR arrays were also clustered based on the spacer content to identify arrays with overlapping spacer sets. The set of 12,850 arrays formed 9,338 clusters with distinct spacer contents at the zero clustering threshold. Weights were assigned to the arrays based on spacer content clusters to account for the non-independence of arrays with similar spacers (see Methods). These weights were used to assess the distribution of isolated CRISPR arrays across CRISPR-Cas types, using repeat clusters that included both isolated and cas-linked CRISPR arrays based on their sequence similarity (Supplementary Table S4). The taxonomic distribution of the arrays was calculated in a similar manner (Supplementary Table S5). These analyses showed that almost 25% of the class 1 arrays are isolated, whereas among the class 2 arrays, only about 7% are isolated. Comparison of the repeats in isolated arrays with the cas-linked repeats showed that 60% of the isolated arrays belonged to class 1 systems and 3% to class 2 systems, whereas the remaining 37% could not be classified by their repeat sequences. A taxonomic survey of the isolated arrays demonstrated the highest fractions in Methanococci (0.70), Aquificae (0.67), Archaeoglobi (0.67) Thermococci (0.58), and Thermotogae (0.52) classes of archaea and bacteria. Most of the genomes in these classes harbor type I and type III CRISPR-Cas systems (Supplementary Table S1). Notably, all these organisms are hyperthermophiles. No taxa showed a conspicuous lack of isolated arrays.
Repeat length distribution for the isolated arrays follows the length distribution for the cas-adjacent arrays, with small excess of short repeats (Supplementary Fig. S4). However, the isolated arrays tend to be shorter than arrays adjacent to cas genes (Supplementary Fig. S5).
Among the isolated arrays, 15% are present in genomes that lack CRISPR-cas loci; 6% account for the only CRISPR arrays in genomes that contain an apparently functional cas operon without an adjacent array; and the rest are in genomes that also carry cas-adjacent arrays (Supplementary Tables S1 and S6).
Potential leader sequences were predicted (see Methods) for 74% of CRISPR arrays that are adjacent to cas genes and 70% of the isolated arrays (Supplementary Tables S1 and S6).
We classified all CRISPR arrays with respect to their adjacency to cas genes and, for the isolated arrays, their similarity (or lack thereof) to cas-adjacent arrays and presence in genomes that possess or lack CRISPR-Cas systems (Fig. 3). Among the isolated arrays, the vast majority are significantly similar (as defined in the permissive clustering procedure; see Methods) to cas-adjacent arrays. The remaining 303 are unique isolated arrays that appear to be of greatest interest from a functional standpoint because they either can function with a heterologous suite of Cas proteins or represent novel CRISPR-Cas types. On a different plane, among both the unique isolated arrays and those that are similar to cas-adjacent ones, the majority reside in genomes that carry cas operons (with or without adjacent arrays) that might be functionally linked to the isolated arrays. By contrast, the remaining 562 arrays (112 unique and 450 similar to cas-adjacent ones) are orphans that were found in genomes lacking CRISPR-cas loci (Fig. 3). The unique orphan arrays are the best candidates for the discovery of unknown CRISPR types.
FIG. 3.
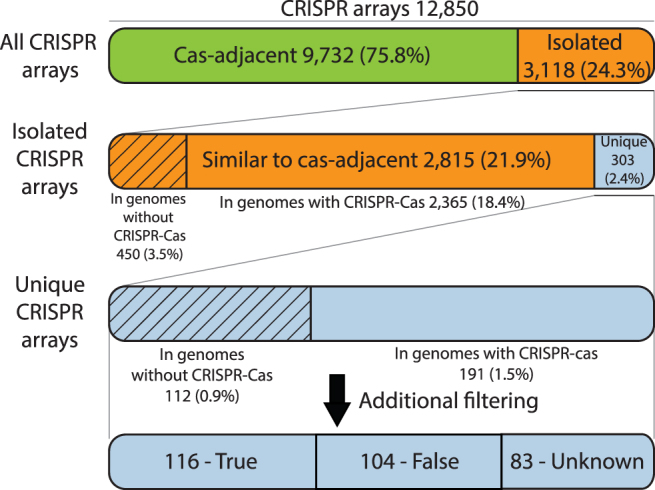
Classification of CRISPR arrays: cas-adjacent, isolated, unique isolated and orphan arrays. The schematic illustrates the partitioning of CRISPR arrays into distinct categories according to their adjacency to cas operons or lack thereof and, for isolated arrays, similarity to cas-adjacent arrays.
Virus-specific spacers in isolated CRISPR arrays
Among the spacers from the isolated arrays, we identified reliable similarity to viral sequences in 13% compared to 18% for spacers from cas-adjacent arrays (Supplementary Table S6). Most of the viral spacer matches (Supplementary Data S3) come from isolated arrays in Bacilli and Gammaproteobacteria (48% and 41% of the matches, respectively). Most of the viruses matching spacers from isolated arrays are incompletely classified (42% have no genus-level taxonomic assignments). Among those that are properly classified, the most common genera are Casadabanvirus (Pseudomonas virus D3112 and its relatives; 15%) and Moineauvirus (Streptococcus virus DT1 and its relatives; 14%).
Analysis of the spacers in the isolated arrays in closely related species showed variability of spacer content for the arrays found in the same position in the genomes. Thus, in ATGC072, the 16 identified isolated arrays had a weight of 14, which is equivalent to 14 arrays with unique spacer content (see Methods for details). In ATGC108, 29 isolated arrays had a weight of 12; and in ATGC127, the 72 isolated arrays had a weight of 18. These findings are most compatible with the possibility that the majority of the isolated arrays are functionally active.
Gene neighborhoods of isolated arrays
Flanking genes of isolated arrays were annotated using COG profiles.37 For comparison, the same number of gene neighborhoods were randomly selected for housekeeping genes, non-housekeeping genes, non-cas defense genes, and random loci from the set of genomes in which isolated arrays were found. Additionally, flanking genes of CRISPR-cas loci6 were analyzed using the same procedure. Analysis for COG categories showed similar distributions of flanking genes for isolated arrays and cas loci (Supplementary Table S2). One of the distinctive features of these two groups was the enrichment for the “X” COG category (mobilome: prophages, transposons) by a factor of 3.7–3.9 compared to random loci (95% confidence interval for enrichment is 1.96–4.43 and 1.92–4.13 for the isolated and cas-adjacent arrays, respectively, Fig. 3 and Supplementary Table S2).
We further investigated the genes in the isolated array neighborhoods by relative enrichment (“icity”) analysis in an attempt to identify genes that might be specifically associated with isolated arrays.7,38 The proteins from these neighborhoods were clustered by sequence similarity, and for each cluster, a PSIBLAST search was performed to identify genes of the same families elsewhere in the entire genomic database. Protein clusters that were highly abundant in the vicinity of isolated arrays and strongly associated with the latter (a strong association was defined as No/N > 0.5, where No is the number of proteins from the given cluster in the vicinity of isolated arrays, and N is the total number of such proteins in the database) were analyzed in detail (Supplementary Table S3). Most of the protein-coding genes that were found to be strongly associated with isolated arrays are not annotated in the current databases and encode short (typically, <100 aa) putative proteins, which seems to indicate false protein predictions or disrupted genes (Fig. 4). Otherwise, the distribution of the functional classes of COGs in the vicinity of isolated arrays closely tracked the distribution around CRISRP-cas loci. In both distributions, MGEs were far more abundant than genes from any other functional class (Supplementary Table S3 and Fig. 3). Thus, we did not identify any functional classes of proteins to be specifically and significantly linked to isolated CRISPR arrays. However, these observations show that isolated arrays are embedded in the same type of genomic neighborhoods that are strongly enriched with mobile elements as bona fide CRISPR-Cas systems. On the one hand, such neighborhoods can be genomic junkyards where insertion of mobile elements is easily tolerated. On the other hand, some of these elements actually might play a role in the dissemination of CRISPR-Cas systems and isolated arrays (see below). These two interpretations are not mutually exclusive.
FIG. 4.

Functional classes of genes in the vicinity of CRISPR-Cas systems and isolated arrays. The box plot shows the enrichment/depletion ratios of genes from different functional classes of COGs in the genomic regions upstream and downstream of CRISPR-cas loci (red boxes) and isolated arrays loci (blue boxes) relative to genes randomly sampled from the same genomes (1,000 bootstrap replications). The boxes shows the 25th/50th/75th percentiles, and black dots shows outliers that fall above 1.5 × interquartile range (IQR). x-Axis: functional classes of COGs (“–” indicates genes that were not recognized by any COG profiles); y-axis: enrichment/depletion ratio. Functional classes of genes are as follows: −, not recognized; C, energy production and conversion; D, cell cycle control, cell division, chromosome partitioning; E, amino acid transport and metabolism; F, nucleotide transport and metabolism; G, carbohydrate transport and metabolism; H, coenzyme transport and metabolism; I, lipid transport and metabolism; J, translation, ribosomal structure, and biogenesis; K, transcription; L, replication, recombination, and repair; M, cell wall/membrane/envelope biogenesis; N, cell motility; O, posttranslational modification, protein turnover, chaperones; P, inorganic ion transport and metabolism; Q, secondary metabolites biosynthesis, transport, and catabolism; R, general function prediction only; S, function unknown; T, signal transduction mechanisms; U, intracellular trafficking, secretion, and vesicular transport; V, defense mechanisms; X, mobilome: prophages, transposons; Z, cytoskeleton. COG, clusters of orthologous gene.
Unique isolated arrays
Among the 3,118 isolated CRISPR arrays, 303 contained repeats that did not share sufficient sequence similarity with repeats from the arrays adjacent to cas genes (hereinafter, unique isolated arrays). Such unique arrays appear to be of particular interest because they might confer function jointly with heterologous Cas proteins, revealing unusual CRISPR-Cas functionality or belonging to novel CRISPR-Cas systems. We clustered the repeats from the unique isolated arrays by sequence similarity to form 197 permissive clusters that were examined on a case-by-case basis. Among these, 34 clusters were found to have similar spacer sequences or diverse repeat sequences, a small number of spacers with different lengths, to contain small ORFs inside, or to possess other features that appear to be incompatible with CRISPR arrays, such as replication initiation genes adjacent to the repeats (in this case, direct repeats could be part of the replication origin42,43). Apparently, these false-positives were missed by our procedure for array identification with the parameters defined above as optimal (Supplementary Table S7). At least 89 clusters including 116 individual arrays appeared to represent bona fide CRISPR arrays because they consisted of repeats in the typical length range (26–36 nt) and/or contained more than five unique spacers (Supplementary Table S7). It remains unclear whether the remaining unique isolated arrays are actual CRISPR. Only two identical arrays in two Streptococcus oralis genomes contain spacers matching a virus genome that confirms their functionality (Supplementary Table S7). For 90% of the unique isolated arrays, potential leader sequences were predicted (see Methods), suggesting that these arrays might be functional.
Notably, among the 89 clusters of unique isolated arrays inferred to be bona fide CRISPR, 21 were contained within MGEs, such as plasmids, prophages, and some specialized elements.44–46 Whether these arrays are involved in the maintenance of these mobile elements or transferred by these vectors to new hosts—or both—remains to be experimentally studied.
Analysis of flanking genes for six unique isolated array clusters resulted in the identification of diverged homologs of effector proteins of subtypes V-K, V-F, and VI that were overlooked in the recent CRISPR-Cas tally6 conceivably because of the low similarity to the respective sequence profiles (Supplementary Table S7). These findings confirm that analysis of unique isolated arrays can lead to the discovery of new CRISPR-Cas variants. However, among the proteins encoded by flanking genes, we failed to identify any candidates for new CRISPR-Cas types, that is, proteins unrelated to currently known Cas proteins but containing domains (primarily, nucleases) that could be implicated in CRISPR functionality. It should be noted, however, that in case such novel Cas proteins are encoded elsewhere in the respective genomes and employ isolated arrays in trans, a new CRISPR-Cas type would have been missed by this analysis.
For the remaining clusters of unique isolated arrays, we did not identify any strongly linked protein-coding genes. Nevertheless, several of these arrays appear to be promising candidates for further investigation. In particular, these include 12 orphan arrays found in genomes that encode no known CRISPR-Cas systems and in genomes that encompass multiple unique isolated arrays. Among the latter, Ktedonobacterales bacterium SCAWS-G2 stands out, with 10 unique isolated arrays (four clusters).
Origins of isolated CRISPR arrays
Multiple, non-exclusive hypotheses for the origin of isolated arrays can be proposed (Fig. 5). The isolated arrays potentially could emerge: (1) from a complete CRISPR-Cas locus as a result of elimination of the cas genes, (2) from insertion of a MGE carrying an array followed by the loss of the MGE, or (3) de novo through insertion of spacers into a random genome location followed by repeat formation as a result of duplication of the flanking sequence. For the isolated arrays with similarity to cas-adjacent ones, the first route appears to be by far most likely, but the situation could be different for the unique arrays.
FIG. 5.
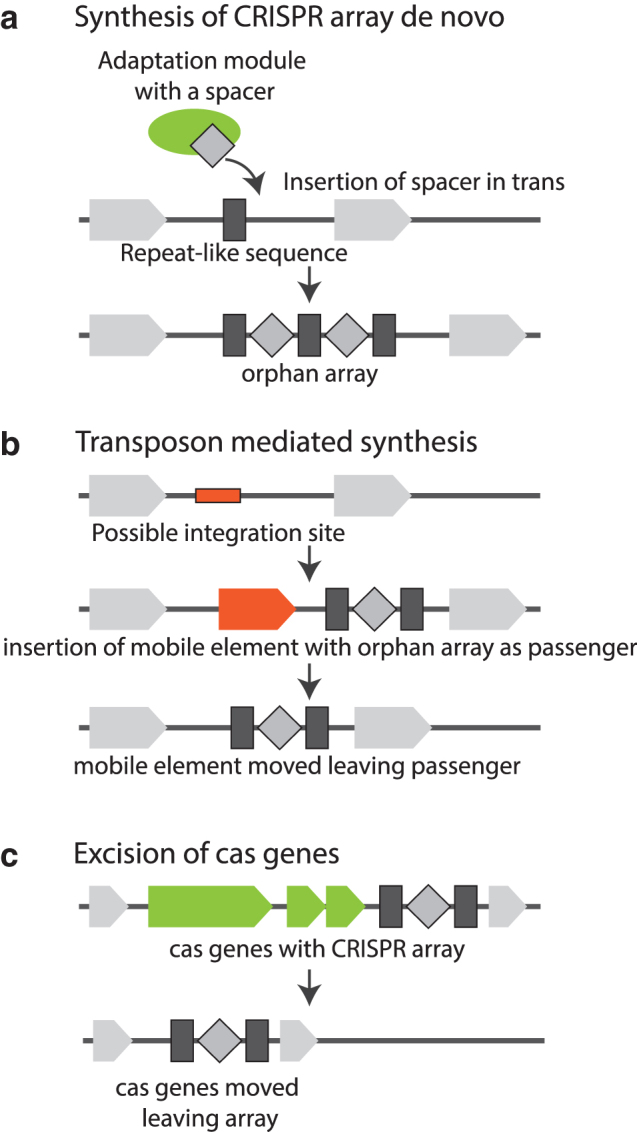
Three scenarios for the origin of isolated CRISPR arrays. CRISPR arrays are shown with dark gray rectangles (repeats) and diamonds (spacers). Flanking non-cas genes are shown with light gray block arrows, transposable elements are shown with orange blocks, and cas genes are shown with green blocks. (a) de novo formation of a CRISPR array; (b) formation of a CRISPR array mediated by transposon insertion; (c) excision of cas genes leaving an isolated array.
To trace possible mechanisms producing isolated CRISPR arrays, we investigated in detail the evolution of these arrays in three ATGCs with the highest content of isolated arrays. For each isolated array and its flanking genes, a BLASTN search was run against all the genomes of the corresponding ATGC in order to identify all instances of the flanking genes, with or without CRISPR arrays.
In ATGC127 (Yersinia), two isolated array loci were detected, both with repeat sequences identical to those in the array associated with type I-F CRISPR-Cas system present in the Yersinia genomes. The functionality of at least one of the isolated arrays in the Yersinia pestis genome manifested in its ability to acquire new spacers has been demonstrated in the classic 2005 work of Pourcel et al.16 Isolated arrays with flanking genes were used as a BLASTN query that retrieved other loci in ATGC127 genomes that contain the query genes but not the isolated arrays (Fig. 6a and Supplementary Fig. S6A). Loci containing an isolated array flanked by rpiR and yagU genes were found in that unique configuration only and thus were not informative with regard to the origin of the isolated array. Another locus that contained an isolated array flanked by sbcB and potE genes was present in three configurations (Fig. 6a): flanking genes and the array; flanking genes without the array; flanking genes, array, and an IS200-like transposable element (Yersinia pseudotuberculosis PA3606 GCF_000834945.1). The sequences between sbcB and potE that lack the CRISPR array (in genomes within the ATGC127 branch that is otherwise enriched with arrays) nevertheless contained a complete or partial single repeat sequence (Supplementary Data S4a), which might be the precursor of the array. The locus containing an IS200-like transposable element has the same DNA sequence as the loci containing isolated arrays except for the sequence of the transposase. These findings are compatible both with de novo formation of an isolated array with a single repeat-like sequence serving as a precursor (even if this precursor itself is in turn a remnant of an earlier array) and with acquisition of an isolated array as a passenger of a mobile element.
FIG. 6.

Evolution of isolated CRISPR arrays. Multiple configurations of gene neighborhoods containing isolated CRISPR arrays and the orthologous loci lacking the arrays are shown for three ATGCs. Flanking non-cas genes are shown as light gray block arrows, with the gene name indicated below the block; mobile elements are shown as orange blocks; cas genes are shown as green blocks; leader sequences shown as blue boxes. An example organism name and locus coordinates in the genome are shown to the right of the schematic depiction of each gene locus. (a) ATGC127: Yersinia; (b) ATGC108: Listeria; (c) ATGC972: Pseudomonas.
Two isolated arrays were identified in ATGC108 (Listeria), both with repeat sequences identical to those in the CRISPR array associated with a subtype I-B CRISPR-Cas system present in these genomes. One isolated array is present in most genomes and is located between prsA and an uncharacterized gene (Fig. 6b). Several genomes from this group contain a single repeat sequence located between these genes, which might be a precursor for the formation of the isolated array (see sequence alignment for Listeria ivanovii londoniensis GCF_000763495.1 and GCF_000763475.1 in Supplementary Data S4b.1). Another isolated array was detected near the first one in two genomes (Fig. 6b and Supplementary Fig. S6B). These arrays are located between His Phos 1 and araJ or proP, where different CRISPR-Cas systems or cas operons lacking CRISPR arrays were apparently inserted or/and excised on multiple occasions (see sequence alignment in Supplementary Data S4b.2). Analysis of the sequence alignment for these loci shows identical array sequences between Listeria ivanovii WSLC3009, Listeria ivanovii sub ivanovii WSLC 3010 (isolated arrays), and Listeria ivanovii sub ivanovii PAM 55 (complete type I-B system), which indicates excision of cas genes, leaving the isolated array.
Two isolated arrays were found in ATGC072 (Pseudomonas) genomes. One of these is located between YHI9 and an uncharacterized gene, along with two additional short uncharacterized genes (Fig. 6c and Supplementary Fig. S6C). The same configuration of these flanking genes without a CRISPR array was observed in another strain. Arrays in these loci have the same repeat sequence as type I-F loci present in the same genomes, and most of the isolated arrays have different spacer sets (Supplementary Data S4c.1), which might indicate insertion of arrays with transposable elements or/and formation of isolated arrays de novo from a precursor repeat-like sequence. Another isolated array is located between SphA and COG3617 genes. Four possible configurations can be found in this gene neighborhood (Fig. 6c and Supplementary Fig. S6C). In two cases, there is an IS3-like transposable element between these genes, which might indicate insertion of a CRISPR-Cas system as a passenger. However, according to the sequence alignment (Supplementary Data S4c.2), these two acquisitions of transposable elements are likely independent events (the IS elements are located in different specific positions in the neighborhood), suggesting prior presence of a CRISPR array in the locus. The sequences of the cas genes in the loci are identical, but the content of CRISPR arrays is different. These observations suggest a scenario for insertion of a CRISPR-Cas system and further excision of cas genes. However, pre-existence of an isolated array followed by insertion of cas genes (with or without a second array) cannot be ruled out either.
In an attempt to gain some insight into the origins of unique CRISPR arrays, these repeat sequences were additionally searched against the NT database (see Methods). Altogether, 1,190 arrays identical or closely similar to the unique ones were detected. Analysis of the flanking genes identified 16 arrays that belonged to I-A, I-C, I-D, I-G, III-A, III-D, V-J, and VI-B2 CRISPR-cas loci (Supplementary Table S7). These observations suggest that some unique isolated arrays originate from rare CRISPR-Cas variants but for the majority of these arrays, the de novo route of origin appears more likely.
Discussion
The CRISPR-Cas systems show a remarkable and growing variety of cas gene compositions and genomic arrangements, which translate into functional diversity that remains to be fully characterized. In addition, however, numerous CRISPR arrays are not associated with cas genes. Repetitive sequences adjacent to cas genes can be assumed to represent functional CRISPR arrays. By contrast, analysis of the (putative) isolated arrays requires much greater caution because in the absence of supporting evidence in the form of the presence of cas genes, artifacts—that is, other types of repetitive structures masquerading as CRISPR—are likely. Here, we developed a dedicated computational pipeline to eliminate most of the false-positives and produced a more accurate census of isolated CRISPR arrays. After eliminating the artifacts, isolated were found to comprise up to 25% of all CRISPR arrays detected in bacterial and archaeal genomes.
Isolated CRISPR arrays are of interest from both functional and evolutionary standpoints. A substantial majority of the isolated arrays consist of repeats that are identical in sequence to the repeats in arrays from the same genomes that are adjacent to cas genes. Therefore, these arrays are likely to be functional and can be employed in trans by the adaptation and effector machineries of the respective CRISPR-Cas systems. When examining the distribution of isolated arrays across the taxonomic diversity of bacteria and archaea, we detected a notable excess of such arrays in several groups of hyperthermophiles—organisms that possess complex repertoires of CRISPR-Cas systems, in particular those of type III that are known for their ability to utilize CRISPR arrays in trans. The excess of isolated arrays in these genomes apparently reflects this feature of type III systems.
However, about 10% of the isolated arrays were found to be unique, with repeat sequences dissimilar from those in known CRISPR-Cas systems. An in-depth examination of these unique isolated arrays identified some additional apparent false-positives, for example repeats that seem to be associated with origins of replication. These cases can be used to refine methods for array identification further. Nevertheless, many clusters of unique isolated arrays possess multiple features of regular CRISPR and thus are likely to be functional. One possibility is that arrays are utilized by promiscuous Cas1 (adaptation) and/or Cas6 (processing) endonucleases.21,47 A more intriguing alternative is that some of the unique isolated arrays, especially unique orphans that are present in genomes lacking any known CRISPR-Cas systems, belong to unknown CRISPR-Cas types that are unrelated or extremely distantly related to the previously described ones. However, the results of the present analysis indicate that if such novel CRISPR-Cas systems exist, they are quite rare. Although we did identify a few variants that have been missed previously, presumably due to the low similarity between the respective Cas proteins and their homologs from known CRISPR-Cas systems, no new types have been discovered. Nevertheless, unique isolated arrays and especially orphans could be a source of novel CRISPR-Cas variants that, although uncommon, might possess unique properties.
The repeats in the majority of isolated arrays are identical in sequence to the repeats in arrays that belong to complete CRISPR-Cas systems in the same or closely related genomes, implying a common origin. Arguably, the default scenario for the emergence of an isolated CRISPR arrays is the loss of the adjacent cas genes. Our comparative analysis of loci containing isolated arrays in closely related bacterial genomes supports this route of evolution, but suggests that two other scenarios might contribute as well: de novo origin of CRISPR arrays and their transfer by MGEs. Off-target spacer integration by CRISPR-Cas adaptation module into sites resembling CRISPR repeats in sequence indeed has been shown to occur in bacteria.48 Our current analysis supports the possibility that this process can generate CRISPR arrays de novo and thus could be an important source of generation of new types of repeats. The third route of isolated array evolution, via transfer by MGE, also appears plausible, given the detection of transposable elements in the vicinity of isolated arrays and the wide presence of CRISPR-Cas systems as well as CRISPR mini-arrays in various MGE. In general, the origins of unique isolated arrays remain uncertain. Further searches in growing genomic and metagenomics databases will show how many of these might be adjacent to known or novel cas genes in some genomes. However, the lack of such adjacency in the genome collection analyzed here suggests that de novo emergence might be the principal route of origin for the unique arrays.
The results of the comparative genomic analyses presented here indicate that isolated arrays are in a dynamic equilibrium with functional CRISPR-Cas systems. On multiple occasions, cas genes are lost, leaving isolated arrays behind. Conversely, arrays emerging de novo or transferred by MGE can become associated with cas genes. Regain of cas genes by an isolated array also appears likely. These processes can contribute to the emergence of new combinations of cas genes with repeats and thus to the functional diversification of CRISPR-Cas systems.
The presence of spacers matching virus genomes, in only a slightly lower proportion than among the arrays from complete CRISPR-cas loci, and the differences in spacer content between orthologous isolated arrays in closely related bacterial genomes suggest that most of the isolated arrays were functionally active recently and at least some remain functional through in trans utilization by Cas proteins, which is in accordance with several previous observations.15,24–26
Supplementary Material
Acknowledgments
The authors thank Koonin group members for helpful discussions.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
S.A.S., Y.I.W., K.S.M., and E.V.K. are funded through the Intramural Research Program of the National Institutes of Health (National Library of Medicine). Research in the K.V.S. lab was supported by the National Institutes of Health Grant GM104071 and Russian Science Foundation grant 19-74-20130 to Sofia Medvedeva.
Supplementary Material
References
- 1. Barrangou R, Horvath P. A decade of discovery: CRISPR functions and applications. Nat Microbiol 2017;2:17092 DOI: 10.1038/nmicrobiol.2017.92. [DOI] [PubMed] [Google Scholar]
- 2. Mohanraju P, Makarova KS, Zetsche B, et al. Diverse evolutionary roots and mechanistic variations of the CRISPR-Cas systems. Science 2016;353:aad5147 DOI: 10.1126/science.aad5147. [DOI] [PubMed] [Google Scholar]
- 3. Hille F, Charpentier E. CRISPR-Cas: biology, mechanisms and relevance. Philos Trans R Soc Lond B Biol Sci 2016;371:20150496 DOI: 10.1098/rstb.2015.0496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hille F, Richter H, Wong SP, et al. The biology of CRISPR-Cas: backward and forward. Cell 2018;172:1239–1259. DOI: 10.1016/j.cell.2017.11.032. [DOI] [PubMed] [Google Scholar]
- 5. Makarova KS, Wolf YI, Koonin EV. The basic building blocks and evolution of CRISPR-cas systems. Biochem Soc Trans 2013;41:392–1400. DOI: 10.1042/BST20130038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Makarova KS, Wolf YI, Iranzo J, et al. Evolutionary classification of CRISPR-Cas systems: a burst of class 2 and derived variants. Nat Rev Microbiol 2020;18:67–83. DOI: 10.1038/s41579-019-0299-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Shmakov SA, Makarova KS, Wolf YI, et al. Systematic prediction of genes functionally linked to CRISPR-Cas systems by gene neighborhood analysis. Proc Natl Acad Sci U S A 2018;115:E5307–E5316. DOI: 10.1073/pnas.1803440115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Westra ER, Buckling A, Fineran PC. CRISPR-Cas systems: beyond adaptive immunity. Nat Rev Microbiol 2014;12:317–326. DOI: 10.1038/nrmicro3241. [DOI] [PubMed] [Google Scholar]
- 9. Faure G, Makarova KS, Koonin EV. CRISPR-Cas: complex functional networks and multiple roles beyond adaptive immunity. J Mol Biol 2019;431:3–20. DOI: 10.1016/j.jmb.2018.08.030. [DOI] [PubMed] [Google Scholar]
- 10. Peters JE, Makarova KS, Shmakov S, et al. Recruitment of CRISPR-Cas systems by Tn7-like transposons. Proc Natl Acad Sci U S A 2017;114:E7358–E7366. DOI: 10.1073/pnas.1709035114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Makarova KS, Karamycheva S, Shah SA, et al. Predicted highly derived class 1 CRISPR-Cas system in Haloarchaea containing diverged Cas5 and Cas7 homologs but no CRISPR array. FEMS Microbiol Lett 2019;366:fnz079 DOI: 10.1093/femsle/fnz079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Deng L, Garrett RA, Shah SA, et al. A novel interference mechanism by a type IIIB CRISPR-Cmr module in Sulfolobus. Mol Microbiol 2013;87:1088–1099. DOI: 10.1111/mmi.12152. [DOI] [PubMed] [Google Scholar]
- 13. Garrett RA, Vestergaard G, Shah SA. Archaeal CRISPR-based immune systems: exchangeable functional modules. Trends Microbiol 2011;19:549–556. DOI: 10.1016/j.tim.2011.08.002. [DOI] [PubMed] [Google Scholar]
- 14. Shah SA, Garrett RA. CRISPR/Cas and Cmr modules, mobility and evolution of adaptive immune systems. Res Microbiol 2011;162:27–38. DOI: 10.1016/j.resmic.2010.09.001. [DOI] [PubMed] [Google Scholar]
- 15. Bernheim A, Bikard D, Touchon M, et al. Atypical organizations and epistatic interactions of CRISPRs and cas clusters in genomes and their mobile genetic elements. Nucleic Acids Res 2020;48:748–760. DOI: 10.1093/nar/gkz1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pourcel C, Salvignol G ,Vergnaud G. CRISPR elements in Yersinia pestis acquire new repeats by preferential uptake of bacteriophage DNA, and provide additional tools for evolutionary studies. Microbiology 2005;151:653–663. DOI: 10.1099/mic.0.27437-0. [DOI] [PubMed] [Google Scholar]
- 17. Barrangou R, Fremaux C, Deveau H, et al. CRISPR provides acquired resistance against viruses in prokaryotes. Science 2007;315:1709–1712. DOI: 10.1126/science.1138140. [DOI] [PubMed] [Google Scholar]
- 18. Li M, Liu H, Han J, et al. Characterization of CRISPR RNA biogenesis and Cas6 cleavage-mediated inhibition of a provirus in the haloarchaeon Haloferax mediterranei. J Bacteriol 2013;195:867–875. DOI: 10.1128/JB.01688-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Soutourina OA, Monot M, Boudry P, et al. Genome-wide identification of regulatory RNAs in the human pathogen Clostridium difficile. PLoS Genet 2013;9:e1003493 DOI: 10.1371/journal.pgen.1003493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hou S, Brenes-Alvarez M, Reimann V, et al. CRISPR-Cas systems in multicellular cyanobacteria. RNA Biol 2019;16:518–529. DOI: 10.1080/15476286.2018.1493330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Reimann V, Ziemann M, Li H, et al. Specificities and functional coordination between the two Cas6 maturation endonucleases in Anabaena sp. PCC 7120 assign orphan CRISPR arrays to three groups. RNA Biol 2020;17:1442–1453. DOI: 10.1080/15476286.2020.1774197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hullahalli K, Rodrigues M, Schmidt BD, et al. Comparative analysis of the orphan CRISPR2 locus in 242 Enterococcus faecalis strains. PLoS One 2015;10:e0138890 DOI: 10.1371/journal.pone.0138890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Touchon M, Rocha EP. The small, slow and specialized CRISPR and anti-CRISPR of Escherichia and Salmonella. PLoS One 2010;5:e11126 DOI: 10.1371/journal.pone.0011126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Almendros C, Guzman NM, Garcia-Martinez J, et al. Anti-cas spacers in orphan CRISPR4 arrays prevent uptake of active CRISPR-Cas I-F systems. Nat Microbiol 2016;1:16081 DOI: 10.1038/nmicrobiol.2016.81. [DOI] [PubMed] [Google Scholar]
- 25. Faure G, Shmakov SA, Yan WX, et al. CRISPR-Cas in mobile genetic elements: counter-defence and beyond. Nat Rev Microbiol 2019;17:513–525. DOI: 10.1038/s41579-019-0204-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Medvedeva S, Liu Y, Koonin EV, et al. Virus-borne mini-CRISPR arrays are involved in interviral conflicts. Nat Commun 2019;10:5204 DOI: 10.1038/s41467-019-13205-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Coordinators NR. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res 2018;46:D8–D13. DOI: 10.1093/nar/gkx1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Steinegger M, Soding J. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat Biotechnol 2017;35:1026–1028. DOI: 10.1038/nbt.3988. [DOI] [PubMed] [Google Scholar]
- 29. Makarova KS, Wolf YI, Koonin EV. Archaeal clusters of orthologous genes (arCOGs): an update and application for analysis of shared features between Thermococcales, Methanococcales, and Methanobacteriales. Life (Basel) 2015;5:818–840. DOI: 10.3390/life5010818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Alkhnbashi OS, Shah SA, Garrett RA, et al. Characterizing leader sequences of CRISPR loci. Bioinformatics 2016;32:i576–i585. DOI: 10.1093/bioinformatics/btw454. [DOI] [PubMed] [Google Scholar]
- 31. Shmakov SA, Wolf YI, Savitskaya E, et al. Mapping CRISPR spaceromes reveals vast host-specific viromes of prokaryotes. Commun Biol 2020;3:321 DOI: 10.1038/s42003-020-1014-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Marchler-Bauer A, Zheng C, Chitsaz F, et al. CDD: conserved domains and protein three-dimensional structure. Nucleic Acids Res 2013;41:D348–352. DOI: 10.1093/nar/gks1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Altschul SF, Madden TL, Schaffer AA, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 1997;25:3389–3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Morgulis A, Coulouris G, Raytselis Y, et al. Database indexing for production MegaBLAST searches. Bioinformatics 2008;24:1757–1764. DOI: 10.1093/bioinformatics/btn322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Shmakov SA, Sitnik V, Makarova KS, et al. The CRISPR spacer space is dominated by sequences from species-specific mobilomes. MBio 2017;8 DOI: 10.1128/mBio.01397-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Database Resources of the National Center for Biotechnology Information. Nucleic Acids Res 2018;46:D8–D13. DOI: 10.1093/nar/gkx1095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Galperin MY, Makarova KS, Wolf YI, et al. Expanded microbial genome coverage and improved protein family annotation in the COG database. Nucleic Acids Res 2015;43:D261–269. DOI: 10.1093/nar/gku1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Shmakov SA, Faure G, Makarova KS, et al. Systematic prediction of functionally linked genes in bacterial and archaeal genomes. Nat Protoc 2019;14:3013–3031. DOI: 10.1038/s41596-019-0211-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kristensen DM, Wolf YI, Koonin EV. ATGC database and ATGC-COGs: an updated resource for micro- and macro-evolutionary studies of prokaryotic genomes and protein family annotation. Nucleic Acids Res 2017;45:D210–D218. DOI: 10.1093/nar/gkw934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Huerta-Cepas J, Serra F, Bork P. ETE 3: reconstruction, analysis, and visualization of phylogenomic data. Mol Biol Evol 2016;33:1635–1638. DOI: 10.1093/molbev/msw046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 2013;30:772–780. DOI: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rajewska M, Wegrzyn K, Konieczny I. AT-rich region and repeated sequences—the essential elements of replication origins of bacterial replicons. FEMS Microbiol Rev 2012;36:408–434. DOI: 10.1111/j.1574-6976.2011.00300.x. [DOI] [PubMed] [Google Scholar]
- 43. Wu Z, Liu J, Yang H, et al. DNA replication origins in archaea. Front Microbiol 2014;5:179 DOI: 10.3389/fmicb.2014.00179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Larsen J, Andersen PS, Winstel V, et al. Staphylococcus aureus CC395 harbours a novel composite staphylococcal cassette chromosome mec element. J Antimicrob Chemother 2017;72:1002–1005. DOI: 10.1093/jac/dkw544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. O'Connor AM, McManus BA, Coleman DC. First description of novel arginine catabolic mobile elements (ACMEs) types IV and V harboring a kdp operon in Staphylococcus epidermidis characterized by whole genome sequencing. Infect Genet Evol 2018;61:60–66. DOI: 10.1016/j.meegid.2018.03.012 [DOI] [PubMed] [Google Scholar]
- 46. Hargreaves KR, Flores CO, Lawley TD, et al. Abundant and diverse clustered regularly interspaced short palindromic repeat spacers in Clostridium difficile strains and prophages target multiple phage types within this pathogen. mBio 2014;5:e01045-01013. DOI: 10.1128/mBio.01045-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hoikkala V, Ravantti J, Diez-Villasenor C, et al. Cooperation between CRISPR-Cas types enables adaptation in an RNA-targeting system. bioRxiv 2020. Feb 20 [Epub ahead of print]; DOI: 10.1101/2020.02.20.957498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Nivala J, Shipman SL, Church GM. Spontaneous CRISPR loci generation in vivo by non-canonical spacer integration. Nat Microbiol 2018;3:310–318. DOI: 10.1038/s41564-017-0097-z [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.