

Abstract
Bone tissue engineering strategies have been developed to address the limitations of the current gold standard treatment options for bone-related disorders. These systems consist of an engineered scaffold that mimics the extracellular matrix and provides an architecture to guide the natural bone regeneration process, and incorporated growth factors that enhance cell recruitment and ingress into the scaffold and promote the osteogenic differentiation of stem cells and angiogenesis. In particular, the osteogenic growth factor bone morphogenetic protein 2 (BMP-2) has been widely studied as a potent agent to improve bone regeneration. A key challenge in growth factor delivery is that the growth factors must reach their target sites without losing bioactivity and remain in the location for an extended period to effectively aid in the formation of new bone. Protein incorporation into nanoparticles can both protect protein bioactivity and enable its sustained release. In this study, a poly(methyl methacrylate-co-methacrylic acid) nanoparticle-based system was synthesized incorporating a custom poly(ethylene glycol) dimethacrylate crosslinker. It was demonstrated that the nanoparticle degradation rate can be controlled by tuning the number of hydrolytically degradable ester units along the crosslinker. We also showed that the nanoparticles had high affinity for a model protein for BMP-2, and optimal conditions for maximum protein loading efficiency were elucidated. Ultimately, the proposed system and its high degree of tunability can be applied to a wide range of growth factors and tissue engineering applications.
Impact Statement
In this study, we developed a novel method of synthesizing nanoparticles with tunable degradation rates through the incorporation of a custom synthesized, hydrolytically degradable crosslinker. In addition, we demonstrated the affinity of the synthesized nanoparticles for a model protein for bone morphogenetic protein 2 (BMP-2). The tunability of these nanoparticles can be used to develop complex tissue engineering systems, for example, for the delivery of multiple growth factors involved at different stages of the bone regeneration process.
Keywords: growth factor delivery, bone regeneration, nanocarriers, tunable degradation, controlled release
Introduction
The increasing occurrence of bone-related defects owing to an increasingly active and aging population poses a significant burden on our societies. The currently available approaches to the repair of critical size bone defects include bone auto- and allografts. However, these solutions suffer from significant drawbacks such as multiple surgery sites, limited source, and the possibility of infection for the former, and disease transmission and donor shortage for the latter.
Bone tissue engineering approaches seek to address these limitations with the use of synthetic grafts based on biodegradable and biocompatible materials such as metal alloys, ceramics, natural or synthetic polymers, and their composites.1–3 Strategies to improve cell response to scaffolds have included surface modifications, as reviewed by Richbourg et al.,4 and the use of molecular recognition.5
Tissue regeneration can be further improved by delivery of the bioactive molecules involved in the natural bone healing process.6 Focus has been directed toward bone morphogenetic protein 2 (BMP-2), which belongs to the family of bone morphogenetic proteins (BMPs) and is considered to be a key osteogenic growth factor widely used in bone tissue regeneration. In fact, the Food and Drug Administration (FDA) first approved its clinical use in bone grafts in 2002.7
Growth factor (GF) delivery for tissue regeneration poses some challenges including the short in vivo half-life of growth factors, particularly of BMP-2,8 and the fact that bone formation relies on the highly coordinated action of multiple growth factors. Encapsulating growth factors in nanoparticles (NPs) have been widely explored as a solution to both protect the payload and prevent the loss of bioactivity and to achieve controlled release profiles.9 In fact, embedding nanoparticles within tissue engineering scaffolds can provide additional benefits to the tissue engineering system as reviewed by Elkhoury et al.10 Furthermore, carrier degradation-mediated release has been explored as a mechanism to tune and obtain the desired growth factor release profile.
In this study, we used a new method to fabricate degradable poly(methyl methacrylate-co-methacrylic acid) (P(MMA-co-MAA)) nanoparticles optimized for the controlled delivery of BMP-2 (Fig. 1). Poly(methyl methacrylate) (PMMA) presented advantages because of its ease of processing and low cost, and has previously been used as a biomaterial in bone tissue engineering systems.11 The hydrophobic nature of PMMA has the potential to increase the affinity of nanoparticles for BMP-2, which presents a hydrophobic exterior surface in vivo. This explains its limited solubility in physiological conditions. In addition, methacrylic acid (MAA) was incorporated into the nanoparticle backbone to introduce reactive carboxyl groups and a negative surface charge. This further increases affinity to BMP-2, and the possibility to bind the particles to a scaffold backbone.
FIG. 1.

The design of PMMA-co-MAA nanoparticles with tunable degradation rates for the controlled, sustained delivery of growth factors in bone tissue engineering applications. P(MMA-co-MAA), poly(methyl methacrylate)-co-methacrylic acid. Color images are available online.
P(MMA-co-MAA) particles were synthesized using a one-pot ultraviolet (UV)-initiated emulsion polymerization scheme and through the incorporation of a poly(ethylene glycol) dimethacrylate (PEGDMA) crosslinker. Subsequently, custom degradable crosslinkers were synthesized by the addition of labile ester bonds in the crosslinker chain. Increasing the number of ester units is believed to increase the hydrolytic degradability of poly(ethylene glycol) (PEG)-based hydrogels and could, therefore, be used to tune the rate of protein release from the particles into the scaffold environment.
Trypsin was used as a model for BMP-2 and the nanoparticles were evaluated for their ability to efficiently load protein. Using a statistical software package, loading conditions were optimized to achieve the maximum protein loading efficiency of the system.
Methods
Materials
Methyl methacrylate (MMA, 99%, Catalog M55909) and MAA (99%, Catalog 155721) were obtained from Sigma-Aldrich Corp. (St. Louis, MO). Polyethylene glycol dimethacrylate with PEG molecular weight (MW) 600 (PEGDMA 600, Catalog 02364) was obtained from Polysciences, Inc. (Warrington, PA). The initiator Irgacure 2959 was obtained from Ciba, Inc. (Basel, Switzerland), whereas the surfactants Brij 30 and myristyl trimethyl ammonium bromide (MyTAB) were obtained from Thermo Fisher Scientific (Waltham, MA), and Sigma-Aldrich, respectively.
d,l-Lactide (3,6-dimethyl-1,4-dioxane-2,5-dione), PEG MW = 400, PEG MW = 200, and methacrylic anhydride (containing 2000 ppm topanol A as inhibitor, 94%) were obtained from Sigma-Aldrich. Hexanes, diethyl ether (99+%, pure, stabilized with BHT), and N,N-dimethylformamide were obtained from Thermo Fisher Scientific, and dichloromethane was obtained from Sigma-Aldrich.
Trypsin from bovine pancreas (powder, Catalog T9201) was obtained from Sigma-Aldrich. Human umbilical vein endothelial cells (HUVECs) (ATCC CRL-1730) and F-12K medium (Kaighn's Modification of Ham's F-12 medium, ATCC 30-2004) were obtained from ATCC (Manassas, VA). Heparin sodium (Catalog AC411210010) and fetal bovine serum (Catalog 35010CV; Corning) were obtained from Thermo Fisher Scientific. Endothelial cell growth supplement from bovine neural tissue (Catalog E2759; ECGS) was obtained from Sigma-Aldrich.
Synthesis of degradable crosslinkers
To fabricate hydrolytically degradable nanoparticles, labile ester groups were incorporated along a PEGDMA crosslinker chain. Custom methacrylate-poly(lactic acid)-b-poly(ethylene glycol)-b-poly(lactic acid)-methacrylate (MA-PLA-b-PEG-b-PLA-MA) crosslinkers were synthesized according to a two-step synthesis originally developed by Hubbell and colleagues12 and adapted from Diederich et al.13 (Fig. 2).
FIG. 2.

Two-step synthesis of the custom MA-PLA-b-PEG-b-PLA-MA crosslinker. (A) The ring opening polymerization of d,l-lactide on PEG using SnOct2 as a catalyst and carried out at 140°C. (B) The dimethacrylation of PLA-b-PEG-b-PLA intermediate using 10-fold excess of methacrylic anhydride. MA-PLA-b-PEG-b-PLA-MA, methacrylate-poly(lactic acid)-b-poly(ethylene glycol)-b-poly(lactic acid)-methacrylate; PEG, poly(ethylene glycol); SnOct2, stannous octoate.
Ring opening polymerization of d,l-lactide on PEG
PEG-co-poly(lactic acid) (PLA) block copolymer intermediates with varying units of lactic acid were synthesized using a ring-opening polymerization of d,l-lactide on PEG (Fig. 2A). Diederich et al. reported a conversion of 70% for the incorporation of lactic acid units along the PEG chain.13 Therefore, to load 4 and 8 lactic acid units per PEG chain, 5.6 and 11.2 units of lactic acid were loaded, respectively. In addition, to maintain an approximately constant crosslinker length, PEG with a lower molecular weight was used for the higher numbers of lactic acid units. The amount of d,l-lactide loaded and the corresponding molecular weights of the PEG used are given in Table 1.
Table 1.
The Ring Opening Polymerization of d,l-Lactide on Poly(Ethylene Glycol)
| Target LA units | Loaded LA units | PEG MW | mol PEG | mol d,l-Lactide | Actual m | Conversion |
|---|---|---|---|---|---|---|
| m = 4 | 5.6 | 400 | 0.0125 | 0.035 | 4.176 | 75% |
| m = 8 | 11.2 | 200 | 0.01 | 0.056 | 10.496 | 94% |
The target lactic acid units, the moles of the reagents used, and the final conversions.
LA, lactic acid; MW, molecular weight; PEG, poly(ethylene glycol).
To load 4 lactic acid units per PEG chain (m = 4), 0.0125 mol PEG MW = 400 (5 g) and 0.035 mol d,l-lactide (5.045 g) were combined in a 50 mL round bottom flask. The solution was purged with nitrogen for 20 min to remove oxidizing species. The round bottom flask was then placed in an oil bath and heated to 140°C. Finally, 0.681 μL of a nitrogen purged stock solution of 10 wt% SnOct2 in toluene was added to achieve a ratio of 0.0012 mol SnOct2/mol d,l-lactide. After 12 h, the reaction was stopped by removing the round bottom flask from the oil bath and the product was allowed to cool to 30°C. Before solidification, the product was dissolved in 7 mL of dichloromethane. To purify the obtained intermediate, the dissolved product was reprecipitated in 400 mL diethyl ether at −80°C for 6 h and redissolved in 7 mL of dichloromethane. After a second precipitation cycle, the intermediate was vacuum-dried overnight at 40°C and 28 mmHg, and subsequently stored at 4°C. The intermediate product was characterized using proton (1H) and carbon (13C) nuclear magnetic resonance (NMR) in deuterochloroform CDCl3 and by Fourier transform infrared (FTIR) spectroscopy.
Dimethacrylation of PLA-b-PEG-b-PLA intermediate
After the synthesis and characterization of the PLA-b-PEG-b-PLA intermediate, a dimethacrylation reaction was carried out (Fig. 2B). The dried intermediate and a 10 M excess of methacrylic anhydride were introduced in a 20 mL capped scintillation vial. The vial was placed in a domestic microwave with the cap loosely twisted. The microwave was set to 5 min at the maximum power. Every 30 s, the vial was removed from the microwave with the cap tightened, and vortexed for 30 s. The cap was then loosened again, and the vial placed back into the microwave. These steps were repeated until the vial was microwaved for a total of 5 min. The vial was then removed from the microwave and allowed to cool to room temperature with the cap loosened. The cooled methacrylated product was dissolved in 3 mL of dichloromethane and precipitated in 400 mL of hexanes at room temperature for 6 h. The precipitated product was collected by vacuum filtration using a Buckner funnel and redissolved in dichloromethane. After a second precipitation step, the product was dried overnight in a vacuum chamber and stored at 4°C. The methacrylated custom crosslinkers were characterized using 1H and 13C NMR in CDCl3 and FTIR spectroscopy.
Synthesis of degradable nanoparticles
The synthesized custom MA-PLA-b-PEG-b-PLA-MA crosslinkers with varying numbers of lactic acid units per chain (m = 4 and m = 10) were incorporated into a UV-initiated emulsion polymerization scheme to fabricate P(MMA-co-MAA) nanoparticles with varying degrees of degradability.14
The prepolymerization mixture consisted of 95 mol% MMA, 4 mol% methacrylic acid, and 1 mol% crosslinker (commercial PEGDMA MW = 600 for the nondegradable m = 0 nanoparticles, and the custom MA-PLA-b-PEG-b-PLA-MA m = 4 and m = 10 crosslinkers for the degradable nanoparticles) in a round bottom flask. Irgacure 2925, a free radical initiator, was added at a ratio of 0.5 wt%. Then, 25.0 mL of deionized water was added. To form an emulsion, Brij 30, a nonionic surfactant, and MyTAB, a cationic surfactant, were added to the aqueous solution at concentrations of 4 and 1.16 mg/mL, respectively. An emulsion was formed by ultrasonication for 20 min and was then purged with nitrogen to eliminate free radical scavengers. The emulsion was then allowed to polymerize by placing the flask under a UV point source with an intensity of 140 mW/cm2 for 2.5 h (BlueWave 200 Spot Lamp System; Dymax Corp., Torrington, CT). The synthesized nanoparticles were precipitated 1:1 in a 6N HCl solution and then further diluted 1:10 in 1 × Dulbecco's phosphate-buffered saline (DPBS). The formed nanoparticle pellet was resuspended in 1 × DPBS and adjusted to a pH of 7.
The synthesized particles were characterized by dynamic light scattering (Zetasizer Nano; Malvern) using 5 × diluted nanoparticle samples in 1 × DPBS (n = 3 per formulation). In addition, the composition of particles with varying number of lactic acid units was compared with the PEGDMA-crosslinked nanoparticles using FTIR spectroscopy.
Degradation of nanoparticles
Accelerated hydrolytic conditions were used for all degradation experiments to correlate the nanoparticle behavior in experimental conditions to their behavior in vivo. Cosgriff-Hernandez and colleagues15 established a correlation between the rate of ester hydrolysis in basic conditions and the corresponding hydrolysis rate in vivo, concluding that the degradation behavior occurring over 1 day at pH 10 and at 37°C in deionized water was equivalent to degradation over 3 weeks in physiological conditions. All experiments were, therefore, carried out in deionized water or deuterium oxide (D2O) at 37°C and maintained at a constant pH of 10.
Nanoparticles count rate studies
Particle degradation was first analyzed with dynamic light scattering studies. The derived count rate corresponds to the theoretical count rate that would be obtained at 100% laser power with zero attenuator and can be used to compare the signal strength between samples. For the present system, a decrease in the count rate can be linked to the loss of signal associated with particle degradation. Particle degradation profiles were evaluated under accelerated hydrolytic conditions at 37°C using dynamic light scattering by measuring the change in the particle size and count rate over time (Zetasizer Nano; Malvern). Nanoparticles with varying degrees of labile ester bonds along the crosslinker (m = 0, 4, 10) were suspended in deionized water at a concentration of 4 mg/mL. Accelerated hydrolytic conditions were achieved by conducting the experiment in an oil bath at 37°C and under alkaline conditions using an autotitrator (MPT-2 Autotitrator; Malvern) to maintain a pH value of 10 for the duration of the degradation experiment. At increasing time points throughout the experiment, samples were analyzed by dynamic light scattering. Because of the aggregation of degradation byproducts, both unfiltered and 0.2 μm filtered samples were analyzed to obtain the count rate for the entire solution and only the nondegraded particles, respectively.
Kinetic nuclear magnetic resonance studies
The nanoparticle degradation mechanism relies on the hydrolysis of the ester bonds within PLA units along the crosslinker chain that produces a lactic acid byproduct. The degradation of the P(MMA-co-MAA) nanoparticles can, thus, be observed through 1H and 13C NMR by analyzing the presence of characteristic peaks for lactic acid in the degradation products, specifically by integrating the doublet centered at 1.21 ppm (range: 1.20–1.22 ppm) characteristic of lactic acid and the quadruplet centered at 1.335 ppm (range: 1.32–1.35 ppm), which corresponds to oligo-lactic acid (OLA).
The particle degradation kinetics were obtained by analyzing the change in the NMR spectra of a degrading nanoparticle sample over time. Solutions of nanoparticles (50 mg/mL) with varying degrees of degradable units on the crosslinker (m = 0, 4, 10) were prepared in a solution of 0.03 v/v% trimethylsilanol (TMS) in deuterated water (D2O) and maintained in accelerated hydrolytic conditions (37°C, pH 10). At given time points (t = 0, 1, 2, 4, 7, 11, 18, 24, 48 h), a sample was obtained from the degradation solution and was analyzed by 1H NMR.
The obtained spectra were analyzed by integrating the TMS peak (0 ppm) and the range of the spectrum, which includes characteristic peaks corresponding to the degradation products (1.55–0.5 ppm). This range was selected to include, in addition to the characteristic peaks for lactic acid and OLA, peaks that correspond to the methyl protons along the P(MMA-co-MAA) backbone at 0.8 ppm and 1.18 ppm. Since these peaks (after Fourier transform) result in a significant amount of drift from the baseline and variability in the degree of baseline drift from sample to sample, integration over this range allows for the normalization of the obtained values. The values obtained by integration were first normalized to the TMS peak. These normalized integral values were then normalized to the maximum peaks at 48 h, and the percent change in peak size was plotted as a function of time, as an indication of the change in degraded mass over time.
Zeta potential studies
To further characterize the mechanism of degradation and the properties of the degraded nanoparticles, the zeta potentials of the nanoparticles before and after degradation were measured (Zetasizer Nano; Malvern) at increasing pH values (n = 3 for each pH value). The changes in the particle zeta potentials with respect to pH were compared between the nondegradable particles and degradable particles before and after degradation.
Imaging of nanoparticles
The morphology of the degraded nanoparticles was determined by imaging the particles using transmission electron microscopy (FEI Tecnai Transmission Electron Microscope; Thermo Fischer Scientific). Solutions of m = 0, 4, 10 nanoparticles both before and after degradation were prepared at 1 mg/mL in distilled water. The samples were negatively stained using 2% phosphotungstic (PTA) at pH 7.0 and prepared on thin bar hexagonal mesh standard thickness, formvar-coated copper grids with 600 mesh. The particles were introduced on the grid for 20 s and then wicked dry, followed by 20 s of incubation with the stain.
Protein loading study
Selection of loading variables
Multiple loading conditions such as the temperature, pH, buffer strength, concentrations, and incubation time have significant effects on both the protein and particle characteristics and could, therefore, have a significant effect during protein loading into nanoparticles. In this study, four key factors were evaluated to determine the optimal loading conditions to maximize protein loading efficiency. First, the pH was varied between 4.5, 6.0, and 7.5. Second, the protein to particle ratio was varied between 3, 6.5, and 10 wt% (mg protein to 100 mg nanoparticles). In addition, protein was incubated for longer times ranging from 2 to 48 h. Finally, the total ionic strength of the loading buffer was varied between 5, 15.4, and 154 mM.
Experimental design
Measuring the loading efficiency for the four selected variables at three levels each and three replicates per condition requires 81 distinct runs and a total of 243 experiments (referred to as 3 × 3 × 3 × 3 full-factorial design). A statistical software was used for a design of experiment analysis (JMP; SAS Institute), to develop a robust model for protein loading ability while minimizing the number of experimental runs required to achieve statistical power. The 3 × 3 × 3 × 3 factorial design was compared with four distinct experimental designs in terms of statistical power per term and per number of runs (Supplementary Fig. S1). The Classical Screening design maintains relatively high predictive power for the different variables including two-factor interactions while requiring only 16 runs and was therefore selected. In addition, to further improve the statistical power, additional runs were included to account for any quadratic terms 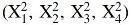 and to provide curvature. Ultimately, 23 loading conditions were tested (Supplementary Table S1).
and to provide curvature. Ultimately, 23 loading conditions were tested (Supplementary Table S1).
Protein loading experiment
BMP-2 has a molecular weight of ∼30 kDa and an isoelectric point >8.5.16 In this study, trypsin was selected as a good model for BMP-2, as it has a similar size of 23.3 kDa, an isoelectric point of 10.8, and similar dimensions and comparable stability behavior, with both proteins being most stable ∼pH 3. P(MMA-co-MAA) particles synthesized with a PEGDMA crosslinker were used for these loading experiments.
Samples for the protein loading study were prepared (Fig. 3) following the formulations outlined in Supplementary Table S1 by combining the different ratios of the protein to the polymer in different buffers and at different pH conditions. The samples were placed on a plate shaker at room temperature and allowed to incubate for the required incubation time (2, 24, 48, and 72 h.) At these respective time points, the corresponding samples were removed from the plate shaker and transferred to a 96-well filter plate (AcroPrep Advance 96 Filter Plate, 100 kDa Omega; Pall.) The membrane cutoff size of 100 kDa was selected such that the particles with bound protein would not be able to traverse the membrane, collecting only the unbound protein. Each loading condition was plated into three wells of the filter plate.
FIG. 3.

The experimental setup for protein loading experiment. (A) The samples were prepared according to the run description provided by JMP, (B) incubated on a plate shaker at room temperature, (C) pipetted onto a filter plate and centrifuged, then (D) the filtrate was pipetted onto a UV plate and the absorbance spectra were obtained, (E) a micro BCA reagent was added to each well of the UV plate, incubated, and absorbance spectra were obtained. BCA, bicinchoninic acid; UV, ultraviolet. Color images are available online.
The plate was centrifuged at 2000 g for 20 min, and the collected samples were transferred to a 96-well UV plate. The UV absorption and micro-bicinchoninic acid (BCA) analyses (n = 9 per loading condition) were carried out using a microplate reader (Cytation 3 Cell Imaging Multi-Mode Reader; BioTek). The UV absorbance values for native trypsin were obtained at 280 nm and 405 nm. Micro BCA analysis was carried out by adding micro-BCA reagent to each well at a 1:1 ratio and incubating at 37°C for 2 h. The absorbance values at 562 nm were obtained to detect the BCA/Cu1+ complex formed between the reagent and the protein present in the samples. The obtained absorption values were then correlated to the values of trypsin concentration using the appropriate calibration curves. The calibration curves for both UV absorption, used for higher concentrations, and micro-BCA, used for the lower concentration range, are given in Figure 4. The measured concentrations correspond to the amount of protein that was not loaded into the nanoparticles and were subtracted from the loaded concentrations to determine the amounts of the loaded protein for each condition.
FIG. 4.

Trypsin concentration calibration curves for (A) UV absorbance at 280 nm and (B) micro-BCA absorbance at 562 nm in 154 mM buffer (blue), 15.4 mM buffer (orange), and 5 mM buffer (gray). Color images are available online.
In vitro studies: cell viability and proliferation
Cell culture
Nonimmortalized HUVECs obtained from ATCC were used to determine the biocompatibility of the degradable nanoparticles in both the nondegraded and fully degraded states. Cells were propagated for two passages using an F-12K medium with 0.1 mg/mL heparin, 1 v/v% endothelial cell growth supplement, and 10 v/v/% fetal bovine serum. Cell culture-treated 96-well plates were coated with 50 μL of 1:100 fibronectin solution in 1 × DPBS and incubated at room temperature for 1 h. The fibronectin solution was then removed from the plate and the wells were rinsed with DPBS. HUVECs were then seeded at 3000 cells per well and were allowed to incubate at room temperature for 1 h. The cells were then incubated for 24 h in a humidified environment at 37°C and 5% CO2.
Solutions of P(MMA-co-MAA) nanoparticles with m = 0, m = 4, and m = 10 crosslinkers and solutions of degraded nanoparticles were prepared. These solutions were diluted in HUVEC media, and 5 μL was added to each well to achieve final concentrations of 0.01, 0.02, 0.5, and 1 mg/mL. Positive and negative controls were prepared by incubating the cells in HUVEC media (positive control) and with both triton and SDS (negative lysis controls). All systems were subsequently cultured for 24 and 48 h in a humidified environment at 37°C and 5% CO2.
MTS cell proliferation assay
Cell proliferation in the presence of the system components after 24 and 48 h was determined using a 3-(4, 5-dimethylthiazol-2-yl)-5 (3-carboxymethonyphenol)-2-(4-sulfophenyl)-2H- tetrazolium (MTS) cellular proliferation assay and used as an indicator of cell health. The CellTiter 96 AQueous One Solution Cell Proliferation Assay (Promega Corp., Madison, WI) was used per manufacturer's instructions, and the resulting absorbance of the plate at 490 and 690 nm was obtained. The relative cellular proliferation was calculated by first subtracting the background absorbance at 690 nm from the absorbance at 490 nm. Subsequently, the obtained values were normalized to the average absorbance of the positive and negative controls (i.e., HUVECs in the HUVEC media and lysis control, respectively) to obtain the average cell proliferation for each condition (n = 6 per condition.)
Lactose dehydrogenase membrane integrity assay
The integrity of the cell membranes in the presence of the system components after 24 and 48 h was determined using a lactose dehydrogenase (LDH) membrane integrity assay as an indicator of cell health. The Promega CytoTox-ONE homogeneous membrane integrity assay (Promega Corp.) was used per manufacturer's instructions. The resulting fluorescence of the samples was measured at an excitation of 560 nm and an emission of 590 nm. The background fluorescence values as a result of the cell culture media were first subtracted from the obtained fluorescence values. These were then normalized to the average maximum LDH release (lysis control) and minimum LDH release (media control) to obtain the average cell viability for each condition (n = 6 per condition).
Results
Nanoparticle-based delivery systems for delivery of growth factors in bone scaffolds were prepared from MMA, methacrylic acid, and a custom MA-PLA-b-PEG-b-PLA-MA crosslinker. We analyze first the nanoparticle structures and their properties, followed by their degradation behavior, ability to load a model protein for BMP-2, and their effect on cell viability and proliferation.
Synthesis of degradable nanoparticles
The custom MA-PLA-b-PEG-b-PLA-MA crosslinkers synthesized according to the procedure in Synthesis of Degradable Crosslinkers section were characterized using 1-H NMR (Fig. 5) to confirm the successful incorporation of PLA units and methacrylation of the end groups of the chains. The PEGDMA 600 MW crosslinker used for the nondegradable particles presented a tall peak at 3.6 ppm (peak e) characteristic of the PEG units.13 This spectrum additionally revealed three characteristic peaks for the two methacrylate ends observed at 1.8, 5.7, and 6.2 ppm (peaks f, b, and a, respectively). Finally, a peak is observed at 4.25 ppm (peak d), which is characteristic of the presence of a carbon bond to the PEG repeating units. Of note, this peak did not appear on the spectrum of the nonmethacrylated PEG, suggesting that this peak appears because of the bond between the repeating unit and the methacrylate ends. The spectra of the m = 4 and m = 10 custom degradable crosslinkers presented these same five peaks with additional peaks at 1.5 ppm (peak g) and 5.2 ppm (peak c), characteristic of the lactic acid units.
FIG. 5.

A comparison of the 1H-NMR spectra for a PEGDMA MW = 600 crosslinker without incorporated degradable units, the custom crosslinker with 4 lactic acid units incorporated per PEG MW = 400 chain, and the custom crosslinker with 10 lactic acid units incorporated per PEG MW = 200 chain. MW, molecular weight; NMR, nuclear magnetic resonance; PEGDMA, poly(ethylene glycol) dimethacrylate. Color images are available online.
From the NMR spectra, a decrease in the height of the PEG peak (peak e) can be observed as the number of degradable units on the crosslinker is increased. This is attributed to the selection and use of 400 and 200 MW PEG for the m = 4 and m = 10 crosslinkers, respectively, to maintain a constant crosslinker length. In addition, the areas under the peaks c and g increased between m = 4 and m = 10 because of the presence of a greater number of lactic acid units. Finally, peaks a and b that correspond to the methacrylate ends of the polymer chains remained similar for all three crosslinker formulations. This is because of the fact that each crosslinker chain, regardless of the number of the lactic acid units or the length of the PEG chain, presents two methacrylate groups, one on each end. Finally, the quantitative analysis of the NMR spectra confirmed the presence of two methacrylate groups per chain, an average of 4.2 lactic acid units per chain for m = 4, and 10.5 lactic acid units per chain for m = 10.
The nanoparticles fabricated with the custom crosslinkers presented similar hydrodynamic diameters to those fabricated with the nondegradable PEGDMA 600 MW (Fig. 6) with average diameters ranging between 57 and 86 nm compared with 65 nm for the nondegradable particles. Batch-to-batch variability remained small, although an increase in the polydispersity indices to ∼0.200 and 0.300 was observed for nanoparticles fabricated with the m = 4 and m = 10 custom crosslinkers, respectively.
FIG. 6.

The dynamic light scattering results of P(MMA-co-MAA) nanoparticles synthesized using a one-pot UV-initiated emulsion polymerization using either: a PEGDMA MW = 600 crosslinker (m = 0), a MA-PLA-b-PEG-b-PLA-MA custom crosslinker with four lactic acid units (m = 4). (A), and summary of average diameter and PDI for each condition (B). PDI, polydispersity index. Color images are available online.
A comparison of the FTIR spectra of the degradable nanoparticles with those of the nondegradable nanoparticles confirms the successful incorporation of the custom crosslinkers (Fig. 7). When comparing the spectra for an increasing number of incorporated lactic acid units, an increase is observed in the peaks at ∼1750 cm−1 that correspond to the carbonyl stretching of the PLA units in the crosslinker. In addition, peaks at ∼1450 and 1350 cm−1 were observed that correspond to the asymmetric and symmetric bending of the methyl groups of the PLA units, respectively. Finally, a peak was identified at ∼1180 cm−1 that corresponds to the C-O stretching of the PLA units. An increase in these peaks for increased number of lactic acid units incorporated in the custom crosslinkers confirms the successful incorporation of these degradable crosslinkers in the synthesis of nanoparticles.
FIG. 7.

The FTIR spectra of nanoparticles fabricated with nondegradable crosslinker (m = 0) and custom degradable crosslinkers with 4 and 10 lactic acid units per chain (m = 4, and m = 10, respectively). FTIR, Fourier transform infrared. Color images are available online.
Degradation of nanoparticles
As demonstrated by the change in relative count rate over time (Fig. 8), nanoparticles fabricated with the custom crosslinkers exhibited a degradation behavior when maintained at pH 10 and 37°C. Indeed, in the first 24 h, a significant reduction in the count rate was observed, with the count rate reduced by half within 4.5 h for the particles fabricated with the m = 4 crosslinker and within 1.5 h for the particles fabricated with the m = 10 crosslinker. In addition, both particle formulations continued to see a decrease in the count rate over time and reached a plateau after ∼48 h for m = 4 and 28 h for m = 10. Overall, a higher rate of reduction in the count rate was observed for the m = 10 nanoparticles. The nondegradable nanoparticles did not exhibit this decrease in the count rate as a function of time and, in fact, showed a slight increase in the count rate after 6 h followed by a plateau, which can be explained by an initial swelling behavior owing to a delayed hydration of the nanoparticles.
FIG. 8.

The plot of the relative count rate as a function of the degradation time for nanoparticles with a nondegradable crosslinker (m = 0, black), m = 4 crosslinker (red), and m = 10 crosslinker (blue) normalized to the initial and final count rate values. Color images are available online.
Degraded nanoparticles showed significant change in the charge behavior as a function of pH (Fig. 9). Both the degraded m = 4 and m = 10 particles exhibited different zeta potential trends as a function of pH compared with the nondegradable particles after being subjected to accelerated degradation conditions for 48 h. The degraded particles began to present negative surface charges at lower pH values, with the observed pKa value being shifted from 4.5 for m = 0 to ∼2.5 for m = 10. The hydrolytic degradation of PLA units results in the formation of acidic byproducts,17 which can explain the onset of the negative charges in the lower pH ranges for the degraded nanoparticles.
FIG. 9.

The zeta potential as a function of pH at 25°C for the degraded nanoparticles with an increasing number of degradable units along the crosslinker chain. Color images are available online.
A change in the nanoparticle morphology was also observed between the initial nondegraded nanoparticles and the nanoparticles subjected to the accelerated degradation conditions. The m = 0, m = 4, and m = 10 nanoparticles all presented similar morphologies in their initial state, as indicated by the white spheres present in the top images of Figure 10. After incubation at 37°C and pH 10, the m = 0 nanoparticles showed no significant change in their morphology. However, a reduction in the number of visible nanoparticles was observed for m = 4 and m = 10, with the appearance of large aggregates of linear polymer and degradation byproducts. In addition, when comparing the degraded m = 4 and m = 10 particles, the m = 4 sample showed the presence of a small number of intact particles, whereas the intact particles for m = 10 were no longer detectable.
FIG. 10.

The transmission electron microscope images of m = 0, m = 4 and m = 10 nanoparticles before and after degradation. Scale bars: 200 μm. Some examples of the intact particles are indicated by dotted lines, whereas the degraded polymer is indicated with arrow. Color images are available online.
Furthermore, the NMR spectra of the nanoparticles subjected to accelerated degradation conditions obtained at different time points throughout the degradation process revealed an increase in the degradation products over time (Fig. 11A). The spectra of the m = 0 nanoparticles showed a minimal change over time with a slight increase in the 1.18 ppm peak over time. This peak increase can be explained by the slow degradation of the ether bonds along the PEG units, which may also appear at the 1.18 ppm peak. On the contrary, the spectra for the m = 4 and m = 10 nanoparticles revealed a distinct change over time, with the appearance of a quadruplet ∼1.33 ppm and a doublet at 1.20 and 1.22 ppm and their visible increase in the height as a function of time. It could also be qualitatively observed that the increase in these peaks is more rapid and significant for the m = 10 nanoparticles. A quantitative analysis of these spectra (Fig. 11B) revealed this difference in the degradation rate. We found that the m = 4 nanoparticles reached 50% degradation after 11 h and 100% degradation after 48 h, whereas the m = 10 nanoparticles reached 50% degradation after ∼6 h and 100% degradation after 24 h. In contrast, the slight degradation behavior observed for the m = 0 nanoparticles occurred on an entirely different time scale, with <10% degradation after 48 h. Comparing these experimentally obtained degradation profiles carried out in the accelerated degradation conditions to the corresponding behavior in vivo as defined by Cosgriff-Hernandez and colleagues,15 it can be estimated that the m = 4 nanoparticles would be fully degraded after 6 weeks in vivo, whereas the m = 10 nanoparticles would be fully degraded after 3 weeks in vivo.
FIG. 11.

The NMR analysis of nanoparticle degradation by comparing the NMR spectra of m = 0, m = 4, m = 10 nanoparticles in the accelerated degradation conditions as a function of time (A), and plotting the relative degradation as a function of time by integration of the peaks between 1.55 and 0.5 ppm (B). Color images are available online.
Protein loading in the nanoparticles
The experimental results of trypsin loading into the P(MMA-co-MAA) nanoparticles showed a high degree of variability between runs (Fig. 12). Several loading conditions led to loading efficiencies of >80%, namely the “−−++” (2 h, pH 4.5, 154 mM, and 10 wt% protein: polymer), “000A” (24 h, pH 6, 79.5 mM, and 13.5 wt% protein: polymer), and “+−++” (48 h, pH 4.5, 154 mM, and 10 wt% protein: polymer) conditions. In addition, certain runs showed very low protein loading such as the “−+−−” (2 h, pH 7.5, 5 mM, and 3 wt% protein: polymer) and “A000” (72 h, pH 6, 79.5 mM, and 6.5 wt% protein: polymer) conditions, which presented loading efficiencies of ∼30% and 10%, respectively.
FIG. 12.

The experimentally obtained protein loading efficiencies for the different loading conditions as defined by the JMP software (see Supplementary Table S1). Color images are available online.
By using these experimental data in the JMP software, we obtained information regarding the relative influences of the different loading variables and the ability to develop a predictive model based on the experimental data. In general, the model developed by JMP had a high ability to accurately predict protein loading efficiency, with a predictive power p < 0.0001 (Fig. 13). In addition, the software identified the loading variables, which had the most significant effects on the protein loading efficiency of the nanoparticle. The most influential factors were the square of the incubation time, the protein:polymer wt% ratios, the ionic strength of the buffer, and the square of the loading buffer pH.
FIG. 13.

(A) A comparison of the actual loading efficiency and the model predicted loading efficiency and (B) the effect summary of the various loading variables where p-values <0.05 correspond to a statistically significant effect on the loading protein loading efficiency. Color images are available online.
The interactions between the different variables considered are described in Figure 14. These contour plots reveal the importance of considering these two-factor interactions because the maximized values for one variable did not lead to maximum loading at all levels of the other variables. For example, although the protein to polymer ratio that leads to the highest loading efficiency is 8 wt%, these high efficiencies are achieved when incubating between 20 and 45 h in buffers >110 mM and at pH <4.7 or >7.2. However, 8 wt% protein: polymer achieves little to no loading, if carried out in buffers <80 mM, at pH ∼6, or for incubation times higher than 50 h.
FIG. 14.

The contour plots obtained from the protein loading analysis and considering two-factor interactions. The areas with Loading Efficiency 100 indicate the conditions that lead to the maximum loading efficiency, as predicted by the model. Color images are available online.
In vitro behavior of human umbilical vein endothelial cells in the presence of degradable nanoparticles
MTS and LDH assays of HUVECs in the presence of both nondegraded and degraded particles show that the P(MMA-co-MAA) nanoparticles did not have significant cytotoxic effects on the cells. For the LDH assay, a value of 80% relative proliferation was considered to be the acceptable threshold value to indicate cellular compatibility. The results of the LDH membrane integrity assays for both the nondegraded and degraded particles indicated that cell viability values remained >90% relative to the media control for concentrations up to 0.2 mg/mL (Figs. 15A and 16A). The relative cell viability decreased for nanoparticle concentrations of 1 mg/mL but remained >80% for both 24- and 48-h exposure times.
FIG. 15.

An evaluation of the cytocompatibility of the nondegraded P(MMA-co-MAA) nanoparticles. (A) The cytocompatibility of HUVECs using an LDH cell membrane integrity assay after 24 h (left) and 48 h exposure (right). (B) The cytocompatibility of HUVECs using an MTS cellular proliferation assay after 24-h (left) and 48-h exposure (right). HUVECs, human umbilical vein endothelial cells; LDH, lactose dehydrogenase; MTS, 3-(4, 5-dimethylthiazol-2-yl)-5 (3-carboxymethonyphenol)-2-(4-sulfophenyl)-2H- tetrazolium. Color images are available online.
FIG. 16.

An evaluation of the cytocompatibility of the degraded P(MMA-co-MAA) nanoparticles. (A) The cytocompatibility of HUVECs using an LDH cell membrane integrity assay after 24-h (left) and 48-h exposure (right). (B) The cytocompatibility of HUVECs using an MTS cellular proliferation assay after 24-h (left) and 48-h exposure (right). Color images are available online.
The results of the cellular proliferation assay indicated that the nondegradable P(MMA-co-MAA) nanoparticles (m = 0) exhibit a concentration-dependent effect on cellular proliferation after 24 h of exposure, with cellular proliferation remaining >80% relative to the media control for concentrations <0.2 mg/mL but decreasing to ∼10% for a concentration of 1 mg/mL (Figs. 15B and 16B).
This concentration-dependent behavior was also observed for the m = 4 and m = 10 degradable particles. However, the proliferation values were significantly higher than those of the media control with up to 700% for the m = 4 nanoparticles and 900% for the m = 10 nanoparticles in both the nondegraded and degraded state. This effect is less pronounced after 48 h of exposure to the nondegraded nanoparticles, with proliferation values remaining ∼200% for all nanoparticles at all concentrations. Of note, after 48 h of exposure, the m = 0 nanoparticles no longer exhibited the concentration-dependent effect, with proliferation values of ∼150% up to concentrations of 1 mg/mL. In addition, after 48 h of exposure, the increased proliferation effect was less pronounced for the degraded m = 4 and m = 10 nanoparticles when compared with the 24-h exposure.
Discussion
Nanoparticles with tunable degradation rates
In the literature, nanoparticle-based approaches for growth factor delivery are considered to be a promising method to achieve sustained release profiles and promote tissue regeneration.18 For example, Neumann et al. developed nanoporous silica nanoparticles ∼50 nm in diameter loaded with BMP-2 and demonstrated osteogenic differentiation.19 Similarly, Park et al. demonstrated that the incorporation of BMP-2 into heparinized poly(l-lysine) nanoparticles led to improved in vitro osteogenic differentiation of human mesenchymal stem cells, thanks to a reduction in BMP-2 burst release and increased BMP-2 bioactivity.20 In this study, P(MMA-co-MAA) nanoparticles were successfully synthesized using a PEGDMA crosslinker. The observed negative surface charges of the particles (Fig. 9) confirm the successful incorporation of methacrylic acid. In addition, the presence of these charges confirms the incorporation of PEGDMA as a crosslinker in the bulk of the particle rather than as a PEG coating on the surface of the particles. Indeed, PEG is often incorporated onto the surface of nanoparticles to reduce the charge-based interactions with proteins and increase circulation time, imparting the so-called “stealth” behavior.21
Many nanoparticle-based approaches for the delivery of growth factors rely on nanoparticle degradation-mediated release of the payload to achieve the desirable sustained release profiles.22 One method to impart degradability to a hydrogel system is through the introduction of labile bonds on the crosslinker.23 In fact, the use of degradable crosslinkers for the release of growth factors such as BMP-2 has been explored before in different systems. For example, Tellier et al. developed heparin-based microparticles for the release of BMP-2 using a hydrolytically degradable crosslinker and showed a slow and sustained release of BMP-2.24 Similarly, Li et al. developed crosslinked micelles for the controlled release of a hydrophobic payload based on a similar PLA/PEG block-copolymer as used in the present system.25 Although in the system developed by Li et al. the PLA/PEG macromolecule is used not as a crosslinker but rather as the bulk nanoparticle material, it was shown that changing the length of the PLA segments allowed for a control of the payload release profile, as is hypothesized in the present design. It was also demonstrated by Diederich et al. that the degradation rate of a PEG-based hydrogel can be increased by increasing the number of the incorporated lactic acid units along the PEG chain.13
In this study, degradable MA-PLA-b-PEG-b-PLA-MA macromolecules were synthesized to subsequently be incorporated as crosslinkers into the P(MMA-co-MAA) nanoparticle synthesis scheme. As given in Figure 6, the degradable nanoparticles have relatively constant hydrodynamic diameters despite being synthesized using different crosslinkers. This can be attributed to the fact that the length of the PEG chain was adapted for each custom crosslinker synthesis scheme such that the final crosslinkers would have lengths comparable with that of PEGDMA MW = 600. In addition, a wider distribution of hydrodynamic diameters for the custom crosslinked nanoparticles was observed and can be explained by a wider distribution in the lengths of the custom crosslinker chains. Indeed, the values obtained by the integration of the NMR spectra (Fig. 5) provide only an average value of the lactic acid unit incorporation. However, it is likely that the number of the lactic acid units incorporated may follow a wide distribution, which would lead to a wide distribution of the crosslinker chain length and, thus, nanoparticle hydrodynamic diameter.
The analysis of the different markers of nanoparticle degradation provides evidence that the custom macromolecules that were synthesized and incorporated as crosslinkers in the nanoparticle fabrication scheme impart a certain degree of degradability to the nanoparticles. In addition, a comparison between the nanoparticles fabricated with varying degrees of lactic acid unit incorporation shows that the developed system presents a degree of tunability, with m = 10 nanoparticles presenting a higher rate of degradation than the m = 4 nanoparticles. This is observed by the difference in the rate of the count rate decrease as a function of time, the amount of the visible intact particles through TEM, and the rate of degradation determined by quantitative NMR analysis. This observation is in line with the hypothesis that increasing the number of degradable bonds along the crosslinker is a viable method to tune the degradability of nanoparticles.23
This tunability of the nanoparticle degradation can be harnessed to optimize the system for the delivery of particular growth factors. It is known that the release kinetics of particulate carriers are highly affected by the rate of degradation of the carrier in question.22 Therefore, fabricating a nanocarrier that degrades more or less rapidly can be adapted for the delivery of growth factors that are relevant at either early or late stages of tissue regeneration. For BMP-2 and bone regeneration specifically, it is known that the regeneration of bone tissue is a highly dynamic process consisting of coordinated cellular responses over several weeks26 with most systems in the literature seeking to optimize their proposed systems for the sustained delivery of BMP-2 for up to 4 weeks.27 In this study, accelerated degradation conditions led to full in vitro degradation of the m = 4 and m = 10 nanoparticles after 48 and 24 h, respectively. This was estimated to correspond to 6 and 3 weeks in vivo, respectively, time scales that are relevant for bone tissue engineering applications. Further in vivo testing would be required to confirm degradation rates of these nanoparticles when delivered into a bone defect.
The results of the in vitro studies indicate that the incorporation of custom degradable crosslinkers in the synthesis of P(MMA-co-MAA) nanoparticles does not result in cytotoxic effects on HUVECs up to concentrations of 1 mg/mL and 48 h of exposure. In addition, cellular proliferation assay results show that, in the presence of degraded nanoparticles, HUVECs exhibit up to a nine-fold increase in proliferation relative to the positive media control, a behavior that is not observed for the nondegradable nanoparticles. This suggests a positive effect of the nanoparticle degradation byproducts on cell proliferation.
As given in Figure 11, NMR data confirm the appearance of lactic acid as a function of time as a result of nanoparticle degradation. In physiological conditions, lactic acid can dissociate into a lactate ion and a hydrogen ion and can, thereby affect the metabolic function of the surrounding cells.28 Specifically, Groussard et al. showed that lactate ions can have antioxidant effects by scavenging superoxide and hydroxyl radicals and limiting lipid peroxidation, thereby protecting cells from oxidative damage.29 Lampe et al. showed that an increase in the concentration of lactic acid up to 0.5 mg/mL results in a 100% increase in the total DNA content compared with the control in fetal forebrain cells, which was attributed to a reduction in cellular redox state.28 In addition, Beckert et al. showed that in HUVECs specifically, an increase in lactate can enhance vascular endothelial growth factor production, resulting in an increase in cellular migration.30 Therefore, the observed positive effects of the degraded P(MMA-co-MAA) nanoparticles on HUVEC proliferation can be explained by the release of lactic acid. This phenomenon was more pronounced after 24 h of exposure, when nanoparticle degradation would be more pronounced. After 48 h of exposure, the increased proliferation is less pronounced, whereas cell viability remains high. This suggests that the impact of lower pH driven by the increase in degradation byproduct does not negatively impact cell viability.
Ultimately, the novelty of this system lies in the ability to adjust the number of the lactic acid units along the crosslinker chains to achieve specific degradation times without significantly altering the other key properties of the carrier system. In addition, the degradation of nanoparticles does not lead to cell lysis and, on the contrary, can promote cell proliferation through the release of lactic acid byproducts.
Protein loading ability of P(MMA-co-MAA nanoparticles)
The proposed system is designed to deliver growth factors relevant for bone tissue engineering in a controlled and sustained way. The synthesized nanoparticles were, therefore, designed to have high affinity for BMP-2, which is a high molecular weight, high isoelectric point, hydrophobic protein, and is commonly used in bone tissue engineering applications. It should be noted that many studies use bovine serum albumin (BSA) as a model for BMP-2.31,32 However, BSA has a much higher molecular weight of ∼66.5 kDa (compared with 30 kDa for BMP-2) and a low isoelectric point of 4.7 (in contrast to 8.5 for BMP-2). Because both, size and electrostatic interactions will greatly influence the partitioning of the protein into the nanoparticles, we used trypsin with a molecular weight of 23.3 kDa and an isoelectric point of 10.1 as a more accurate model to understand the ability of the selected system to load growth factors such as BMP-2.
The results of the protein loading study pointed to the excellent ability of the synthesized P(MMA-co-MAA) nanoparticles to load the model protein. Indeed, loading efficiencies of up to 100% were observed for certain loading conditions. In addition, the experimental results pointed to the importance of choosing the appropriate loading conditions, because loading efficiencies ranged between 10% and 100% simply by changing certain loading variables. In fact, all four loading conditions were shown to have a significant effect on the protein loading efficiency, with incubation time and pH having second-order effects, whereas the protein to polymer ratio and the buffer strength exhibited first-order effects.
First, loading time was found to have an important effect on protein loading. In fact, increased loading time leads to increased chances of the protein partitioning into the particles. However, as the system reaches equilibrium, very long loading times could have the reverse effect and result in lower protein incorporation into particles. This phenomenon was observed in this study, with a 24.8-h incubation leading to maximum loading, and times >48 h leading to a significant loss of the loading efficiency. It was also shown that an ionic strength of 154 mM is desirable. This buffer strength corresponds to that at which proteins similar to BMP-2 are most stable. In fact, it is known that the buffer ionic strength influences the BMP-2 solubility, with buffer strengths up to 150 mM significantly increasing the solubility of the protein, and >500 mM leading to the precipitation of the protein.16
As determined by zeta potential measurements, at pH 4.5, the synthesized particles exhibited significant degrees of aggregation. At pH 6, the particles possessed slight negative charges, whereas at pH 7.5, the particles presented a net negative surface charge. Of interest, a pH of 4.5 was found to lead to the highest loading efficiency, which, despite the aggregation of the particles in this low pH conditions, can be explained by the high stability of the protein in such acidic environments.16 The pH-dependent changes in the loading behavior point to the important pH-related tradeoffs with regard to increasing the protein loading efficiency.
Finally, higher values of protein:polymer ratios allow for improved loading. A range of protein to particle ratios has recently been reported in the literature with doses varying between 1 and 200 μg/mL.33,34 In contrast, the BMP-2 dose currently approved for human use ranges between 1.5 and 2.0 mg/mL.35 However, it has been shown that these higher doses can lead to inflammation and diminished quality of the regenerated bone.35,36 Evaluating the ability of the particles to load protein at a wide range of weight ratios would allow both for the optimization of the present system and providing an indication of the range of the doses that can be delivered by this system for different applications.
Ultimately, this work demonstrates the excellent ability of the synthesized nanoparticles to load a model protein for BMP-2. This result underlines the importance of understanding the protein properties and polymer selection in designing nanocarriers. However, these results can be further supported by performing loading experiments with BMP-2.
In addition, a statistical software was used to further quantify the effect of loading variables on protein loading efficiency. We found that all the factors had a statistically significant effect and can be optimized for the system at hand to achieve the maximum loading efficiency. Finally, a key advantage in the use of nanoparticles for growth factor delivery is the ability to tune the delivered dose to achieve the desired tissue response. Through understanding the protein loading efficiency of the nanoparticles, the number of the nanoparticles loaded into the scaffold carrier can be tuned to control the dose delivered to the site of injury.
Conclusions
The results of this study show that P(MMA-co-MAA) nanoparticles designed to have high affinity for high isoelectric point, large molecular weight proteins such as BMP-2 can be fabricated in a simple and reproducible way through a one-pot UV-initiated emulsion polymerization scheme. In addition, this system was capable of loading a model protein for BMP-2 at efficiencies of up to 100% and the loading conditions to maximize the protein loading efficiency were identified.
Furthermore, a degradable crosslinker was custom synthesized and incorporated into the P(MMA-co-MAA) nanoparticles to introduce varying degrees of degradability. Indeed, it was shown that by varying the number of the hydrolytically degradable units along the crosslinker chain, the rate of degradation could be tuned to meet the desired protein release profile.
Overall, this tunable platform can be developed for a variety of growth factor delivery applications. This system can potentially be adapted to achieve a controlled, sequential delivery of multiple growth factors by simply optimizing the degradation kinetics of the nanoparticles. In fact, sequential delivery systems have been identified as essential to the highly complex bone regeneration process and have the potential to, for example, simultaneously promote osteogenesis and angiogenesis for the formation of vascularized bone.37,38 The incorporation of these nanoparticle carriers into scaffold structures could further improve bone tissue regeneration by delivering growth factors to the injury site while providing structural support to the surrounding tissue. In particular, the proposed nanoparticle delivery system can be covalently bound to a chitosan scaffold backbone to limit diffusion out of the scaffold pores, as demonstrated in our previous study.39,40
Supplementary Material
Acknowledgments
The authors acknowledge helpful technical discussions with Julia Vela Ramirez and John R Clegg. This work is published in a special issue in honor of Prof. Antonios G. Mikos of Rice University who just celebrated his 60th birthday. The authors thank him for his tremendous impact on tissue engineering through his pioneering work over 30 years. One of the authors (N.A.P.) thanks Dr. Mikos for having spent 37 years interacting with him (at Purdue University, Rice University, and the University of Texas at Austin) and continuing to produce important work in the field. Dr. Mikos' dedication to fundamental and translational research in regenerative medicine has transformed the field and has made it mature and scientifically exciting.
Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported in part by a grant from the National Institutes of Health (Grant R01-EB022025) and by the UT-Portugal Collaborative Research Program (CoLAB) “Intelligent Scaffolds for Molecular Recognition of Advanced Applications in Regenerative Medicine.”
Supplementary Material
References
- 1. Bose S., Roy M., and Bandyopadhyay A.. Recent advances in bone tissue engineering scaffolds. Trends Biotechnol 30, 546, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Porter J.R., Ruckh T.T., and Popat K.C.. Bone tissue engineering: a review in bone biomimetics and drug delivery strategies. Biotechnol Prog 25, 1539, 2009 [DOI] [PubMed] [Google Scholar]
- 3. Oldenkamp H.F., Vela Ramirez J.E., and Peppas N.A.. Re-evaluating the importance of carbohydrates as regenerative biomaterials. Regen Biomater 6, 1, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Richbourg N.R., Peppas N.A., and Sikavitsas V.I.. Tuning the biomimetic behavior of scaffolds for regenerative medicine through surface modifications. J Tissue Eng Regen Med 13, 1275, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Clegg J.R., and Peppas N.A.. Molecular recognition with soft biomaterials. Soft Matter 16, 856, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wechsler M., Clegg J., and Peppas N.. The interface of drug delivery and regenerative medicine. In: Reis R., ed. Encyclopedia of Tissue Engineering and Regenerative Medicine. Vol. 1 San Diego, CA: Academic Press - Elsevier, pp. 77–86 [Google Scholar]
- 7. McKay W.F., Peckham S.M., and Badura J.M.. A comprehensive clinical review of recombinant human bone morphogenetic protein-2 (INFUSE® Bone Graft). Int Orthop 31, 729, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Azevedo H.S., and Pashkuleva I.. Biomimetic supramolecular designs for the controlled release of growth factors in bone regeneration. Adv Drug Deliv Rev 94, 63, 2015 [DOI] [PubMed] [Google Scholar]
- 9. De Witte T.-M., Fratila-Apachitei L.E., Zadpoor A.A., et al. Bone tissue engineering via growth factor delivery: from scaffolds to complex matrices. Regen Biomater 5, 197, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Elkhoury K., Russell C.S., Sanchez-Gonzalez L., et al. Soft-nanoparticle functionalization of natural hydrogels for tissue engineering applications. Adv Healthc Mater 8, 1900506, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Han J., Ma G., and Nie J.. A facile fabrication of porous PMMA as a potential bone substitute. Mater Sci Eng C 31, 1278, 2011 [Google Scholar]
- 12. Sawhney A.S., Pathak C.P., and Hubbell J.A.. Bioerodible hydrogels based on photopolymerized poly(ethylene glycol)-co-poly(.alpha.-hydroxy acid) diacrylate macromers. Macromolecules 26, 581, 1993 [Google Scholar]
- 13. Diederich V.E.G., Villiger T., Storti G., et al. Modeling of the degradation of poly(ethylene glycol)-co-(lactic acid)-dimethacrylate hydrogels. Macromolecules 50, 5527, 2017 [Google Scholar]
- 14. Fisher O.Z., and Peppas N.A.. Polybasic nanomatrices prepared by UV-initiated photopolymerization. Macromolecules 42, 3391, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Browning M.B., Cereceres S.N., Luong P.T., et al. Determination of the in vivo degradation mechanism of PEGDA hydrogels. J Biomed Mater Res A 102, 4244, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. King W.J., and Krebsbach P.H.. Growth factor delivery: how surface interactions modulate release in vitro and in vivo. Adv Drug Deliv Rev 64, 1239, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Samarajeewa S., Shrestha R., Li Y., et al. Degradability of poly(lactic acid)-containing nanoparticles: enzymatic access through a cross-linked shell barrier. J Am Chem Soc 134, 1235, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Corona-Gomez J., Chen X., and Yang Q.. Effect of nanoparticle incorporation and surface coating on mechanical properties of bone scaffolds: a brief review. J Funct Biomater 7, 18, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Neumann A., Christel A., Kasper C., et al. BMP2-loaded nanoporous silica nanoparticles promote osteogenic differentiation of human mesenchymal stem cells. RSC Adv 3, 24222, 2013 [Google Scholar]
- 20. Park K.-H., Kim H., Moon S., et al. Bone morphogenic protein-2 (BMP-2) loaded nanoparticles mixed with human mesenchymal stem cell in fibrin hydrogel for bone tissue engineering. J Biosci Bioeng 108, 530, 2009 [DOI] [PubMed] [Google Scholar]
- 21. Jokerst J.V., Lobovkina T., Zare R.N., et al. Nanoparticle PEGylation for imaging and therapy. Nanomedicine (Lond) 6, 715, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Vo T.N., Kasper F.K., and Mikos A.G.. Strategies for controlled delivery of growth factors and cells for bone regeneration. Adv Drug Deliv Rev 64, 1292, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hennink W.E., and van Nostrum C.F.. Novel crosslinking methods to design hydrogels. Adv Drug Deliv Rev 64, 223, 2012 [DOI] [PubMed] [Google Scholar]
- 24. Tellier L.E., Miller T., McDevitt T.C., et al. Hydrolysis and Sulfation Pattern Effects on Release of Bioactive Bone Morphogenetic Protein-2 from Heparin-Based Microparticles. J Mater Chem B Mater Biol Med 3, 8001, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Li J., Guo S., Wang M., et al. Poly(lactic acid)/poly(ethylene glycol) block copolymer based shell or core cross-linked micelles for controlled release of hydrophobic drug. RSC Adv 5, 19484, 2015 [Google Scholar]
- 26. Hettiaratchi M.H., Rouse T., Chou C., et al. Enhanced in vivo retention of low dose BMP-2 via heparin microparticle delivery does not accelerate bone healing in a critically sized femoral defect. Acta Biomater 59, 21, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Krishnan L., Priddy L.B., Esancy C., et al. Delivery vehicle effects on bone regeneration and heterotopic ossification induced by high dose BMP-2. Acta Biomater 49, 101, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lampe K.J., Namba R.M., Silverman T.R., et al. Impact of lactic acid on cell proliferation and free radical induced cell death in monolayer cultures of neural precursor cells. Biotechnol Bioeng 103, 1214, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Groussard C., Morel I., Chevanne M., et al. Free radical scavenging and antioxidant effects of lactate ion: an in vitro study. J Appl Physiol 89, 169, 2000 [DOI] [PubMed] [Google Scholar]
- 30. Beckert S., Farrahi F., Aslam R.S., et al. Lactate stimulates endothelial cell migration. Wound Repair Regen 14, 321, 2006 [DOI] [PubMed] [Google Scholar]
- 31. Yilgor P., Tuzlakoglu K., Reis R.L., et al. Incorporation of a sequential BMP-2/BMP-7 delivery system into chitosan-based scaffolds for bone tissue engineering. Biomaterials 30, 3551, 2009 [DOI] [PubMed] [Google Scholar]
- 32. Liu T., Wu G., Zheng Y., et al. Cell-mediated BMP-2 release from a novel dual-drug delivery system promotes bone formation. Clin Oral Implants Res 25, 1412, 2014 [DOI] [PubMed] [Google Scholar]
- 33. Choi J.W., Jeong W.S., Yang S.J., et al. Appropriate and effective dosage of BMP-2 for the ideal regeneration of calvarial bone defects in beagles. Plast Reconstr Surg 138, 64e, 2016 [DOI] [PubMed] [Google Scholar]
- 34. Ben-David D., Srouji S., Shapira-Schweitzer K., et al. Low dose BMP-2 treatment for bone repair using a PEGylated fibrinogen hydrogel matrix. Biomaterials 34, 2902, 2013 [DOI] [PubMed] [Google Scholar]
- 35. Zara J.N., Siu R.K., Zhang X., et al. High doses of bone morphogenetic protein 2 induce structurally abnormal bone and inflammation in vivo. Tissue Eng Part A 17, 1389, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. James A.W., LaChaud G., Shen J., et al. A review of the clinical side effects of bone morphogenetic protein-2. Tissue Eng Part B Rev 22, 284, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hankenson K.D., Dishowitz M., Gray C., et al. Angiogenesis in bone regeneration. Injury 42, 556, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Farokhi M., Mottaghitalab F., Shokrgozar M.A., et al. Importance of dual delivery systems for bone tissue engineering. J Control Release 225, 152, 2016 [DOI] [PubMed] [Google Scholar]
- 39. De Witte T.-M., Wagner A.M., Fratila-Apachitei L.E., et al. Immobilization of nanocarriers within a porous chitosan scaffold for the sustained delivery of growth factors in bone tissue engineering applications. J Biomed Mater Res A 2020. [Epub ahead of print]; DOI: 10.1002/jbm.a.36887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. De Witte T.-M. Development of a two-phase scaffold-nanoparticle system for the sustained delivery of growth factors in bone tissue engineering applications [M.S. thesis]. Department of Biomechanical Engineering, Delft University of Technology, Delft, The Netherlands, 2018 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.