

Abstract
Targeted mass spectrometry methods produce high quality quantitative data in terms of limits of detection and dynamic range, at the cost of a substantial compromise in throughput compared to methods such as data independent and data dependent acquisition. The logistical and experimental issues inherent to maintaining assays of even several hundred targets are significant. Prominent among these issues is the drift in analyte retention time as liquid chromatography (LC) columns wear -- forcing targeted scheduling windows to be much larger than LC peak widths. If these problems could be solved, proteomics assays would be capable of targeting 1000’s of peptides in an hour-long experiment, enabling large cohort studies to be performed without sacrificing sensitivity and specificity. We describe a solution in the form of a new method for real-time chromatographic alignment and demonstrate its application to a 56-minute LC-gradient HeLa digest assay with 1489 targets. The method is based on the periodic acquisition of untargeted survey scans in a reference experiment and alignment to those scans during subsequent experiments. We describe how the method enables narrower scheduled retention time windows to be used. The narrower scheduling windows enables more targets to be included in the assay or proportionally more time to be allocated to each target -- improving the sensitivity. Finally, we point out how the procedure could be improved and how much additional target multiplexing could be gained in the future.
Graphical Abstract
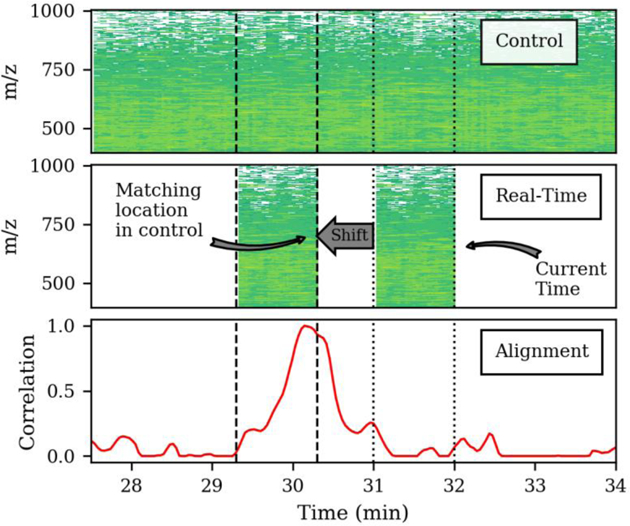
Introduction
The promise of proteomics is that the effect of a disease or perturbation on the cell or an organism can be assessed by the accurate and precise quantification of proteins and their modifications. Proteomics technologies based on tandem mass spectrometry have advanced significantly over the last decade, but the methods still frequently suffer from relatively low throughput methods, requiring hours per sample, and relatively low numbers of samples across conditions, samples, and specimens. Additionally, discovery methods, while capable of profiling peptides from thousands of proteins in a sample, the methods rarely assess quantitative figures of merit for the measured analytes1,2.
Historically, quantitative assays require defining the analytes to be measured and then validating the quantitative figures of merit. In contrast, discovery proteomics experiments define the peptide composition on the fly and attempt to define differences using the measurements made between conditions and groups. While powerful, many of these discovery methods underestimate the magnitude of quantitative differences between conditions at low intensity3–5 – a result of one of the conditions being outside the linear measurement range. Despite knowing the importance of quantitative linearity in the accuracy of a quantitative assay, it is rare that a proteomics assay will define figures of merit like limit of detection (LOD) and limit of quantification (LOQ) on the per analyte level2. Because of this, discovery experiments done on relatively limited numbers of samples or “well-founded hunches”6 are often followed up using targeted proteomics assays with much greater assay validation1,7,8. Despite the promise of targeted proteomics, there are limitations in the number of peptides that can be measured resulting in significant compromises that balance the number of analytes that can be tested versus the sensitivity and precision of the measurement.
Retention time scheduling, which limits the number of targets measured at each point in time, is done to increase the number of peptides that can be measured per run. Traditional targeted MS analyses, especially for nano-flow applications, commonly use scheduled acquisition segments for each peptide spanning 2–5 minutes, despite targeting chromatographic peak widths at of around 15–30 seconds (at the base). This inefficiency has been necessary because of fluctuations in analyte retention times from run to run and a systematic shift as the column degrades. For example, a targeted assay could be developed on one column over the course of several days or weeks, and during that time, changes in elution time of several minutes can occur. When the column must be replaced, the retention time of the analytes would revert to near their original times. Therefore, either the acquisition segments need to be quite large, or the user is forced to monitor the drift in elution time every few days and adjust the acquisition segment scheduling accordingly.
An alternative has been to sacrifice the selectivity of the measurement and intentionally multiplex the MS/MS acquisition using data independent acquisition (DIA). This comes at the expense of specificity and dynamic range. The method has significantly reduced data acquisition complexity but a huge increase in the computational analysis complexity. Significant artifacts of the qualitative peak assignment can occur if care is not taken.9
Several solutions to this problem in the framework of targeted analyses have been developed already, most of which involve the monitoring of internal standards. Sanghvi et. al. described a method that utilizes the Pierce Retention Time Calibration mixture, a set of heavy isotope labeled yeast peptides with varying hydrophobicities10. Each internal standard is monitored within a 5-minute scheduled acquisition window, and when detected, the time shift is updated and used to determine the active targets. This method is straight-forward and effective. Its disadvantages are that it requires the user to manage the parameters of the internal standards, including retention times and triggering thresholds, and that the time shifts are applied in large discrete steps, limiting how narrow the acquisition segments can be made.
Lemoine and coworkers developed a method called “Scout MRM”, in which each analyte target is associated with one or more internal standards11,12. The detection of an internal standard triggers the continuous acquisition of all the analytes associated with that standard. The advantage of the method is that once associated with an internal standard, the retention times of the internal standards and analytes no longer need to be maintained as method parameters. The user can do the internal standard association step with a very fast experiment, and then apply the results to a longer experiment. The disadvantage of the technique is that triggering thresholds for the standards must be maintained, and again the resolution of the retention time alignment is limited by the number of internal standards, in some examples to around an order of magnitude larger than the LC peak width11.
A related method is “internal standard parallel reaction monitoring” (IS-PRM)13, an implementation of which is also known as “SureQuant”. This method is developed with a heavy-labeled internal standard for every analyte spiked into the sample at high concentration. Detection of an internal standard in a high-throughput, low-sensitivity scanning mode triggers acquisition of both the internal standard and endogenous analytes in a low-throughput, high-sensitivity scanning mode. This method eliminates retention time considerations for the user and provides for very high experiment level duty cycle, albeit divided by a factor of ~2 when the internal standards and monitoring scans are accounted for. A disadvantage of the method is that the acquisition of the target analyte does not begin until after the trigger, resulting in the absence of part of the chromatographic peak – complicating background subtraction. A second disadvantage of the method is that internal standards are required for every analyte, increasing cost and limiting its feasibility for very large analyte panels. On the other hand, assay kits are becoming more common and when available, method development is greatly simplified.
Here we combine the use of a modified Fusion Lumos mass spectrometer and novel real-time alignment capabilities that enables the targeted acquisition MS/MS data on 1000s of targets in a single 56 min long chromatographic run. Unlike most prior cases of parallel reaction monitoring, these experiments are performed at unit resolution using a linear ion trap and improved rod drivers to gain an MS/MS acquisition rate of ~65 Hz. This strategy provides an opportunity for high sensitivity and high selectivity targeted data acquisition in an easy- to-use platform for targeted proteomics. This enables rapid quantitative re-analysis without the complexities of retention time scheduling, nor the sacrifices of limiting follow up experiments to a few critical peptides.
Experimental Procedures
Chromatogram Library Generation
To define which peptides were present in each sample type, DIA data were collected using multiple injections with each injection spanning a narrow subset of the combined precursor range. This gas-phase fractionation strategy is based off of methods originally described by the Goodlett lab14,15 and refined more recently16–18. The method used here was a modification of the method described by Searle et al17, adapted to an instrument with a faster acquisition speed.
HeLa protein digest was obtained from Thermo Fisher Scientific™ (Pierce™ P/N 88328) in 20 μg vials, to which was added 40 μL water with 0.1% formic acid. The 0.5 ug/μL sample was injected onto a 50 cm × 0.075 mm column (Thermo P/N ES803A) which was heated to 45 C. Liquid chromatography was performed with a RSLC Ultimate 3000. The flow rate was 300 nl/min, and gradient buffers A and B were water and acetonitrile with 0.1% formic acid. The gradient program was as follows, given in terms of %B: (2–22%, 0–50 min), (22–32%, 50–55 min), (32–90%, 55–56 min), (90%, 56–65 min), (90–2%, 65–66 min), (2%, 66–102 min). The mass spectrometer was a modified research version of an Orbitrap Fusion Lumos™ (see the supporting information (S.I.). for a discussion of the differences from production systems). The sample was characterized with DIA over the precursor range m/z 400–1200 in 4 experiments that each covered 200 Th with 1 Th isolation using the linear ion trap for the product ion collection. The 4 experiments characterized the precursor ranges m/z 400–600, 600–800, 800–1000, 1000–1200. The actual precursor masses were chosen so that the edges of the isolation windows are in regions of low peptide m/z probability, due to the characteristic peptide mass defects19,20. The precursor lists can be found in the S.I. Thermo Instrument Control version 3.4.293 method editor was used to create method files. The main parameters were quadrupole isolation with 1 Th isolation width, linear ion trap mass analysis with Turbo scan rate, range m/z 200–1600, RF Lens 30%, HCD activation with normalized collision energy 27%, AGC target “Standard” (1e4 charges), maximum injection time mode “Auto”, and default charge state 2.
The four raw files generated from the gas phase fractioned DIA analysis using 1 Th precursor isolation were analyzed with Proteome Discover 2.3 using SEQUEST and Percolator. Of note is that the precursor mass tolerance was 1.0 Da, and fragment mass tolerance was 0.5 Da. Additionally, the spectrum selector node specified that the Unrecognized Charge Replacements were set to 2+ and 3+. This, together with an instrument control modification that set the Charge State field in the spectrum scan headers to 0 instead of the default of 2, meant that each spectrum was searched as both a 2+ and 3+ precursor, improving the detection of 3+ peptides. Peptide spectral matches were validated by Percolator with a 0.01 FDR. There were 22413 unique peptides identified from the combination of the four raw files.
Selection of Peptides for Targeted Analysis
The unique peptides were filtered for characteristics that enable high quality targeted analysis. A computer program was created to assign metrics to each peptide for filtering. For each peptide, the spectrum with the highest Xcorr was used to select b and y ions above an arbitrary relative intensity threshold of 0.01. The intensities of the fragments were extracted in the 19 adjacent spectra (in retention time) from the same precursor isolation window. The intensities for each fragment were separately normalized, the median fragment intensity was computed for each spectrum, and the time correlation of each fragment to the median was computed. See the S.I. for a Jupyter notebook that illustrates this procedure in more detail. Peptides were required to have at least 5 fragments with minimum correlation 0.95, minimum relative area 0.30, and summed area greater than 3e4. These filters removed all except 5299 peptides. Finally, a “load-balancing” step was applied to lower the number of peptides acquired at any one time. To do this, a desired cycle time of 1.15 seconds was chosen, an acquisition segment duration of 3 minutes, and a minimum injection time/analysis time of 10 ms. Combining this information, the active targets were added to an acquisition list at each experimental time until the instrument time required to perform them exceeded the desired cycle time. This process resulted in a final list of 1489 peptides for targeted analysis.
Targeted Mass Spectrometry
The targeted analysis method consisted of two cycles of scans. The first cycle performed DIA over the range m/z 400–1000 in 31 scans, with isolation width 20 Th. The spectra generated by this cycle are referred to as “alignment spectra”. The mass range was m/z 200–1000, maximum injection time mode was Auto, and Loop Control was set to Time mode with 2 seconds. The instrument control software was modified such that this time corresponded to the minimum period between cycles, instead of the time between inserting a full MS1 prescan for predictive automatic gain control (AGC). The prescan for a targeted cycle is normally not saved to the rawfile, but for these experiments it was saved, and was used in the alignment procedures described below. The second cycle contained the 1489 targets, with scheduled start and end times specified so that a subset of the total targets would be “active” during each cycle. We refer to the scheduling window, which is the time range over which the instrument is directed to acquire targeted MS/MS spectra for a specific analyte, as the “acquisition segment”. These time windows are a key complexity of methods that implement “scheduled PRMs”. The segment duration was set to a constant value for an experimental run, with replicates performed using durations 0.5, 0.75, 1.0, 3.0, and 5.0 minutes. The quadrupole isolation width was 0.7 Th, mass range was m/z 200–1450, maximum injection time mode was “Dynamic” with a minimum of 9 points across the expected LC peak width of 15 s (cycle time of 1.7 seconds). The targeted precursor lists can be found in the S.I.
Real-Time Chromatographic Alignment
The spectra from the first cycle of the targeted experiment were used for chromatogram alignment. A control targeted experiment was run, and the spectra from the first cycle were processed to produce a data file which could be loaded onto the instrument embedded computer for real-time chromatographic alignment in later runs. The retention times of the targets from this experiment were determined by Skyline and used to create a final list of precursors and retention times. The centroided alignment spectra were projected onto a fixed space grid with spacing 0.250125 Da, producing vectors of size 3198. The Haar wavelet transform21 was applied to the spectra to yield vectors of size 128. In this way, the DIA spectra in the control data set were compressed to a total size of 16.7 MB. Despite the compression, the signal to noise ratio of the cross-correlations between experiments are similar relative to using uncompressed data (Figure S1), which are too large to load onto the limited memory of the embedded processor.
The average period between DIA cycles was computed to be 2.327 seconds, and the compressed spectra were projected onto a fixed time grid with that spacing. The spectra were serialized and loaded to the embedded computer. A targeted method was initiated with the injection of the sample to the LC column, and the procedure outlined in Figure 1 was continuously carried out. Each cycle began with the acquisition of an MS prescan and 31 DIA MS/MS spectra. As each spectrum was acquired, it was Haar compressed to n = 128 points. Independently for each DIA scan, a buffer of size q was maintained with the most recent spectra projected onto the same time grid as the control data. These spectra were cross correlated with the control data in the region around the current experimental time (Eq 1). This cross-correlation rXY of control data matrix X and real-time data matrix Y was performed for the time shift index i = {0, 1, …, (p − q)}, where p is the number of columns (time points) in X, q is the number of columns in Y, and k is the row, or m/z variable.
Figure 1.
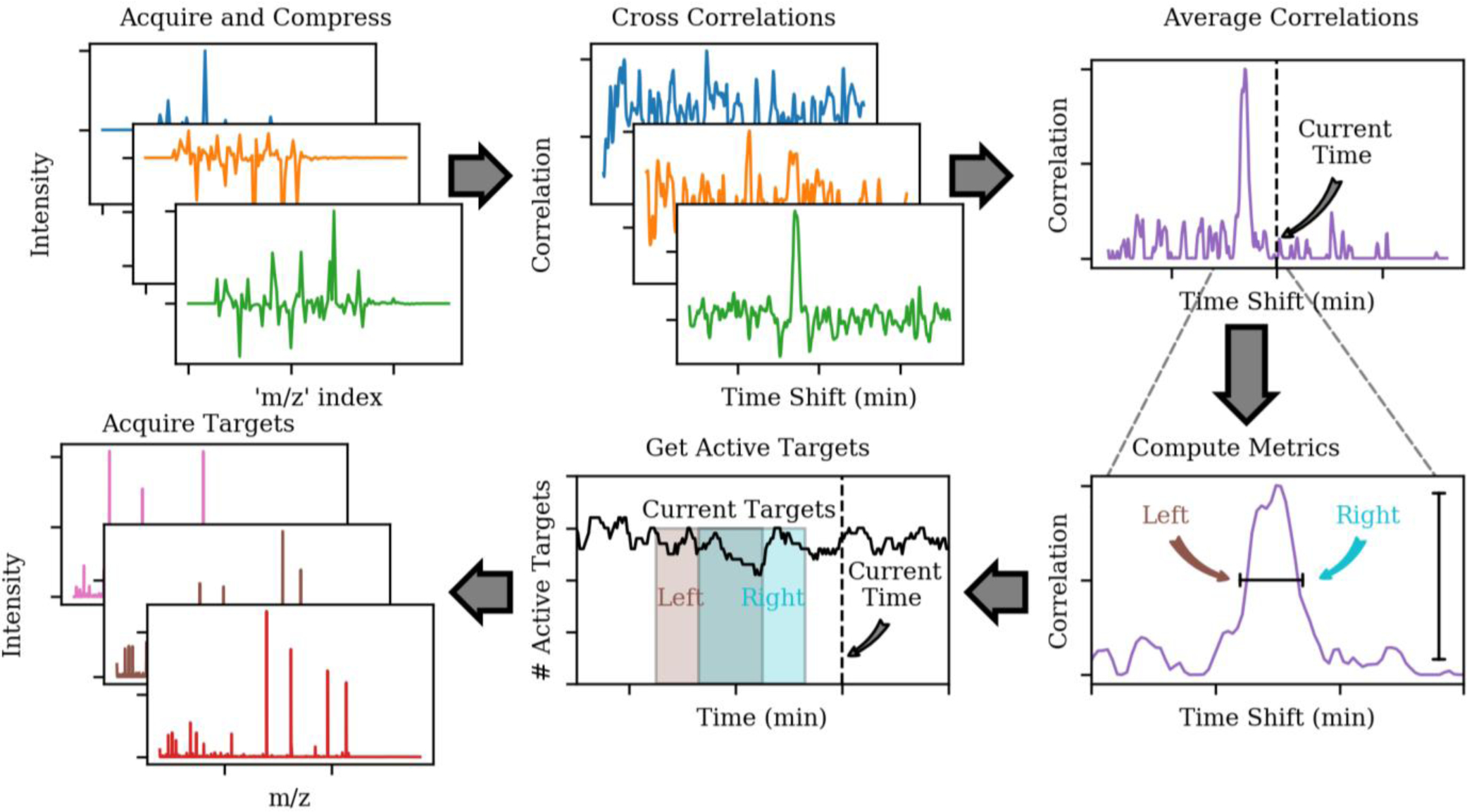
Real-time chromatographic alignment workflow. DIA alignment spectra are acquired, compressed, cross correlated with a control data set separately for each spectrum, and averaged together. The averaged cross-correlation is characterized, and the amount of retention time shift with respect to the control data set is calculated, which determines the set of currently active analytical targets. Finally, targeted MS/MS spectra are acquired for the active targets.
| (1) |
The p columns of X are taken from the larger control set of data to avoid unnecessarily computing cross-correlations across the entire data set. Therefore, the time shift i = 0 corresponds to the current time minus cycles. The size of the interrogated time region is set to +/− 8 minutes by the value of p = 417 and the time spacing of 2.327 s. The number of most recent spectra was set to q = 3. While slightly larger values of q are generally desirable due to the averaging affect they impart, 3 was deemed to be the largest number that did not incur an unacceptable computational time-penalty on the embedded computer. The cross-correlations are normalized by the norms of X and Y to account for changes in intensity between control and experimental data, and to protect against vectors with very small or large numbers. The norm of X is denoted with the subscript i in Equation 1 to indicate that it only includes those columns used in the calculation at time shift i, that is, {i, i + 1, …, i + q − 1}.
As the DIA spectra are acquired, compressed, and cross correlated with the control data set, they are summed together to form an averaged, final cross-correlation. The final cross-correlation was characterized by centroiding it and determining the area α of all of its η peaks. The position δt of the largest peak for cycle iteration t nominally represents the amount of retention time shift between the current experiment and the control data set. A set of quality filters were implemented to protect against spurious results, which typically show up at the beginning and end of the experiments when no sample elutes, and are described in more detail in the S.I. Having computed a robust time shift δt between control and real-time data, the adjusted time t′ is calculated (Eq 2) and the set of currently active targets are determined in the normal manner, as the m scans whose acquisition segment start and stop times t0 and tf bracket the time t′ (Eq 3).
| (2) |
| (3) |
To increase the robustness and account for small shifts in the peptide elution order between runs, the left and right sides of the cross-correlation peak was used to determine sets of active targets (Eq 2, 3), and their superset was taken to be the set of active targets for the cycle. Finally, MS/MS spectra were acquired for these active targets, completing the workflow for a cycle outlined in Figure 1. Raw files were imported into Skyline22 for analysis of the actual peptide retention times. The Skyline document and RAW files can be found on Panorama Public at https://panoramaweb.org/rt_alignment.url and has been assigned the ProteomeXchange ID PXD018675.
Results and Discussion
We have developed a new method capable of real-time chromatographic alignment against a previously collected run. Here the method is demonstrated for targeted proteomics where the real-time alignment enables narrower scheduling windows (aka acquisition segments) without fear of targets shifting outside of the windows over many injections. These narrower windows reduce the number of targets measured at each time, enabling longer ion trap fill times and higher sensitivity MS/MS analysis. The method uses the signals generated by unspecified background matrix molecules in the sample to perform retention time alignment relative to a control experiment. A proof of concept of this method was described previously in an American Society for Mass Spectrometry poster where periodic unit-resolution MS1 scans were used for alignment. The proof of concept was performed post-acquisition for an unscheduled targeted assay of 15 compounds23. Here, the entire process was integrated into the instrument embedded computer to estimate the retention time shifts relative to a control on-the-fly and adjust the active targets in real-time.
We built a list of peptides that can be detected in our HeLa digest using gas-phase fractionated DIA data collected from four injections. The precursor isolation was 1 Th spanning the entire precursor range. The data were searched with SEQUEST using the precursor tolerance of the entire precursor isolation window and post-processed using Percolator. This resulted in a list of 22413 peptides (q-value <0.01) from 4676 protein groups. We further filtered the data to only those peptides that had 5 product ion transitions that covaried over time and correlated to the median signal intensity using a similar method described by Searle et al.17. This resulted in a target list of 1489 peptides that could be detected and provided sufficient interference free transitions for selective quantitative measurements. There are many more quantifiable peptides in the sample than 1489; however, this number of peptides was chosen because it can be analyzed with enough points across the LC peak in the largest (5 minute) acquisition segment studied here.
A single control run was performed using a PRM experiment that included an additional periodic cycle of unit-resolution DIA spectra with 20 Th precursor isolation. The retention times of the 1489 peptides were determined for the control run and held as constant method file inputs for subsequent runs. The spectra from the DIA cycles in the control run were compressed and stored on the instrument embedded computer.
Following that control run, subsequent runs were collected using scheduled PRM acquisition on our target list of 1489 peptides, again including an additional cycle of DIA spectra. The real-time chromatogram alignment methodology was used to adjust the location of scheduling window segments and the active list of peptide targets. Experiments were performed using a series of different scheduled acquisition segment widths, to demonstrate the value of real-time chromatogram alignment on the number of active targets at each time, total injection time for each target, and quantitative precision.
The use of DIA MS/MS spectra for alignment were found to enhance the selectivity and robustness of the retention time shift estimates relative to using only MS1 cycles (Figure S2). Each DIA linear ion trap spectrum used a reduced product ion mass range of m/z 200–1000 to save time in the acquisition of each spectrum. In total, the 32 DIA alignment scans took 0.5 seconds to complete. After collecting each of the 32 DIA MS/MS spectra, the embedded computer incurred about 7 ms processing overhead. After the final DIA spectrum was collected, the average of the 32 cross-correlations was used to determine the retention time shift relative to the control run and adjust the active target list. While 7 ms per alignment spectrum is not insignificant, the current computational overhead is small relative to the gain in time provided by reducing the number of precursor targets at any point in time.
We use an average of the cross-correlation from the 32 DIA spectra against the control to provide information about the spread in retention time shifts of the various peptides eluting at any time. An example set of averaged cross-correlations versus time is shown in Figure 2a, where an active run was compared to a control run collected 16 days prior. Light regions on the contour plot represent high correlation, and dark regions represent low correlation. The location of the maximum cross-correlation along the y-axis denotes a shift in retention times, where negative shifts indicate an earlier elution time in the control. The cyan lines are the full width half maximum positions of the cross-correlation maxima. In the first 10 minutes of the experiments where only solvent and contaminants are observed, the cross-correlation is non-specific, and all shifts are equally likely. As compounds start to elute into the instrument, the cross-correlations become more specific, and during the middle of the run they are typically about 30 seconds wide.
Figure 2.
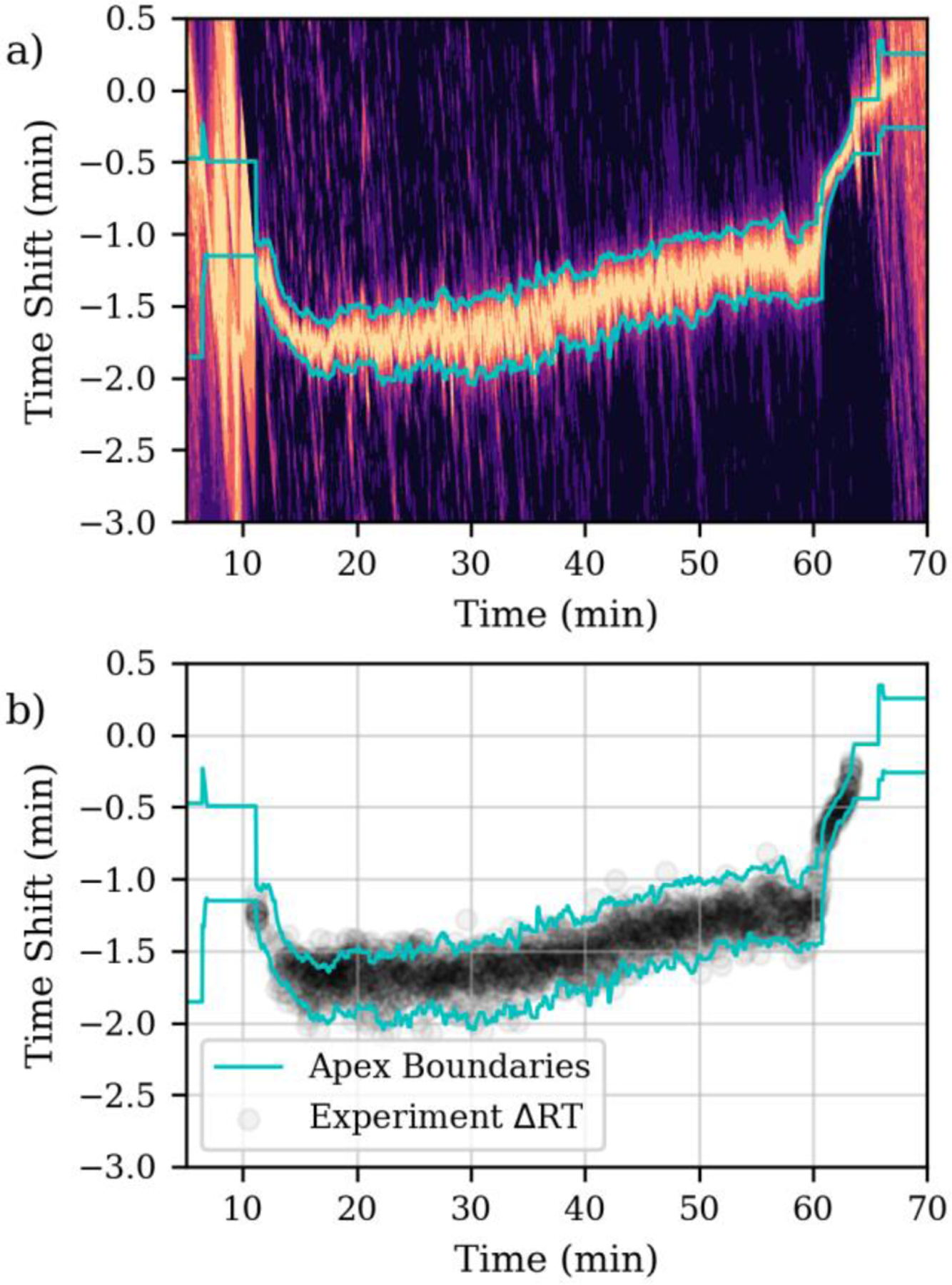
a) Cross-correlations versus time for a 1-minute acquisition segment width targeted assay aligned to a control data set acquired 16 days prior. Lighter colors denote high similarity to the control, while dark regions denote low similarity. The cyan lines denote boundary positions of the apex peak in the cross-correlation. b) Experimentally measured retention time shifts.
Despite run-to-run differences in the absolute chromatographic retention time, the elution order of peptides on the same chromatographic column remains similar. Nevertheless, minor re-ordering of peptides from different runs prevent the determination of an exact match of the location of a peptide between runs. Interestingly, the alignment performed in real-time here produces a similar alignment precision shown previously between DIA data performed on the same column9. In the present experiments, the re-ordering of retention times between runs were accounted for by measuring it and widening the acquisition segments appropriately, as described in the Methods. For the nominally 1-minute acquisition segment duration experiment, 90.6% of the measured retention time shifts fall within the estimated full width half maximum positions of the cross-correlation maxima (Fig 2b).
The ability of the real-time chromatographic alignment to accurately adjust the time range where precursors were triggered for MS/MS was assessed. The start and end of each acquisition segment was recorded and subtracted from the analyte peak apex position, as determined by Skyline, and displayed in Figure 3 for the 1-minute segment duration replicate. The raw, relative positions of the acquisition edges are labeled “Left Edge” and “Right Edge” in Figure 3a, where the white space around Y=0 demonstrates that the peak apices were fully acquired. Accurate peak picking of each peptide was also manually verified. For the 1-minute nominal segment condition, every analyte had a sufficient margin of time between its apex and the start or end of the acquisition segment that the entire LC peak was baseline resolved (Fig 3b). The actual acquisition segment duration was typically about 30 seconds wider than the nominal duration, due to the technique used to account for the spread in retention time shifts (Fig 3c). The analyses using nominal segment durations of 0.75 and 0.50 minutes had just 2 and 6 targets with an apex within 0.1 minutes of an acquisition segment edge.
Figure 3.
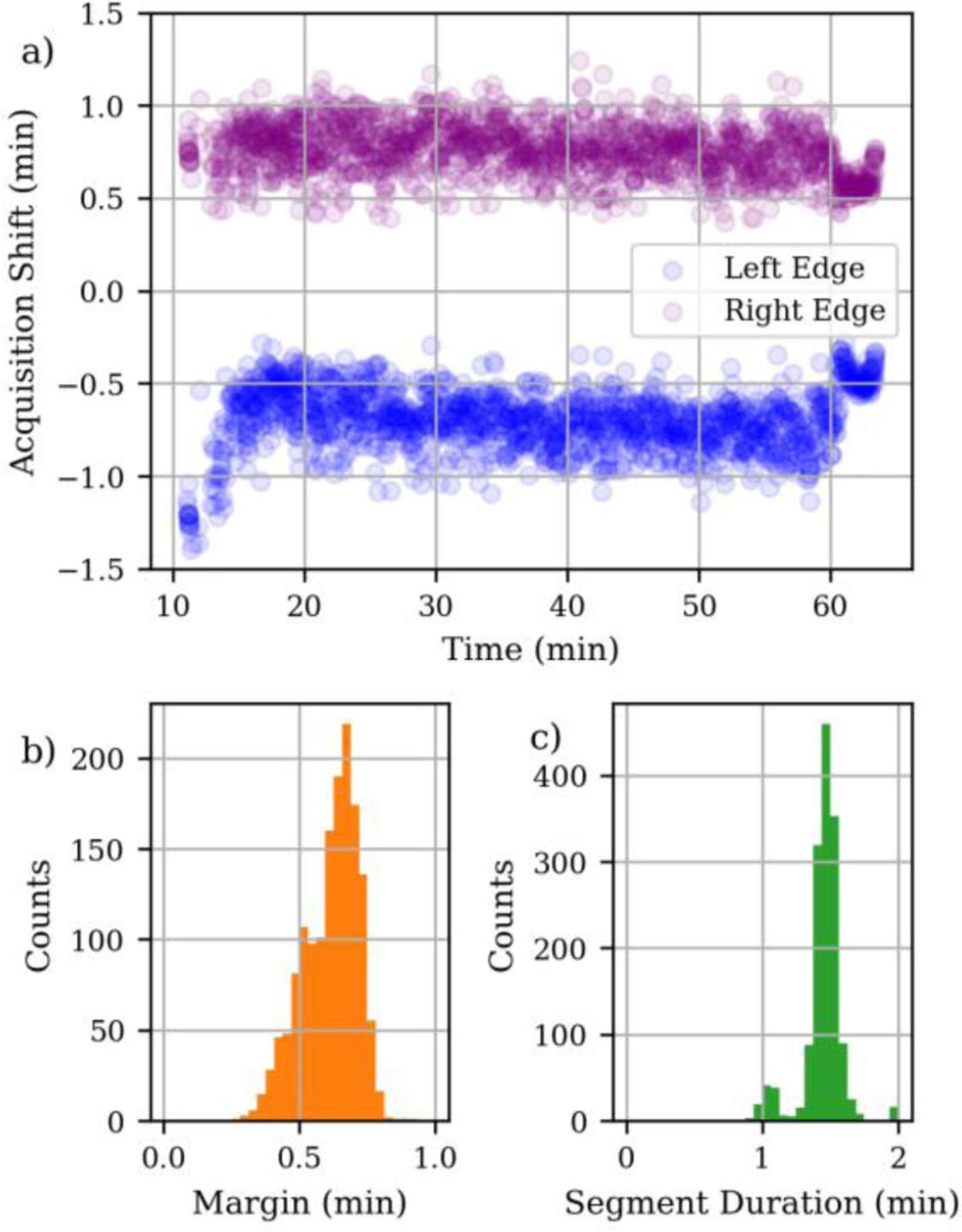
Analysis of acquisition segment positions for an aligned LC-MS run using nominal segment duration 1 min. a) Positions of the left and right sides of the acquisition segment relative to the actual LC peak apex. b) Histogram of minimum time between the LC peak apex and an acquisition edge. c) Histogram of total size of the acquisition segments.
In contrast to previous real-time chromatogram alignment methods, here the signals from the sample matrix are used for alignment. Therefore, analyte retention times must be measured in the matrix for the control experiment, but this is not usually burdensome. For peptide analysis, retention times are often already known from discovery experiments before the development of a targeted assay is begun. For small molecule analysis like pesticides, standards are usually readily available, and part of the normal development process is to characterize their retention times in matrix, since the matrix compounds can alter the analyte retention times. We have observed that the cross-correlation metric is quite robust to differences between samples. For example, proof of principle alignment was demonstrated for an experiment with a high field asymmetric waveform ion mobility (FAIMS) device installed, relative to a control experiment with no FAIMS23. We also observed that data from a control experiment using human plasma could align with samples diluted with chicken plasma down to around 6% human (Figure S3). These observations give some confidence that the method will work when applied across samples from different subjects, patients, disease states, cell lines and/or lots, but this has yet to be demonstrated.
The peptide peak areas from the 5 replicates were detected and integrated using Skyline22,24. The coefficient of variation (CV) of the 5 replicates is plotted in Figure 4 as a histogram (blue bars) and cumulative distribution (red dashed line). Most of the targets, 84%, had CV < 10%, while 98% of the targets had CV < 20%. Of those targets with CV > 20%, a few were the peptide targets where the boundaries of the chromatographic peak were not fully captured by the narrowest acquisition segment. We also investigated the CV distributions as a function of retention time (Figure S4). Many of the peptides with poor precision eluted either at the beginning or end of the LC run. These time regions represent a range where the real-time alignment is suboptimal because of poor chromatographic reproducibility and low signal from the background sample matrix. These peptides are not ideal quantitative measurements and should be excluded from future assays. In any case peptides with sub-optimal precision represent a small fraction (<2%) of the total targets.
Figure 4.
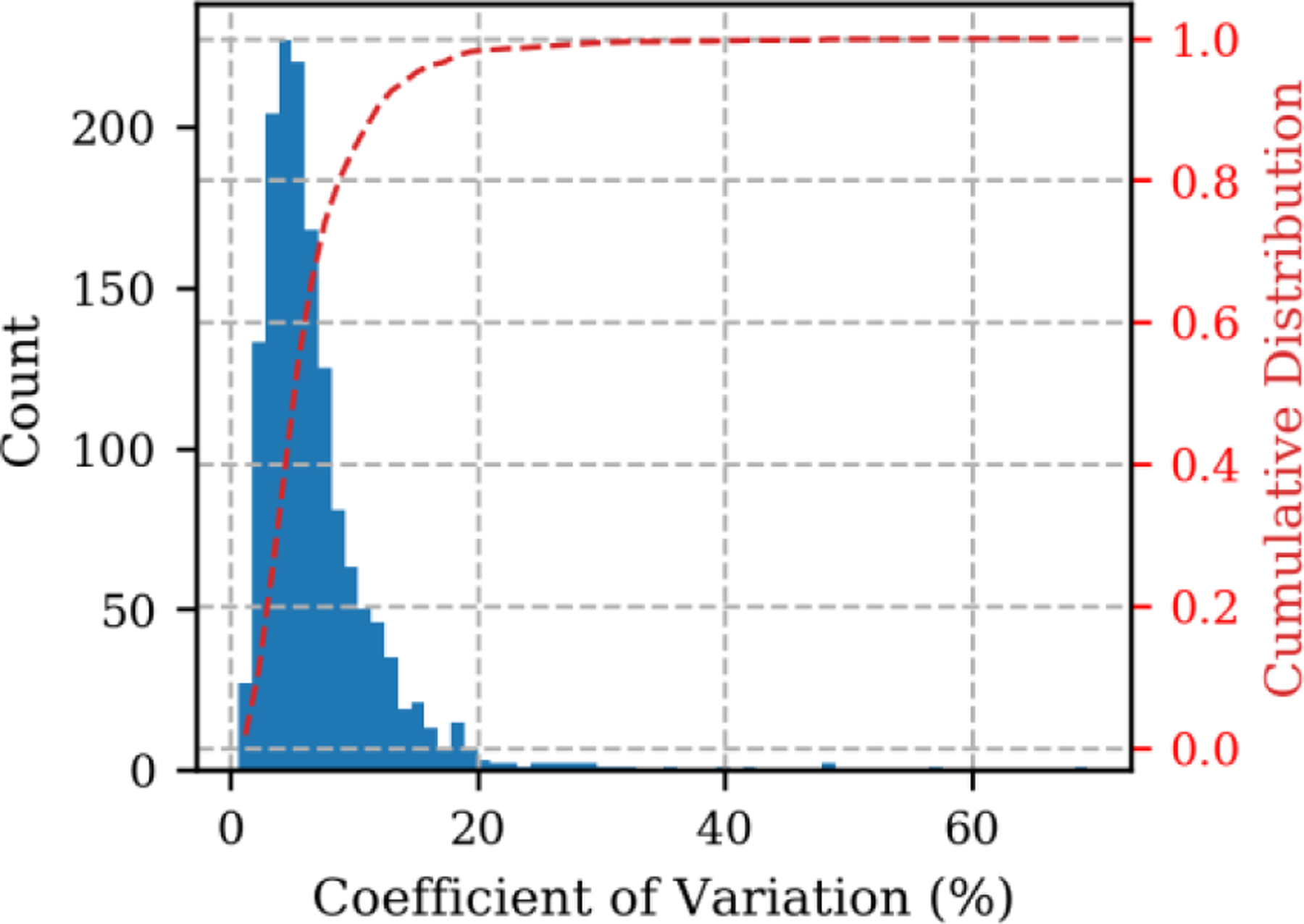
Coefficient of variation (CV) of 5 replicate injections for 1489 peptides.
The main motivation for developing the real-time alignment procedure is to enable narrower acquisition segments, increasing the number of peptide targets that can be sampled concurrently in the same cycle, and/or increasing the maximum injection time – improving the sensitivity. The effect of the acquisition segment duration (aka scheduling window time) on the number of active targets (of the total 1489) per iterative acquisition cycle was measured and is shown in Figure 5. The actual number of active scans (blue circles) includes the 32 alignment MS/MS spectra per cycle, while the no-alignment data (yellow squares) subtracts them. The difference between data points is less than the 32 subtracted DIA alignment spectra for the narrower segment durations, because the alignment was performed every 2.3 seconds, and therefore multiple cycles of MS/MS spectra on active peptide targets may occur between alignments.
Figure 5.
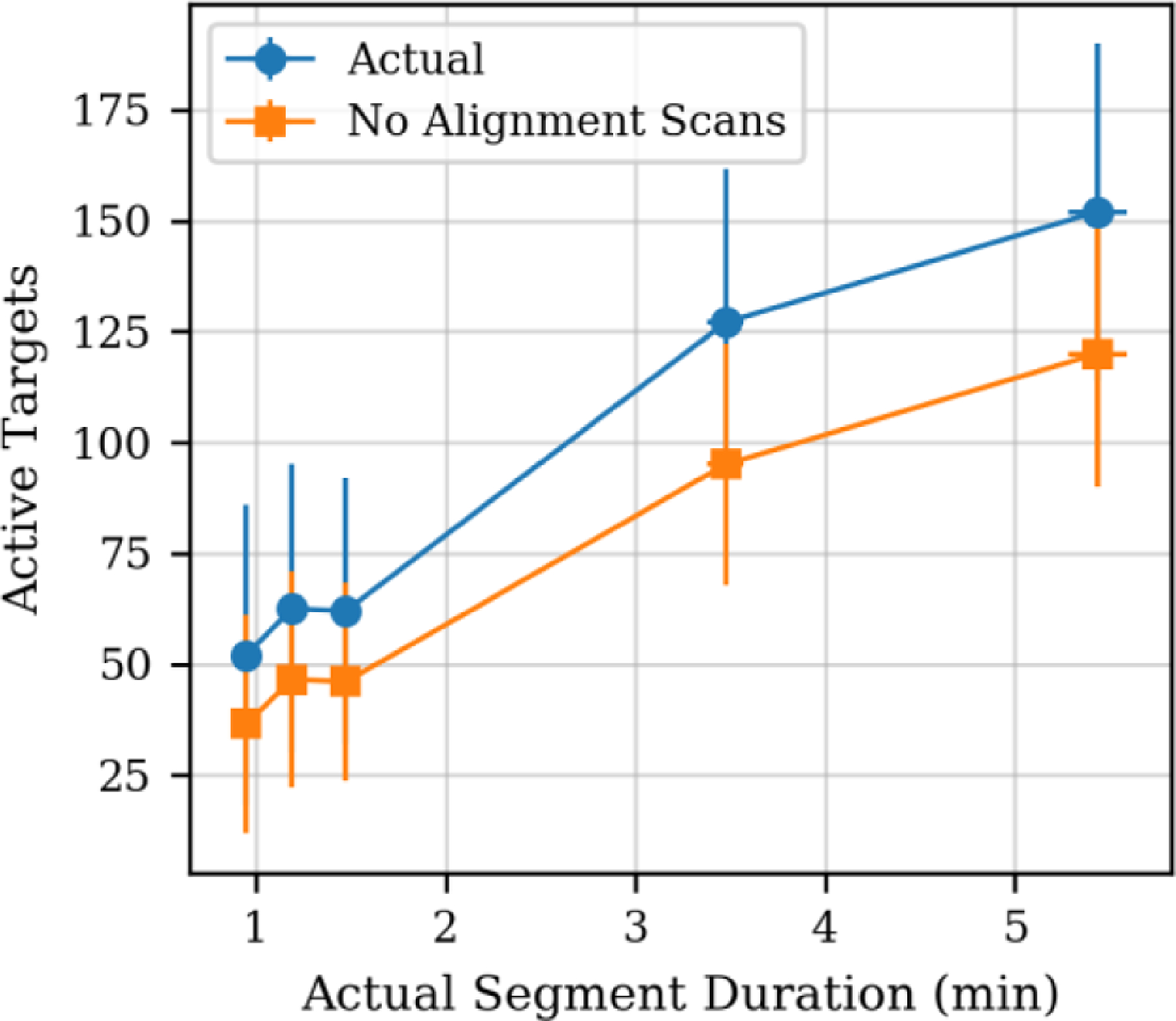
Median number of active targets per cycle versus median segment duration (aka scheduling window width). Data labeled “Actual” are from experiment, while “No Alignment Scans” refers to a fictitious situation where the alignment scans were eliminated.
The data in Figure 5 illustrates that going from a loosely scheduled targeted PRM acquisition method with 5.5 min scheduling windows to ~1.0 min scheduling windows reduced our mean active targets from 152 +/− 38 to 52 +/− 34 – a reduction of about 300%. This indicates that for ~1-minute scheduling segments, the instrument currently has capacity to extend our targeted PRM assay from 1489 targets to 4347 total targets. In an optimal (theoretical) case where we could perform the alignment without the overhead of the DIA MS/MS spectra, the reduction in concurrent active targets would be even greater. If the alignment overhead was completely removed, then we would be able to add 32 peptide targets per cycle and our targeted PRM assay of 1489 could be extended to approximately 6000 targets. Note that these estimates assume the current fixed MS2 spectral scan range of m/z 200–1450 is used, but with a customized scan range per peptide the instrument could scan faster and analyze more targets, albeit with less than the current minimum of ~13 ms injection time per target afforded by the current scan range. See the S.I. for a discussion of the target capacity of a production Lumos system.
The fact that more time per precursor is available as acquisition segment is reduced can also be observed in the number of times a chromatographic peak is sampled, and the injection time used per precursor. Figure 6a shows distributions of the number of spectra acquired per a 25 second LC peak width for each of the nominal acquisition segment durations. The average number of points acquired per LC peak increases linearly as acquisition segment duration is decreased, from a minimum of 9.5 scans/peak for the largest nominal RT segment (5 min) up to a maximum of 34 scans/peak smallest nominal RT segment (0.5 min). The mean number of ions per spectrum, given in red dots with the right axis of Figure 6a, shows that for all analyses, regardless of the scheduling window/segment duration, the ion population was near the target of 10,000 charges.
Figure 6.
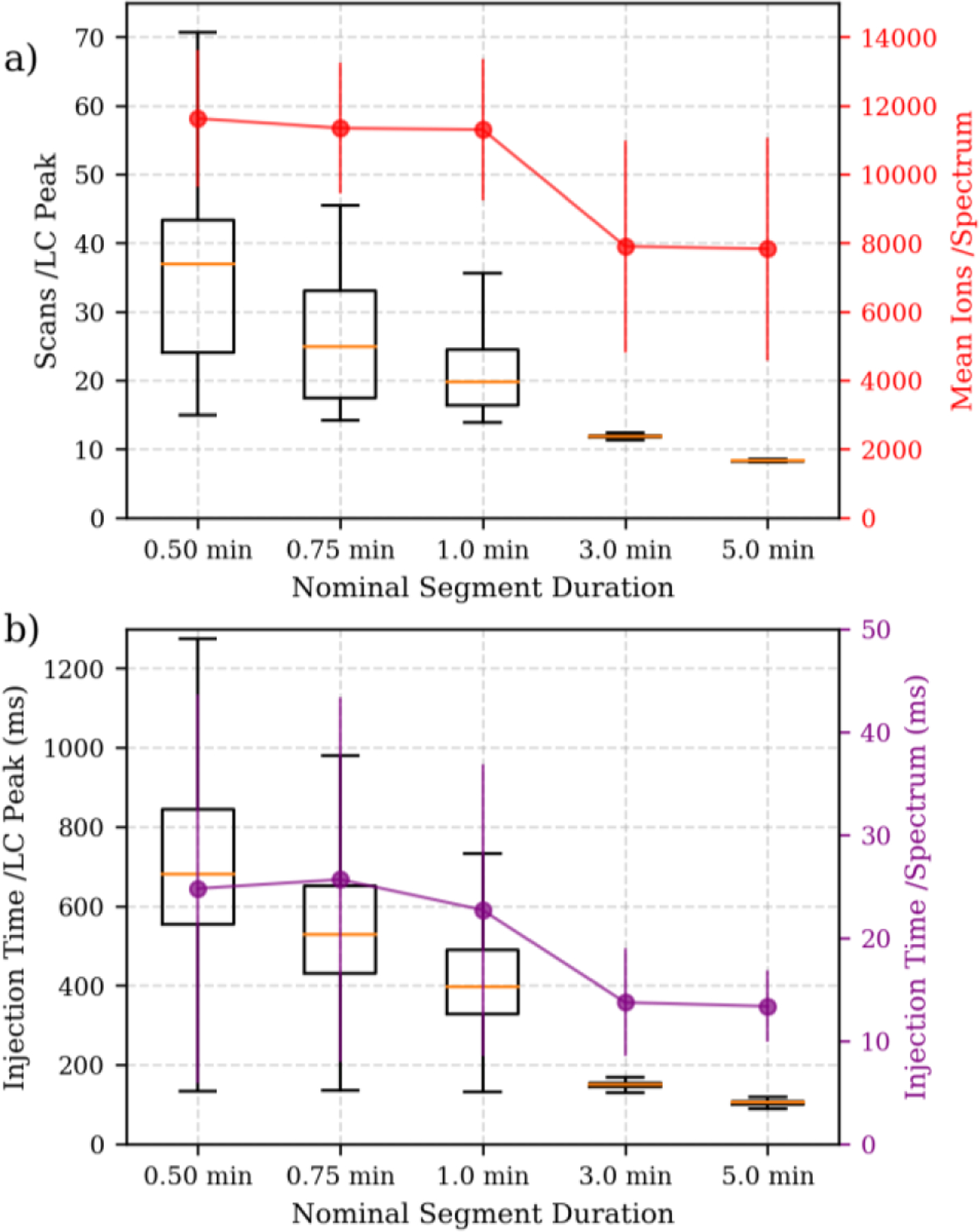
Acquisition metrics related to assay limits of detection. a) Distributions of number of scans per LC peak width on the left axis, in box and whiskers. Mean number of ions per spectrum with standard deviation error bars for the right axis. b) Distributions of total injection time per analyte per LC peak width on the left axis, in box and whiskers. Mean injection time per spectrum with standard deviation error bars on the right axis.
The use of Automatic Gain Control (AGC)25 is a powerful method to achieve a target population of ions per spectrum – maximizing sensitivity while minimizing the effects of space charging. The decrease in the acquisition segment duration provides more time to collect ions and fill the trap. However, Figure 6b (purple dots and right axis) shows that the increase in injection time is limited because the target of 10,000 charges is reached. In Figure 6b (left axis, boxes), we plotted the sum of all the injection times for all spectra across the chromatographic peak. The mean of the total injection time for each peptide LC peak does scale linearly as segment duration decreases.
We observed that the total injection time for all spectra across the chromatographic peak averaged 130 ms at the largest nominal segment (5.0 min) to 755 ms for the smallest nominal segment (0.5 min). The increased injection time per precursor increases the fraction of spectra that achieve the AGC target of 10,000 charges and is directly related to the sensitivity.
In addition to the sensitivity, the increased injection time and resulting increased number of ions measured will also improve the precision. There are numerous contributing sources of signal variation, such as variation in the amount of sample injected to the LC and in the electrospray ionization efficiency. However, from the perspective of the MS operation, the limiting factor comes from “shot noise” which is derived from the statistics of counting discrete events. Thus, the precision is limited by Poisson counting statistics and the CV of n ions is given by . From this perspective, for a given increase in signal intensity, CV decreases at a slower rate. All other things being equal, when the acquisition segment durations are reduced by a factor of 5, the CV’s and LOD’s achievable for this assay of 1489 peptides should decrease by . While we do not demonstrate this directly, the relationship between the total number of ions measured and precision has been well established in older literature26,27 and demonstrated in the context of linear ion traps.28,29
Summary and conclusions
We demonstrated application of a real-time chromatographic alignment method applied to a targeted proteomics assay that was able to reduce the acquisition segment duration to around 1 minute, compared to the 5-minute duration that would be required using traditional methods. The smaller segments led to a reduction in target density of about 3x and a 5x increase in amount of injection time per target. The implications for this assay are that the decreased segment durations could be used to analyze 3x more targets with the same limits of detection as the 5 minute segment durations, or alternatively achieve about 2.4x better limits of detection with the current number of peptide targets.
Our current implementation has a 0.5 second cost of performing the alignment scans, part of which is a ~7 ms processing overhead incurred by each scan. Future developments will focus on reducing the processing time and determining the minimum number of alignment scans needed to make robust estimates. Although using a DIA protocol for alignment does provide for additional analysis opportunities, the alignment scans would not necessarily need to use DIA, but rather could be targeted to specific background species or m/z windows identified in a prior experiment. As long as the determination of the scans can be automated, based on data from the acquisition of discovery or survey experiments, the user does not have to be burdened with any of the details of the procedure. If the alignment cost could be completely reduced, the additional instrument capacity would allow for up to 4x the number of targets. Any reduction of the alignment cost will also increase the suitability of the procedure for chromatography with narrower LC peaks, such as those encountered in micro and high flow methods.
An experimental platform with the capacity to perform targeted MS/MS quantitation for thousands of analytes in a short amount of time will find utility in a number of applications, such as biomarker discovery or re-analysis of clinical specimens including tissue or fluids. These are applications where it may be possible to quantify multiple peptides from every detectable protein in one targeted assay, with good quantitative figures of merit. The increased throughput afforded by such an assay would be a valuable tool for researchers in these fields. Additionally, because this method dynamically compensates for retention time shifts on-the-fly, we have established a prototype method that can effectively eliminate peptides drifting outside of statically scheduled retention time windows – increasing the robustness of scheduled PRM assays.
Supplementary Material
ACKNOWLEDGEMENTS
This work is supported in part by National Institutes of Health Grants P41 GM103533, R01 GM103551, and U19 AG065156.
Footnotes
SUPPORTING INFORMATION
A document with details about cross correlations; estimation quality filters; MS1-only based chromatogram alignment; the human plasma diluted with chicken study; experimental duty cycle. A zip file with spreadsheets of precursor lists. A zip file containing a Jupyter notebook, accompanying data file, and HTML copy of the notebook that describe the peptide filtering algorithm.
DATA AVAILABILITY
The RAW data and processed data (i.e. Skyline documents) from these experiments are available at Panorama Public (https://panoramaweb.org/rt_alignment.url) under ProteomeXchange ID PXD018675.
COMPETING FINANCIAL INTERESTS
P.M.R and P.Y. are employees of Thermo Fisher Scientific, the manufacturer of the instrumentation used in this research. The MacCoss Lab at the University of Washington has a sponsored research agreement with Thermo Fisher Scientific. Additionally, M.J.M. is a paid consultant for Thermo Fisher Scientific.
REFERENCES:
- (1).Grant RP; Hoofnagle AN From Lost in Translation to Paradise Found: Enabling Protein Biomarker Method Transfer by Mass Spectrometry. Clin. Chem 2014, 60 (7), 941–944. 10.1373/clinchem.2014.224840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Pino LK; Searle BC; Yang H-Y; Hoofnagle AN; Noble WS; MacCoss MJ Matrix-Matched Calibration Curves for Assessing Analytical Figures of Merit in Quantitative Proteomics. J. Proteome Res 2020, 19 (3), 1147–1153. 10.1021/acs.jproteome.9b00666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Ow SY; Salim M; Noirel J; Evans C; Rehman I; Wright PC ITRAQ Underestimation in Simple and Complex Mixtures: “The Good, the Bad and the Ugly.” J. Proteome Res 2009, 8 (11), 5347–5355. 10.1021/pr900634c. [DOI] [PubMed] [Google Scholar]
- (4).Karp NA; Huber W; Sadowski PG; Charles PD; Hester SV; Lilley KS Addressing Accuracy and Precision Issues in ITRAQ Quantitation. Mol. Cell. Proteomics 2010, 9 (9), 1885–1897. 10.1074/mcp.M900628-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).DeSouza LV; Romaschin AD; Colgan TJ; Siu KWM Absolute Quantification of Potential Cancer Markers in Clinical Tissue Homogenates Using Multiple Reaction Monitoring on a Hybrid Triple Quadrupole/Linear Ion Trap Tandem Mass Spectrometer. Anal. Chem 2009, 81 (9), 3462–3470. 10.1021/ac802726a. [DOI] [PubMed] [Google Scholar]
- (6).Marx V Targeted Proteomics. Nat. Methods 2013, 10 (1), 19–22. 10.1038/nmeth.2285. [DOI] [PubMed] [Google Scholar]
- (7).Whiteaker JR; Halusa GN; Hoofnagle AN; Sharma V; MacLean B; Yan P; Wrobel JA; Kennedy J; Mani DR; Zimmerman LJ; Meyer MR; Mesri M; Rodriguez H; Clinical Proteomic Tumor Analysis Consortium (CPTAC); Paulovich, A. G. CPTAC Assay Portal: A Repository of Targeted Proteomic Assays. Nat. Methods 2014, 11 (7), 703–704. 10.1038/nmeth.3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Carr SA; Abbatiello SE; Ackermann BL; Borchers C; Domon B; Deutsch EW; Grant RP; Hoofnagle AN; Hüttenhain R; Koomen JM; Liebler DC; Liu T; MacLean B; Mani DR; Mansfield E; Neubert H; Paulovich AG; Reiter L; Vitek O; Aebersold R; Anderson L; Bethem R; Blonder J; Boja E; Botelho J; Boyne M; Bradshaw RA; Burlingame AL; Chan D; Keshishian H; Kuhn E; Kinsinger C; Lee JSH; Lee S-W; Moritz R; Oses-Prieto J; Rifai N; Ritchie J; Rodriguez H; Srinivas PR; Townsend RR; Van Eyk J; Whiteley G; Wiita A; Weintraub S Targeted Peptide Measurements in Biology and Medicine: Best Practices for Mass Spectrometry-Based Assay Development Using a Fit-for-Purpose Approach. Mol. Cell. Proteomics MCP 2014, 13 (3), 907–917. 10.1074/mcp.M113.036095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Pino LK; Just SC; MacCoss MJ; Searle BC Acquiring and Analyzing Data Independent Acquisition Proteomics Experiments without Spectrum Libraries. Mol. Cell. Proteomics MCP 2020. 10.1074/mcp.P119.001913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Sanghvi V; Abbatiello SE; Blackburn M; Song Q; Sikora JW; Zheng Y; Thomas PM; Kelleher N Demonstration of Automated On-The-Fly Retention Time Updating and SRM Method Visualization for Targeted Peptide Quantitation. ASMS Annual Conference on Mass Spectrometry and Allied Topics San Antonio, TX., 2016. [Google Scholar]
- (11).Rougemont B; Bontemps Gallo S; Ayciriex S; Carrière R; Hondermarck H; Lacroix JM; Le Blanc JCY; Lemoine J Scout-MRM: Multiplexed Targeted Mass Spectrometry-Based Assay without Retention Time Scheduling Exemplified by Dickeya Dadantii Proteomic Analysis during Plant Infection. Anal. Chem 2017, 89 (3), 1421–1426. 10.1021/acs.analchem.6b03201. [DOI] [PubMed] [Google Scholar]
- (12).Ayciriex S; Carrière R; Bardet C; Blanc JCYL; Salvador A; Fortin T; Lemoine J Streamlined Development of Targeted Mass Spectrometry-Based Method Combining Scout-MRM and a Web-Based Tool Indexed with Scout Peptides. PROTEOMICS 2020, 20 (2), 1900254 10.1002/pmic.201900254. [DOI] [PubMed] [Google Scholar]
- (13).Gallien S; Kim SY; Domon B Large-Scale Targeted Proteomics Using Internal Standard Triggered-Parallel Reaction Monitoring (IS-PRM). Mol. Cell. Proteomics MCP 2015, 14 (6), 1630–1644. 10.1074/mcp.O114.043968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Panchaud A; Scherl A; Shaffer SA; von Haller PD; Kulasekara HD; Miller SI; Goodlett DR Precursor Acquisition Independent from Ion Count: How to Dive Deeper into the Proteomics Ocean. Anal. Chem 2009, 81 (15), 6481–6488. 10.1021/ac900888s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Panchaud A; Jung S; Shaffer SA; Aitchison JD; Goodlett DR Faster, Quantitative, and Accurate Precursor Acquisition Independent from Ion Count. Anal. Chem 2011, 83 (6), 2250–2257. 10.1021/ac103079q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Ting YS; Egertson JD; Bollinger JG; Searle BC; Payne SH; Noble WS; MacCoss MJ PECAN: Library-Free Peptide Detection for Data-Independent Acquisition Tandem Mass Spectrometry Data. Nat. Methods 2017. 10.1038/nmeth.4390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Searle BC; Pino LK; Egertson JD; Ting YS; Lawrence RT; MacLean BX; Villén J; MacCoss MJ Chromatogram Libraries Improve Peptide Detection and Quantification by Data Independent Acquisition Mass Spectrometry. Nat. Commun 2018, 9 (1), 5128 10.1038/s41467-018-07454-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Searle BC; Swearingen KE; Barnes CA; Schmidt T; Gessulat S; Küster B; Wilhelm M Generating High Quality Libraries for DIA MS with Empirically Corrected Peptide Predictions. Nat. Commun 2020, 11 (1), 1548 10.1038/s41467-020-15346-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Frahm JL; Howard BE; Heber S; Muddiman DC Accessible Proteomics Space and Its Implications for Peak Capacity for Zero-, One- and Two-Dimensional Separations Coupled with FT-ICR and TOF Mass Spectrometry. J. Mass Spectrom. JMS 2006, 41 (3), 281–288. 10.1002/jms.1024. [DOI] [PubMed] [Google Scholar]
- (20).Mann M Useful Tables of Possible and Probable Peptide Masses. ASMS Annual Conference on Mass Spectrometry and Allied Topics, Atlanta, GA., 1995. [Google Scholar]
- (21).Haar A Zur Theorie der orthogonalen Funktionensysteme. Math. Ann 1910, 69 (3), 331–371. 10.1007/BF01456326. [DOI] [Google Scholar]
- (22).MacLean B; Tomazela DM; Shulman N; Chambers M; Finney GL; Frewen B; Kern R; Tabb DL; Liebler DC; MacCoss MJ Skyline: An Open Source Document Editor for Creating and Analyzing Targeted Proteomics Experiments. Bioinforma. Oxf. Engl 2010, 26 (7), 966–968. 10.1093/bioinformatics/btq054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Remes PM; Yip P; Huguet R Retention Time Correction Method Utilizing Unspecified Peaks in MS Scans. ASMS Annual Conference on Mass Spectrometry and Allied Topics Atlanta, GA., 2019. [Google Scholar]
- (24).Pino LK; Searle BC; Bollinger JG; Nunn B; MacLean B; MacCoss MJ The Skyline Ecosystem: Informatics for Quantitative Mass Spectrometry Proteomics. Mass Spectrom. Rev 2017. 10.1002/mas.21540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Remes PM; Senko MW; Blethrow J Methods for Predictive Automatic Gain Control for Hybrid Mass Spectrometers. US9165755B2, October 20, 2015.
- (26).Peterson DW; Hayes JM Signal to Noise Ratios in Mass Spectroscopic Ion-Current-Measurement Systems In Contemporary Topics in Analytical and Clinical Chemistry; Hercules DM, Hieftje GM, Evenson MA, Eds.; Plenum Press: New York, 1978. [Google Scholar]
- (27).MacCoss MJ; Toth MJ; Matthews DE Evaluation and Optimization of Ion-Current Ratio Measurements by Selected-Ion-Monitoring Mass Spectrometry. Anal. Chem 2001, 73 (13), 2976–2984. [DOI] [PubMed] [Google Scholar]
- (28).Blackler AR; Klammer AA; MacCoss MJ; Wu CC Quantitative Comparison of Proteomic Data Quality between a 2D and 3D Quadrupole Ion Trap. Anal. Chem 2006, 78 (4), 1337–1344. 10.1021/ac051486a. [DOI] [PubMed] [Google Scholar]
- (29).Schwartz JC Measuring Ion Number and Detector Gain. US7109474B2, September 19, 2006.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.