

Abstract
As a serious public health concern, alcohol-related liver disease is associated with dysregulations in the intestinal barrier function and the gut microbiota. The liver and gut communicate via the gut-liver axis, through which microbial products and metabolites translocate to the liver. Here, we review the current knowledge of various microbial products and metabolites which contribute to the alcohol-related liver diseases, including bile acids, indole-3-acetic acid, butyrate, long-chain fatty acids, endotoxin, cytolysin, β-glucan and candidalysin. Some of these might serve as therapeutic targets for alcohol-related liver disease.
Keywords: alcoholic liver disease, microbiome, mycobiome
Graphical Abstract
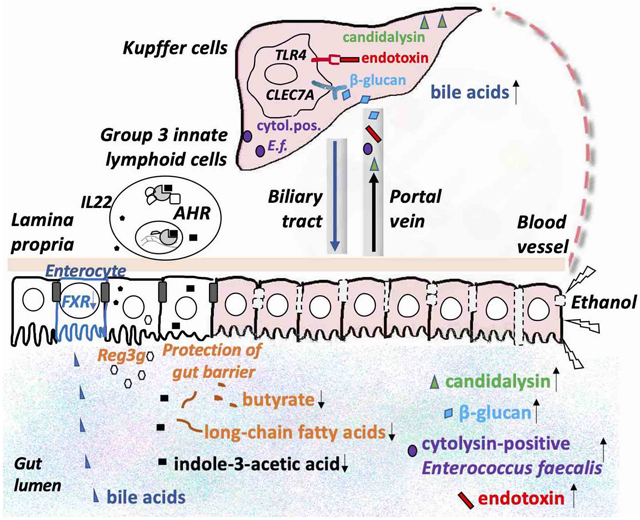
Excessive alcohol consumption is a major healthcare burden worldwide. Alcohol is oxidized mainly in hepatocytes with ethanol-oxidizing enzymes alcohol dehydrogenase and cytochrome P450, family 2, subfamily e, polypeptide 1 (Cyp2e1) [1]. As the primary site for ethanol metabolism, the liver sustains the earliest and greatest degree of injury [2]. Common consequences of chronic and excessive alcohol consumption contribute to the development of various liver damage, ranging from simple steatosis to steatohepatitis with inflammation, fibrosis and cirrhosis [2]. In addition to liver disease, alcohol consumption also contributes to gut barrier dysfunction, increased intestinal permeability and dysregulation of the gut microbiota [3-8].
Pathophysiology of alcohol-related liver disease involves many factors, such as genetic and gender [9-11]. In the last decades, emerging evidence suggested the gut microbiota plays an essential role in alcohol-related liver disease [12,13]. Qualitative and quantitative changes of gut microbiota are associated with chronic alcohol consumption [14-19]. At the crossroads between the gut microbiota and the host, microbial products from the intestine travel through the portal vein and enter the liver [20]. In this article, we briefly discuss the gut-liver communication axis and the intestinal barrier, and then summarize microbial metabolites and products which contribute to alcohol-related liver diseases and the strategies targeting the gut microbiota in alcohol-related liver diseases. The changes of the gut microbiota in alcohol-related liver disease, and the role of microbial metabolites in non-alcohol-related liver disease have been extensively described in other publications and reviews [21-23].
Gut-liver axis
The bidirectional communication between gut and liver is via portal vein, biliary tract and systemic circulation [24]. Liver is unique as it receives dual blood supply from both hepatic artery and portal vein. Contributing to 70-75% of the blood supply, the portal vein drains blood from the mesenteric veins of the small and large intestine [25]. Through the portal vein, endogenous and exogenous molecules produced by the host and microbes in the intestine translocate to the liver. Meanwhile, the liver releases bile acids and other bioactive mediators into the biliary tract and the systemic circulation, which influence the intestine and the gut microbiota.
Gut barrier dysfunction
The intestinal barrier separates the host from luminal microbes and restricts the movement of microbiota and molecules from the gut lumen. This barrier comprises 1) physical barrier including the mucins and underlying intestinal epithelial cells linked by tight junction proteins, 2) biochemical barrier such as antimicrobial proteins, and 3) immunological barrier such as immune cells [26]. Gut barrier dysfunction leads to the translocation of the microbes and microbial metabolites and products from the gut lumen to the portal vein and systemic circulation. Increased intestinal permeability was found in patients with alcohol-related liver disease [6-8].
Bile acids
Bile acids are synthesized from cholesterol in the pericentral hepatocytes and released in the biliary tract to facilitate the intestinal absorption and transport of cholesterol, dietary fat and fat-soluble vitamins by emulsifying them into micelles [27]. Before excretion into bile, bile acids are conjugated to glycine and to a less extent taurine in humans. 95% of the bile acids are reabsorbed in the ileum and recirculate to the liver through the portal vein [28]. The remaining bile acids are deconjugated and dehydroxylated to form secondary bile acids by the gut microbiota [29]. Bile acid homeostasis is tightly regulated by bile acid synthesis in the liver and biotransformation in the intestine to protect liver and other tissues and cells from cholesterol and bile acid toxicity [29]. Bile acids function as signaling molecules for nuclear receptors such as farnesoid X receptor (FXR) [30].
The bile acid pool size and composition are influenced by the gut microbiota. Compared with germ free mice, the bile acid pool size was reduced in conventional mice. Reduced levels of taurine-conjugated beta muricholic acid (TβMCA) and increased levels of taurine-conjugated cholic acid (TCA) and TαMCA were found in the liver of conventional mice, while TωMCA and taurodesoxycholic acid (TDCA) were exclusively present in the liver of conventional mice [31]. Lower bile acid diversity was also found in the liver of germ-free and antibiotic-treated (streptomycin/penicillin) rats compared with conventional rats [32].
In a cross-sectional study, a significant increase in the secondary bile acid formation was found in active alcohol use patients with cirrhosis compared with abstinent alcoholic cirrhotics and nonalcoholic cirrhotics [33]. The bile acid pool also changes with the severity of alcohol-related liver disease in patients [34]. Higher serum levels of total and conjugated bile acids were found in patients with alcoholic hepatitis, as well as fibroblast growth factor 19 (FGF19), which is a major regulator for bile acid synthesis [35].
In a rat model, chronic alcohol consumption has been observed to increase the bile acid pool and decrease their excretion [36]. Increased expression of genes involved in bile acid biosynthesis and efflux transport, as well as decreased expression of genes involved in bile acid influx into the liver were reported in rats fed with modified Lieber-DeCarli diet [37]. In another mouse model fed Lieber-DeCarli ethanol diet, supplementation of an FXR agonist WAY-362450 rescued the impaired FXR activity and suppressed the upregulation of Cyp2e1 and oxidative stress induced by ethanol in the liver of ethanol-fed mice [38]. In another study, Hartmann et al. demonstrated chronic ethanol feeding disrupted the bile acid enterohepatic circulation and reduced the intestinal FXR activity, and supplementation of an intestine-restricted FXR agonist fexaramine protected mice from ethanol-induced liver injury [39]. Although bile acid synthesis and interconversion was minimally affected by fexaramine, it repressed lipid synthesis and enhanced lipid catabolism in the liver of ethanol-fed mice [39]. In addition to FXR activation by fexaramine, overexpression of human fibroblast growth factor (FGF)-15-variant M52 in mice showed similar beneficial metabolic effects and mitigated ethanol-induced liver injury [39]. Influence of bile acids on the alcohol-induced liver disease is mainly through FXR-mediated pathways including FGF-15 secretion.
Indole-3-acetic acid
Derived from tryptophan by microbial enzymes tryptophan monooxygenase and indole-acetamide hydrolase, indole-3-acetic acid is an agonist for transcriptional aryl hydrocarbon receptor (AhR) [40,41]. Intestinal level of indole-3-acetic acid was reduced in patients with alcoholic hepatitis compared with non-alcoholic controls and in mice fed ethanol [42]. To investigate the therapeutic effect of indole-3-acetic acid supplementation, Hendrikx et al. supplemented the mice with indole-3-acetic acid by daily oral administration (20mM) over a 10-day ethanol feeding period and the authors found that indole-3-acetic acid attenuated ethanol-induced liver injury and restored expression of Il22 in type 3 innate lymphoid cells (ILC3) and regenerating islet-derived 3 gamma (Reg3g) mRNAs expression in small intestine epithelial cells [42]. Reg3 molecules are important guards to maintain the inner mucus layer devoid of bacteria. Impaired function leads to increased bacterial translocation and progression of ethanol-induced liver disease in mice [43]. Indole-3-acetic acid plays a protective role in ethanol-induced liver injury through the activation of AhR-dependent IL22 and Reg3g expression, which guards the inner mucus layer from bacteria translocation.
Butyrate
Butyrate is produced by gut bacteria during the fermentation of partially and nondigestible polysaccharides [44]. Lower butyrate concentrations in fecal samples were found in patients with alcohol overconsumption and in mice and rats fed with ethanol [45-47]. Cresci et al. demonstrated that tributyrin supplementation (0.83 to 10mM) in diet protected C57BL/6 mice from acute ethanol-induced liver injury [48]. Chronic-binge ethanol feeding impaired intestinal tight junction protein co-localization staining, depleted tight junctions and transepithelial electrical resistance in Caco-2 monolayers, and induced hepatic toll-like receptor and tumor necrosis factor-alpha protein expression [49]. These effects were significantly dampened by tributyrin supplementation [49]. In a follow-up study, the authors found tributyrin supplementation preserved immune responses and reduced oxidative stress in the proximal colon induced by ethanol exposure [50]. In another study, chronic-binge ethanol feeding induced liver injury, steatosis, and losses in tight junction protein expression were attenuated in mice supplemented with a designer synbiotic supplementation consisting of a butyrate-producing bacterium (Faecalibacterium prausnitzii) and a butyrate-yielding prebiotic (potato starch) in mice [51]. The intestinal barrier protects the host from the invasion of bacteria and toxins. Butyrate plays a key role in the protection of tight junction protein and gut barrier function, preventing the translocation of bacteria and microbial toxins. Through the protection of gut barrier, butyrate attenuates alcohol-induced liver lesions.
Long-chain fatty acids
Bacterial genes involved in fatty acid biosynthesis (fabF and fabG) were significantly lower in the feces from patients with alcohol use disorder compared with non-alcoholic controls [52]. Bacterial biosynthesis of saturated long chain fatty acids in mice were also reduced after chronic ethanol feeding [52]. Dietary supplementation of saturated long chain fatty acids (12% palmitic acid and 85% stearic acid) prevented ethanol-induced gut leakiness and ameliorated ethanol-induced liver injury in mice [52]. Long-chain fatty acids play a protective in alcohol-induced liver damage through the protection of gut barrier function which blocks bacterial translocation.
Endotoxin
Endotoxin is part of the outer membrane of the cell wall of Gram-negative bacteria. Endotoxin concentrations were increased more than 5-fold in the plasma of patients with alcohol use disorder compared with non-alcoholic subjects [8]. Alcohol administration impaired the function of the intestinal barrier, increased intestinal permeability, and facilitated the absorption and systemic translocation of endotoxin in rats [53]. Plasma endotoxin levels were strongly correlated with the severity of ethanol-induced liver injury in rats [54]. Bacterial endotoxin production was reduced by polymyxin B and neomycin treatment, which prevent the long-term ethanol exposure induced liver injury in rats [55]. When endotoxin crosses the gut barrier and enters the circulation, it interacts with and activates the Kupffer cells in the liver, which generates superoxide and TNF-α leading to liver damage [56].
Cytolysin
Cytolysin is a two-component exotoxin secreted by Enterococcus faecalis (E. faecalis). Cytolysin is toxic to a broad range of gram-positive bacteria and has lytic activity against eukaryotic cells [57]. The presence of cytolysin-positive E. faecalis correlated with the severity of liver disease and the mortality in alcoholic hepatitis patients [14], but not in patients with non-alcoholic fatty liver disease [58]. Duan et al. precisely edited the gut microbiota by bacteriophages to reduce cytolytic E. faecalis and to decrease cytolysin in the liver, which abolished ethanol-induced liver disease in humanized mice colonized with fecal bacteria from cytolysin-positive alcoholic hepatitis patients [14]. Cytolysin did not affect the function of the intestinal barrier [14]. Alcohol-induced changes in the gut barrier are essential for the translocation of cytolysin-positive E. faecalis from gut to the liver, which contributes to liver damage.
β-glucan
As part of the gut microbiota, the intestinal fungi also contribute to the development of alcohol-related liver disease [59]. Although the immune system develops tolerance to commensal fungi, it defends against the invasion of fungi [60]. When the gut barrier is dysregulated, the intestine serves as a source for systemic fungal infection and fungal products [61]. As a systemic immune response to fungi and fungal products, higher serum level of anti-Saccharomyces cerevisiae antibodies was shown in patients with alcoholic hepatitis [62]. Mycobiota populations and translocation of fungal β-glucan into systemic circulation were observed in mice after 8-week chronic administration of an ethanol containing Lieber DeCarli diet [59]. Subsequently, liver inflammation was induced by β-glucan through the C-type lectin-like receptor CLEC7A on Kupffer cells and IL-1β expression and secretion was elevated, which contributed to hepatocyte damage and ethanol-induced liver injury [59]. Antifungal agent, nonabsorbable amphotericin B, reduced intestinal fungal overgrowth, decreased β-glucan translocation, and ameliorated ethanol-induced liver damage in mice [59]. In another study, paeonal, a Chinese medicine derived from the dried root bark of Paeonia Suffruticoas Andr, reduced fungal abundance and β-glucan translocation, attenuated the production of CLEC7A and IL-1β in Kupffer cells and ameliorates ethanol-induced hepatic lipogenesis and inflammatory liver injury in mice [63]. Translocating across ethanol-induced damaged gut barrier to the liver, β-glucan binds to receptor CLEC7A on Kupffer cells and liver macrophages, and induces the expression and secretion of IL-1β, which contributes to liver damage.
Candidalysin
Lower fungal diversity was found in patients with alcohol-associated liver disease, with pronounced overgrowth of Candida [62]. Candidalysin is a peptide toxin secreted by Candida albicans. Candidalysin was associated with liver disease severity and mortality in patients with alcoholic hepatitis [64]. Candidalysin also exacerbated ethanol-induced liver disease in mice and damages primary hepatocytes in a dose-dependent manner in vitro [64]. Candidalysin does not affect the intestinal permeability and it enhances alcohol-associated liver disease independently of the β-glucan receptor CLEC7A on bone marrow-derived cells [64]. Ethanol-induced gut barrier dysfunction is necessary for the translocation of candidalysin from the intestine to the liver, where it can cause direct hepatocyte damage.
Modulation of gut microbiota
Strategies targeting the gut microbiota, including fecal microbiota transplantation, probiotics and antibiotics treatment have been evaluated in patients with alcohol-related liver disease. Fecal microbiota transplantation from healthy donor to steroid-ineligible patients with severe alcoholic hepatitis improved mortality [65]. Llopis et al. transplanted human microbiota from alcoholic patients to germ-free mice followed by 5 weeks of Lieber-DeCarli diet containing 3% ethanol, they found that the gut microbiota contributed to the individual susceptibility of alcohol-induced liver disease [26642859]. In addition to fecal microbiota transplantation, probiotics may have a therapeutic benefit in patients with alcohol-related liver disease [66-70]. In animal studies, Lactobacillus rhamnosus GG supplementation was found to ameliorate ethanol-induced liver injury in both mice and rats [71,72]. In another study, supplementation of depleted Akkermansia muciniphila by ethanol protected the host against ethanol-induced gut leakiness, enhanced mucus thickness and tight-junction expression, and ameliorated ethanol-induced hepatic injury and neutrophil infiltration in mice [5]. In a double-blind, placebo-controlled trial, 4-week treatment of antibiotic paromomycin did not show significant effect on liver function or endotoxin concentration in patients with alcohol-related liver disease [73]. In a non-randomized clinical trial, 8-week rifaximin treatment significantly improved endotoxemia and cognition in patients with cirrhosis and minimal hepatic encephalopathy [74].
Conclusions
Various microbial products and metabolites contribute to alcohol-related liver disease. This is facilitated by an ethanol-mediated disruption of the intestinal barrier function. Observations in patients confirm these findings. Microbiota-based therapeutic intervention studies in animal models that mimic alcohol-related liver disease in humans further support an important role of intestinal dysbiosis for alcohol-associated liver diseases. However, well-designed, larger and longitudinal clinical studies are needed to get additional and confirmatory insight into alcohol-associated changes in the intestinal microbiota. It will be important to use humanized gnotobiotic rodents for preclinical intervention studies. Such findings will be more relevant for human disease and are expected to be easier translatable to patients with alcohol-associated diseases. Precise modulation of the gut microbiota, microbial products and metabolites holds promise for the development of personalized therapeutics for alcohol-related liver disease.
Acknowledgements:
This study was supported in part by a Biocodex Microbiota Foundation Grant, NIH grants R01 AA24726, R01AA020703, U01 AA026939, by Award Number BX004594 from the Biomedical Laboratory Research & Development Service of the VA Office of Research and Development and services provided by the San Diego Digestive Diseases Research Center P30 DK120515.
Abbreviations:
- AhR
aryl hydrocarbon receptor
- FXR
farnesoid X receptor
- ILC3
type 3 innate lymphoid cells
- Reg3g
regenerating islet-derived 3 gamma
Footnotes
Conflicts of interest: B.S. has been consulting for Ferring Research Institute, Intercept Pharmaceuticals, HOST Therabiomics and Patara Pharmaceuticals. B.S.’s institution UC San Diego has received grant support from BiomX, NGM Biopharmaceuticals, CymaBay Therapeutics, Synlogic Operating Company and Axial Biotherapeutics.
References:
- [1].Zakhari S, Li T-K, Hepatology 2007, 46, 2032–2039. [DOI] [PubMed] [Google Scholar]
- [2].Osna NA, Donohue TM, Kharbanda KK, Alcohol Res 2017, 38, 147–161. [PMC free article] [PubMed] [Google Scholar]
- [3].Ciocan D, Voican CS, Wrzosek L, Hugot C, Rainteau D, Humbert L, Cassard A-M, Perlemuter G, Aliment. Pharmacol. Ther 2018, 48, 961–974. [DOI] [PubMed] [Google Scholar]
- [4].Puri P, Liangpunsakul S, Christensen JE, Shah VH, Kamath PS, Gores GJ, Walker S, Comerford M, Katz B, Borst A, Yu Q, Kumar DP, Mirshahi F, Radaeva S, Chalasani NP, Crabb DW, Sanyal AJ, TREAT Consortium, Hepatology 2018, 67, 1284–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Grander C, Adolph TE, Wieser V, Lowe P, Wrzosek L, Gyongyosi B, Ward DV, Grabherr F, Gerner RR, Pfister A, Enrich B, Ciocan D, Macheiner S, Mayr L, Drach M, Moser P, Moschen AR, Perlemuter G, Szabo G, Cassard AM, Tilg H, Gut 2018, 67, 891–901. [DOI] [PubMed] [Google Scholar]
- [6].Rainer F, Horvath A, Sandahl TD, Leber B, Schmerboeck B, Blesl A, Groselj-Strele A, Stauber RE, Fickert P, Stiegler P, Møller HJ, Grønbaek H, Stadlbauer V, Aliment. Pharmacol. Ther 2018, 47, 657–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Schäfer C, Parlesak A, Schütt C, Bode JC, Bode C, Alcohol Alcohol. 2002, 37, 81–86. [DOI] [PubMed] [Google Scholar]
- [8].Parlesak A, Schäfer C, Schütz T, Bode JC, Bode C, J. Hepatol 2000, 32, 742–747. [DOI] [PubMed] [Google Scholar]
- [9].Sato N, Lindros KO, Baraona E, Ikejima K, Mezey E, Järveläinen HA, Ramchandani VA, Alcohol. Clin. Exp. Res 2001, 25, 40S–45S. [DOI] [PubMed] [Google Scholar]
- [10].Monzoni A, Masutti F, Saccoccio G, Bellentani S, Tiribelli C, Giacca M, Mol. Med 2001, 7, 255–262. [PMC free article] [PubMed] [Google Scholar]
- [11].Tian C, Stokowski RP, Kershenobich D, Ballinger DG, Hinds DA, Nat. Genet 2010, 42, 21–23. [DOI] [PubMed] [Google Scholar]
- [12].Bluemel S, Williams B, Knight R, Schnabl B, Am. J. Physiol. Gastrointest. Liver Physiol 2016, 311, G1018–G1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Brenner DA, Paik Y-H, Schnabl B, J. Clin. Gastroenterol 2015, 49 Suppl 1, S25–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Duan Y, Llorente C, Lang S, Brandl K, Chu H, Jiang L, White RC, Clarke TH, Nguyen K, Torralba M, Shao Y, Liu J, Hernandez-Morales A, Lessor L, Rahman IR, Miyamoto Y, Ly M, Gao B, Sun W, Kiesel R, Hutmacher F, Lee S, Ventura-Cots M, Bosques-Padilla F, Verna EC, Abraldes JG, Brown RS, Vargas V, Altamirano J, Caballería J, Shawcross DL, Ho SB, Louvet A, Lucey MR, Mathurin P, Garcia-Tsao G, Bataller R, Tu XM, Eckmann L, van der Donk WA, Young R, Lawley TD, Stärkel P, Pride D, Fouts DE, Schnabl B, Nature 2019, 575, 505–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Yan AW, Fouts DE, Brandl J, Starkel P, Torralba M, Schott E, Tsukamoto H, Nelson KE, Brenner DA, Schnabl B, Hepatology 2011, 53, 96–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Hartmann P, Chen P, Wang HJ, Wang L, McCole DF, Brandl K, Stärkel P, Belzer C, Hellerbrand C, Tsukamoto H, Ho SB, Schnabl B, Hepatology 2013, 58, 108–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Fouts DE, Torralba M, Nelson KE, Brenner DA, Schnabl B, J. Hepatol 2012, 56, 1283–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Mutlu E, Keshavarzian A, Engen P, Forsyth CB, Sikaroodi M, Gillevet P, Alcohol. Clin. Exp. Res 2009, 33, 1836–1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Mutlu EA, Gillevet PM, Rangwala H, Sikaroodi M, Naqvi A, Engen PA, Kwasny M, Lau CK, Keshavarzian A, Am. J. Physiol. Gastrointest. Liver Physiol 2012, 302, G966–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Stärkel P, Leclercq S, de Timary P, Schnabl B, Clin. Sci 2018, 132, 199–212. [DOI] [PubMed] [Google Scholar]
- [21].Hartmann P, Seebauer CT, Schnabl B, Alcohol. Clin. Exp. Res 2015, 39, 763–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Caussy C, Hsu C, Lo M-T, Liu A, Bettencourt R, Ajmera VH, Bassirian S, Hooker J, Sy E, Richards L, Schork N, Schnabl B, Brenner DA, Sirlin CB, Chen C-H, Loomba R, Genetics of NAFLD in Twins Consortium, Hepatology 2018, 68, 918–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Chu H, Duan Y, Yang L, Schnabl B, Gut 2019, 68, 359–370. [DOI] [PubMed] [Google Scholar]
- [24].Tripathi A, Debelius J, Brenner DA, Karin M, Loomba R, Schnabl B, Knight R, Nat Rev Gastroenterol Hepatol 2018, 15, 397–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Abdel-Misih SRZ, Bloomston M, Surg. Clin. North Am 2010, 90, 643–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Mu Q, Kirby J, Reilly CM, Luo XM, Front Immunol 2017, 8, 598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Hofmann AF, Borgstroem B, J. Clin. Invest 1964, 43, 247–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Wahlström A, Sayin SI, Marschall H-U, Bäckhed F, Cell Metab. 2016, 24, 41–50. [DOI] [PubMed] [Google Scholar]
- [29].Chiang JYL, Ferrell JM, Gene Expr. 2018, 18, 71–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Chiang JYL, J. Lipid Res 2009, 50, 1955–1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Sayin SI, Wahlström A, Felin J, Jäntti S, Marschall H-U, Bamberg K, Angelin B, Hyötyläinen T, Orešič M, Bäckhed F, Cell Metab. 2013, 17, 225–235. [DOI] [PubMed] [Google Scholar]
- [32].Swann JR, Want EJ, Geier FM, Spagou K, Wilson ID, Sidaway JE, Nicholson JK, Holmes E, Proc. Natl. Acad. Sci. U.S.A 2011, 108 Suppl 1, 4523–4530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Kakiyama G, Hylemon PB, Zhou H, Pandak WM, Heuman DM, Kang DJ, Takei H, Nittono H, Ridlon JM, Fuchs M, Gurley EC, Wang Y, Liu R, Sanyal AJ, Gillevet PM, Bajaj JS, Am. J. Physiol. Gastrointest. Liver Physiol 2014, 306, G929–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Ciocan D, Voican CS, Wrzosek L, Hugot C, Rainteau D, Humbert L, Cassard A-M, Perlemuter G, Aliment. Pharmacol. Ther 2018, 48, 961–974. [DOI] [PubMed] [Google Scholar]
- [35].Brandl K, Hartmann P, Jih LJ, Pizzo DP, Argemi J, Ventura-Cots M, Coulter S, Liddle C, Ling L, Rossi SJ, DePaoli AM, Loomba R, Mehal WZ, Fouts DE, Lucey MR, Bosques-Padilla F, Mathurin P, Louvet A, Garcia-Tsao G, Verna EC, Abraldes JG, Brown RS, Vargas V, Altamirano J, Caballería J, Shawcross D, Stärkel P, Ho SB, Bataller R, Schnabl B, J. Hepatol 2018, 69, 396–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Lefevre AF, DeCarli LM, Lieber CS, J. Lipid Res 1972, 13, 48–55. [PubMed] [Google Scholar]
- [37].Xie G, Zhong W, Li H, Li Q, Qiu Y, Zheng X, Chen H, Zhao X, Zhang S, Zhou Z, Zeisel SH, Jia W, FASEB J. 2013, 27, 3583–3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Wu W, Zhu B, Peng X, Zhou M, Jia D, Gu J, Biochem. Biophys. Res. Commun 2014, 443, 68–73. [DOI] [PubMed] [Google Scholar]
- [39].Hartmann P, Hochrath K, Horvath A, Chen P, Seebauer CT, Llorente C, Wang L, Alnouti Y, Fouts DE, Stärkel P, Loomba R, Coulter S, Liddle C, Yu RT, Ling L, Rossi SJ, DePaoli AM, Downes M, Evans RM, Brenner DA, Schnabl B, Hepatology 2018, 67, 2150–2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Agus A, Planchais J, Sokol H, Cell Host Microbe 2018, 23, 716–724. [DOI] [PubMed] [Google Scholar]
- [41].Hendrikx T, Schnabl B, J. Intern. Med 2019, 286, 32–40. [DOI] [PubMed] [Google Scholar]
- [42].Hendrikx T, Duan Y, Wang Y, Oh J-H, Alexander LM, Huang W, Stärkel P, Ho SB, Gao B, Fiehn O, Emond P, Sokol H, van Pijkeren J-P, Schnabl B, Gut 2019, 68, 1504–1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Wang L, Fouts DE, Stärkel P, Hartmann P, Chen P, Llorente C, DePew J, Moncera K, Ho SB, Brenner DA, Hooper LV, Schnabl B, Cell Host Microbe 2016, 19, 227–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Tan J, McKenzie C, Potamitis M, Thorburn AN, Mackay CR, Macia L, Adv. Immunol 2014, 121, 91–119. [DOI] [PubMed] [Google Scholar]
- [45].Bjørkhaug ST, Aanes H, Neupane SP, Bramness JG, Malvik S, Henriksen C, Skar V, Medhus AW, Valeur J, Gut Microbes 2019, 10, 663–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Forsyth CB, Shaikh M, Bishehsari F, Swanson G, Voigt RM, Dodiya H, Wilkinson P, Samelco B, Song S, Keshavarzian A, Alcohol. Clin. Exp. Res 2017, 41, 2100–2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Xie G, Zhong W, Zheng X, Li Q, Qiu Y, Li H, Chen H, Zhou Z, Jia W, J Proteome Res 2013, 12, 3297–3306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Cresci GA, Bush K, Nagy LE, Alcohol. Clin. Exp. Res 2014, 38, 1489–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Cresci GA, Glueck B, McMullen MR, Xin W, Allende D, Nagy LE, J. Gastroenterol. Hepatol 2017, 32, 1587–1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Glueck B, Han Y, Cresci G. a. M., J Immunol Res 2018, 2018, 9671919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Roychowdhury S, Glueck B, Han Y, Mohammad MA, Cresci GAM, Nutrients 2019, 11, DOI 10.3390/nu11010097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Chen P, Torralba M, Tan J, Embree M, Zengler K, Stärkel P, van Pijkeren J-P, DePew J, Loomba R, Ho SB, Bajaj JS, Mutlu EA, Keshavarzian A, Tsukamoto H, Nelson KE, Fouts DE, Schnabl B, Gastroenterology 2015, 148, 203–214.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Tamai H, Kato S, Horie Y, Ohki E, Yokoyama H, Ishii H, Alcohol. Clin. Exp. Res 2000, 24, 390–394. [PubMed] [Google Scholar]
- [54].Nanji AA, Khettry U, Sadrzadeh SM, Yamanaka T, Am. J. Pathol 1993, 142, 367–373. [PMC free article] [PubMed] [Google Scholar]
- [55].Adachi Y, Moore LE, Bradford BU, Gao W, Thurman RG, Gastroenterology 1995, 108, 218–224. [DOI] [PubMed] [Google Scholar]
- [56].Wheeler MD, Alcohol Res Health 2003, 27, 300–306. [PMC free article] [PubMed] [Google Scholar]
- [57].Tang W, van der Donk WA, Nat. Chem. Biol 2013, 9, 157–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Lang S, Demir M, Duan Y, Martin A, Schnabl B, Liver Int. 2020, DOI 10.1111/liv.14377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Yang A-M, Inamine T, Hochrath K, Chen P, Wang L, Llorente C, Bluemel S, Hartmann P, Xu J, Koyama Y, Kisseleva T, Torralba MG, Moncera K, Beeri K, Chen C-S, Freese K, Hellerbrand C, Lee SM, Hoffman HM, Mehal WZ, Garcia-Tsao G, Mutlu EA, Keshavarzian A, Brown GD, Ho SB, Bataller R, Stärkel P, Fouts DE, Schnabl B, J. Clin. Invest 2017, 127, 2829–2841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Iliev ID, Underhill DM, Curr. Opin. Microbiol 2013, 16, 366–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Miranda LN, van der Heijden IM, Costa SF, Sousa API, Sienra RA, Gobara S, Santos CR, Lobo RD, Pessoa VP, Levin AS, J. Hosp. Infect 2009, 72, 9–16. [DOI] [PubMed] [Google Scholar]
- [62].Lang S, Duan Y, Liu J, Torralba MG, Kuelbs C, Ventura-Cots M, Abraldes JG, Bosques-Padilla F, Verna EC, Brown RS, Vargas V, Altamirano J, Caballería J, Shawcross D, Lucey MR, Louvet A, Mathurin P, Garcia-Tsao G, Ho SB, Tu XM, Bataller R, Stärkel P, Fouts DE, Schnabl B, Hepatology 2020, 71, 522–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Wu J, Wu D, Ma K, Wang T, Shi G, Shao J, Wang C, Yan G, J. Leukoc. Biol 2020, DOI 10.1002/JLB.3MA0120-325RR. [DOI] [PubMed] [Google Scholar]
- [64].Chu H, Duan Y, Lang S, Jiang L, Wang Y, Llorente C, Liu J, Mogavero S, Bosques-Padilla F, Abraldes JG, Vargas V, Tu XM, Yang L, Hou X, Hube B, Stärkel P, Schnabl B, J. Hepatol 2020, 72, 391–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Philips CA, Pande A, Shasthry SM, Jamwal KD, Khillan V, Chandel SS, Kumar G, Sharma MK, Maiwall R, Jindal A, Choudhary A, Hussain MS, Sharma S, Sarin SK, Clinical Gastroenterology and Hepatology 2017, 15, 600–602. [DOI] [PubMed] [Google Scholar]
- [66].Loguercio C, Federico A, Tuccillo C, Terracciano F, D’Auria MV, De Simone C, Del Vecchio Blanco C, J. Clin. Gastroenterol 2005, 39, 540–543. [DOI] [PubMed] [Google Scholar]
- [67].Stadlbauer V, Mookerjee RP, Hodges S, Wright GAK, Davies NA, Jalan R, J. Hepatol 2008, 48, 945–951. [DOI] [PubMed] [Google Scholar]
- [68].Lata J, Novotný I, Príbramská V, Juránková J, Fric P, Kroupa R, Stibůrek O, Eur J Gastroenterol Hepatol 2007, 19, 1111–1113. [DOI] [PubMed] [Google Scholar]
- [69].Dhiman RK, Rana B, Agrawal S, Garg A, Chopra M, Thumburu KK, Khattri A, Malhotra S, Duseja A, Chawla YK, Gastroenterology 2014, 147, 1327–1337.e3. [DOI] [PubMed] [Google Scholar]
- [70].Kirpich IA, Solovieva NV, Leikhter SN, Shidakova NA, Lebedeva OV, Sidorov PI, Bazhukova TA, Soloviev AG, Barve SS, McClain CJ, Cave M, Alcohol 2008, 42, 675–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Forsyth CB, Farhadi A, Jakate SM, Tang Y, Shaikh M, Keshavarzian A, Alcohol 2009, 43, 163–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Wang Y, Kirpich I, Liu Y, Ma Z, Barve S, McClain CJ, Feng W, Am. J. Pathol 2011, 179, 2866–2875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Bode C, Schäfer C, Fukui H, Bode JC, Alcohol. Clin. Exp. Res 1997, 21, 1367–1373. [PubMed] [Google Scholar]
- [74].Bajaj JS, Heuman DM, Sanyal AJ, Hylemon PB, Sterling RK, Stravitz RT, Fuchs M, Ridlon JM, Daita K, Monteith P, Noble NA, White MB, Fisher A, Sikaroodi M, Rangwala H, Gillevet PM, PLoS ONE 2013, 8, e60042. [DOI] [PMC free article] [PubMed] [Google Scholar]