

Summary
Patient-derived tumor organoid cultures are an essential and innovative methodology for translational research. However, current techniques to establish these cultures are cumbersome, expensive, and often require irreplaceable clinical tissue from surgery or core biopsies. Fine-needle aspiration (FNA) provides a minimally invasive biopsy technique commonly performed in clinical settings. Here, we provide a protocol for FNA. We have found that FNA provides a cost-effective, rapid, and streamlined method for tissue acquisition for cancer organoid culture.
For complete details on the use and execution of this protocol, please refer to Lee et al. (2020) and Vilgelm et al. (2020).
Subject Areas: cell culture, cell isolation, cancer, organoids
Graphical Abstract
Highlights
-
•
Fine-needle aspiration (FNA) to obtain patient-derived organoid cultures
-
•
FNA offers a cost-effective and minimally invasive method to obtain tissue
-
•
FNA-based organoid cultures can be used for many downstream applications
Patient-derived tumor organoid cultures are an essential and innovative methodology for translational research. However, current techniques to establish these cultures are cumbersome, expensive, and often require irreplaceable clinical tissue from surgery or core biopsies. Fine-needle aspiration (FNA) provides a minimally invasive biopsy technique commonly performed in clinical settings. Here, we provide a protocol for FNA. We have found that FNA provides a cost-effective, rapid, and streamlined method for tissue acquisition for cancer organoid culture.
Before you begin
Matrigel preparation
Timing: 6–8 h
-
1.Aliquot Matrigel
-
a.Remove stock bottle of Matrigel from −20°C storage and thaw at 4°C for 6–8 h.
-
b.Place stock bottle of Matrigel on ice in a sterile environment, as it will be used for tissue culture.
-
c.Aliquot 1 mL of Matrigel into labeled microcentrifuge tubes and immediately place aliquots on ice.
-
d.Store aliquots at −20°C for long-term storage.
-
a.
Note: Aliquots can be thawed at 4°C and we have kept aliquots at 4°C for up to a week to prevent additional freeze-thaw or waste.
Media preparation
-
2.Media Component Aliquot Preparation
-
a.Aliquot fetal bovine serum (FBS)
-
i.Thaw 500 mL bottle of FBS at 4°C.
-
ii.Aliquot 45 mL of FBS into labeled 50 mL conical tubes.Note: If more than 45 mL of FBS is aliquoted into a 50 mL conical, upon refreezing, the FBS can leak.
-
i.
-
b.Aliquot L-glutamine
-
i.Thaw bottle of L-glutamine at 4°C for 12–24 h for next-day aliquoting or quickly in 37°C water bath. Aliquot 6 mL of L-glutamine into labeled 15 mL conical tubes.
-
i.
-
c.Aliquot penicillin-streptomycin (pen/strep)
-
i.Thaw bottle of pen/strep at 4°C for 12–24 h for next-day aliquoting or quickly in a 37°C water bath.
-
ii.Label appropriate amount of 15 mL conical tubes.
-
iii.Aliquot 6 mL of pen/strep into labeled 15 mL conical tubes.
-
i.
-
d.Store all aliquots at −20°C.Note: Once prepared, aliquots can be thawed for 12–24 h at 4°C for next-day use, or in a 37°C water bath for immediate use.
-
a.
-
3.
Prepare Complete DMEM and Complete Media as described in the Materials and Equipment section.
Pre-warm plate and complete media
-
4.
Place a 6-well plate in the incubator to warm for at least 20 min before use.
-
5.
Aliquot 20 mL of Complete Media and place in a 37°C water bath for at least 20 min before use.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, peptides, and recombinant proteins | ||
| Dulbecco’s modified Eagle’s medium (DMEM) | Sigma | D6429-500ML |
| BioWhittaker RPMI-1640 cell culture media, Lonza (RPMI) | VWR | 12001-534 |
| Nutrient mixture F-12 Ham (Ham’s F12) | Sigma | N4888-500ML |
| MCDB 105 medium, unmodified | Cell Applications | 117-500 |
| Penicillin-streptomycin | Sigma | P4333-100ML |
| L(+)-Glutamine 200 mM 100× | VWR | VWRL0131-0100 |
| Normocin | Invivogen | ant-nr-1 |
| Fetal bovine serum (FBS) | Thermo Fisher Scientific | 26140079 |
| B-27 supplement | Gibco | 17504044 |
| Matrigel | Fisher Scientific | CB-40234 |
| Trypsin (EDTA 0.25%) | Gibco | 25200056 |
| Dimethyl sulfoxide (DMSO) | Sigma | D8418 |
| Phosphate-buffered saline (PBS) | Corning | MT21040CV |
| Other | ||
| 24-well Corning Costar ultra-low-attachment microplates, sterile | Fisher Scientific | 07-200-602 |
| 6-well CELLSTAR cell culture multiwell plates, polystyrene, Greiner Bio-One, sterile | VWR | 82050-842 |
| VWR T75 cell culture flasks, sterile | VWR | 10062-860 |
| Low-retention filter tips, 1,000 μL, sterile | VWR | 89174-530 |
| Low-retention filter tips, 200 μL, sterile | VWR | 89174-526 |
| Low-retention filter tips, 20 μL, sterile | VWR | 89174-524 |
| Syringe holder, 10 mL | Belpro Medical | 938 |
| BD PrecisionGlide single-use needles: regular bevel, regular wall, 25-gauge | Fisher Scientific | 14-821-13D |
| 10 mL Fisherbrand sterile syringes for single use | Fisher Scientific | 14-955-459 |
| VWR centrifuge tube, flatcap, sterile, 50 mL | VWR | 76176-952 |
| VWR disposable aspirating pipets, individually wrapped, sterile | VWR | 414004-265 |
| VWR disposable plugged 5 mL serological pipets, individually wrapped, sterile | VWR | 89130-896 |
| VWR disposable plugged 10 mL serological pipets, individually wrapped, sterile | VWR | 89130-898 |
| VWR disposable plugged 25 mL serological pipets, individually wrapped, sterile | VWR | 89130-900 |
| Wobble-not plugged 50 mL serological pipets, individually wrapped, sterile | VWR | 75993-434 |
| CryoCooler freeze controller (Mr. Frosty) | VWR | 414004-284 |
| Corning cell scrapers | Fisher Scientific | 07-200-365 |
| 50 mL conical tubes | N/A | N/A |
| 15 mL conical tubes | N/A | N/A |
| Petri dishes | N/A | N/A |
| Forceps, sterile | Sklar | 96-2917 |
| Cryovials | VWR | 10018-754 |
| Hemocytometer | N/A | N/A |
| Specimen cup | N/A | N/A |
| CO2 incubator | N/A | N/A |
| Water bath | N/A | N/A |
| P1000 | N/A | N/A |
| P200 | N/A | N/A |
| P20 | N/A | N/A |
| Easypet | N/A | N/A |
| Centrifuge | N/A | N/A |
| Biosafety cabinet | N/A | N/A |
| −80°C freezer | N/A | N/A |
| Liquid nitrogen freezer | N/A | N/A |
Materials and equipment
Complete DMEM
| Reagent | Final concentration (mM or μM) | Amount |
|---|---|---|
| DMEM | 44% | 250 mL |
| Ham’s F12 | 22% | 125 mL |
| MCDB | 22% | 125 mL |
| FBS | 10% | 56 mL |
| L-Glutamine | 0.9% | 5.6 mL |
| Penicillin-streptomycin | 0.9% | 5.6 mL |
| Normocin | 0.2% | 1.2 mL |
| Total | 100% | 568.4 mL |
CRITICAL: Certain cell types (i.e., gastrointestinal and pancreaticobiliary) may require the use of Advanced DMEM with supplements that assist with growth (i.e., Glutamax, Epidermal Growth Factor (EGF), Fibroblast Growth Factor (FGF), Insulin-Transferrin-Selenium (ITS)).
Note: Normocin is added to prevent mycoplasma contamination.
Note: Instead of penicillin-streptomycin, Antibiotic-Antimycotic (100×) (Thermo Fisher, 15240062) can be substituted to help prevent fungal growth that is common in animal FNAs.
Note: Complete DMEM should be stored at 4° C and can be used for up to one month after preparation.
Complete media
| Reagent | Final concentration (mM or μM) | Amount |
|---|---|---|
| Complete DMEM | 98% | 515 mL |
| B27 | 2% | 10.5 mL |
| Total | 100% | 525.5 mL |
Note: Store complete media at 4˚ C when not in use. The media expires approximately 1 month after it is freshly made.
Step-by-step method details
Ex vivo specimen fine-needle aspiration (FNA)
Fine-needle aspiration is a technique commonly used in a clinical setting to obtain cells for cytopathology and genetic testing. Here, we apply it to a research setting to obtain cells and cell clumps that will generate organoid cultures. FNA is advantageous over traditional digestion methods because it is much more gentle, allowing for increased cell survival.
-
1.
Attach a sterile 25-gauge beveled needle to a sterile 10 mL luer-lock syringe and insert into a syringe holder.
-
2.
If using surgical tissue, transfer specimen to a sterile petri dish within a laminar flow biosafety cabinet. If performing FNA in vivo, perform in a procedure room with sterile technique.
-
3.
Immobilize the specimen/nodule with sterile-gloved fingers or sterile forceps and using your other hand, insert the needle with the syringe holder perpendicular to the specimen/nodule.
-
4.
Apply backward pressure to the syringe using the syringe holder, while rapidly agitating back and forth in a “saw-like” motion within the specimen at a rate of 2–3 strokes per second (Methods Video S1). This motion loosens tumor and other cells from the stromal matrix. One “needle pass” typically involves 10–20 forward strokes (excursions). For optimal tumor sampling, reangle the needle slightly with each excursion so that sampling occurs in a fan shape. The needle should not come out of the specimen/nodule until the 10–20 excursions have been completed and material is visible in the needle hub. The needle should not protrude from the opposite side of the specimen at any point. Troubleshooting 1
Note: Traditionally, FNA is performed on patients when the nodule is 1 cm or greater in greatest dimension. However, ex vivo FNA has been successfully performed on specimens as small as 2 mm with adequate yield for organoid culture, including heavily fibrotic tumors.
-
5.
Once material is visible in the hub of the needle, release suction and remove the needle from the specimen.
-
6.
Following this process (a “needle pass”), rinse the needle in 20 mL of sterile, pre-warmed, RPMI or DMEM in a 50 mL conical tube. Expel needle contents into the conical tube and rinse needle 2–3 times.
Note: Extensive rinsing (more than 3–4 times) will lead to increased cell shearing and lower yields.
-
7.
Repeat steps c–f 3–5 times.
-
8.
Dispose of syringe and needle in appropriate sharps container.
Cell growth protocol
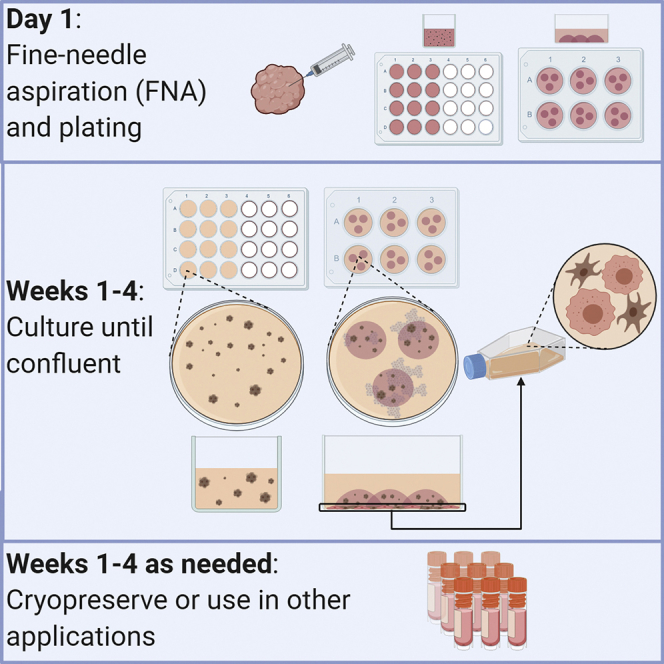
Once you have obtained the cells from the FNA, they can be immediately plated using one of two culture methods. Semi-solid culture is useful for longer culture periods and free movement of cells. Disc culture can be beneficial for more compact organoid growth and allows for generation and propagation of two-dimensional cultures. We recommend using both semi-solid and disc culture methods for new organoid cultures, as the growth needs vary from sample to sample. See Figure 1. For a summary of steps used in FNA-based organoid culture.
-
9.
Centrifuge the FNA material in the conical tube at 340 × g for 5 min at 20°C–25°C.
-
10.
Aspirate the supernatant and resuspend the pellet in Complete Media.
Note: If using only one plating method, we recommend resuspending the FNA material in 300 μL. If both plating methods are to be used, we recommend resuspending the FNA material in 600 μL and dividing into two 300 μL aliquots.
-
11.Cells can be plated in semi-solid culture to obtain organoids, or in disc culture to obtain both organoids and 2D cell cultures.
-
a.Semi-solid culture
-
i.Pipet and mix 300 μL of the FNA cell suspension with 5% Matrigel in complete media with an additional 2% FBS.
-
ii.Pipet 550 μL cell suspension in a 24-well low-attachment plate following a figure 8 motion while gently expelling cell suspension.
-
iii.Place the cells in the incubator.
-
iv.Replenish 200 μL per well of fresh complete media, 5% Matrigel, and 2% FBS dropwise weekly.
-
v.Passage cells as needed. Troubleshooting 2.CRITICAL: In this type of culture, the low percentage of Matrigel makes the matrix fragile and is more susceptible to media evaporation. Troubleshooting 3.
-
i.
-
b.Disc culture (Figure 2)
-
i.Pipet and mix 300 μL of the FNA cell suspension with 75% Matrigel and 25% complete media.
-
ii.Add three 50 μL drops (disc) to each well of a pre-warmed 6-well plate. Alternatively, you can plate a single 150–200 μL disc per well of the 6-well plate.
-
iii.Place the plate in a tissue culture incubator for 10 min to allow the Matrigel to solidify.
-
iv.Cover cells with warmed complete media by slowly pipetting the media down the side of the well until the discs are completely covered (~2.5 mL).
-
v.Place cells back in the incubator.
-
vi.Replace media every 2–3 days by aspirating off the old media and replacing it with pre-warmed complete media following the step 11b.v. Troubleshooting 4
-
i.
-
c.2-dimensional (2D) cell culture (Figure 2)
-
i.Follow the disc culture protocol until 2D cells have undermined the disc.
-
ii.Freeze the disc down following the freezing cells protocol.
-
iii.Add 2 mL of 20°C–25°C complete media to each well after removing disc. Make sure media is either 4°C or 20°C–25°C, as 37°C media could shock the cells.
-
iv.While in a 6-well plate, replace media with fresh, pre-warmed media every 2–3 days.
-
v.Once cells are confluent, passage them from the 6-well plate to a T75 flask following step 2a in the passaging cells protocol.
-
vi.In the T75, replace three-fourths (~11 mL) of complete media with fresh complete media weekly.
-
vii.Passage cells as needed.Note: If 2D culture is not desired, plate in a low-attachment plate in the semi-solid format. Low-attachment plates prevent the matrix disrupting 2D cells from growing underneath the organoids.
-
i.
-
a.
Figure 1.

Summary of steps for plating cells from FNA
Collect cells via FNA. After the FNA, half the cells are plated in semi-solid culture in a 24-well ultra-low attachment plate. The other half of the cells are plated in a 6-well plate in the 75% Matrigel disc culture.
Figure 2.

Two-dimensional cultures can be derived from disc culture
(A) Animated figure of disc culture organoids with 2Ds undermining the disc.
(B) Bright-field image of a patient-derived melanoma organoid 6 days after plating in disc culture with 2Ds undermining the disc culture at 10×. Scale bar, 100 μm.
Passaging cells
Cells should be passaged when media needs replaced or replenished more often than twice a week or when organoids are harvested for experimental use.
-
12.Semi-solid culture (Figure 3)
-
a.Pipet the organoid suspension with a P1000.
-
b.In order to break up the Matrigel/organoids, draw a small amount of organoid suspension up into the pipet and then expel it slowly while moving the pipet in a circular motion in the plate. Repeat this 2–4 times.
-
c.Transfer the cell suspension to a 50 mL conical tube.
-
d.Centrifuge the cell suspension at 340 × g for 5 min at 20°C–25°C and aspirate off the supernatant.
-
e.Resuspend the pelleted cells in complete media.
-
f.Pipet out the desired amount of the cell suspension and replate following step 12b in the cell growth protocol.
-
g.Either discard the remaining cell suspension or save for other uses (e.g., freezing, plating for experiments, etc.)
-
a.
-
13.Disc culture
-
a.Remove media and add 1 mL ice-cold PBS to each well.
-
b.Scrape discs gently off of the plate with a cell scraper.
-
c.Pipet ice-cold PBS to break apart discs and collect into a 1.5 mL microfuge tube.
-
d.Centrifuge cells at 340 × g for 5 min at 20°C–25°C then remove PBS and Matrigel overlying the pellet.
-
e.Wash pellet gently with 1 mL 4°C PBS and centrifuge 340 × g for 5 min at 20°C–25°C.
-
f.Remove PBS and resuspend cell pellet in 3× Matrigel (for a 1:3 split) then plate in pre-warmed 6-well plate.
-
a.
-
14.2D cell culture
-
a.Passaging from a 6-well to a T75 flask
-
i.Aspirate off all the media.
-
ii.Add 1 mL of trypsin to each well.
-
iii.Incubate at 37°C for 3 min.
-
iv.Pipet trypsinized cells from each well into a conical tube with 20 mL of DMEM or RPMI.
-
v.Centrifuge the cell suspension at 340 × g for 5 min at 20°C–25°C and aspirate off the supernatant.
-
vi.Resuspend cell pellets in 1 mL of complete media then transferred to a T75 containing 14 mL of complete media.
-
vii.Rock the T75 back and forth until the bottom is covered with cell suspension.
-
viii.Place in the incubator.
-
i.
-
a.
Note: Depending on the number of cells, multiple T75s might be needed.
Figure 3.

Passaging of semi-solid cultures
(A) Once the organoids are centrifuged, resuspend and aliquot (B) the desired amount for replating then use the rest for freeze downs. (C) For a 1:4 passage resuspend in 2,800 μL and pipet 700 μL in a new conical (to replate). The remaining 2,100 μL is sufficient for 3 freeze downs.
Freezing cells
As these cultures come from ex vivo tissue, they are not easily replaceable or replicable. We recommend freezing down multiple vials of any organoid cultures before beginning experiments, as these cultures can be fragile and do change over time. Freeze downs enable preservation of the cultures should there be a contamination or error in propagation.
-
15.Obtaining Cells
-
a.Disc Culture
-
i.Aspirate off media.
-
ii.Pipet 1 mL of 4˚C PBS directly on to the disc in each well.
-
iii.Use a cell scraper to gently scrape off any remaining disc.
-
iv.Pipet cell suspension into a 1.5 mL microfuge tube.
-
v.Pipet up and down several times to dissolve the Matrigel.
-
i.
-
b.Semi-solid Culture
-
i.Follow the relevant steps in the Passaging Cells protocol and save the remaining cell suspension.
-
i.
-
c.2D culture
-
i.Follow relevant steps in the Passaging Cells protocol and save the remaining cell suspension.
-
ii.Pipet 20 μL into a hemocytometer to count the number of cells.
-
i.
-
a.
-
16.
Centrifuge the cell suspension at 340 × g for 5 min.
-
17.Aspirate off the supernatant and resuspend in complete DMEM.
-
a.For disc and semi-solid culture, deposit approximately 2 wells per cryovial.
-
b.For 2D culture, deposit 300,000–1,000,000 cells per cryovial.
-
a.
Note: For disc culture and semi-solid culture, the number of wells per cryovial can vary depending on amount and size of organoids.
-
18.
Pipet 700 μL of cell suspension into each appropriately labeled cryovial.
-
19.
Pipet 200 μL of FBS into each cryovial.
-
20.
Pipet 100 μL of DMSO into a cryovial.
-
21.
Screw the cap on tightly and invert 2–3 times.
-
22.
Place directly into a Mr. Frosty.
-
23.
Repeat steps 20–22 until all the cryovials are complete.
-
24.
Tighten the lid on the Mr. Frosty and place directly into a −80˚ C freezer.
-
25.
Keep cells in Mr. Frosty for at least 24 h and then they can be transferred to a box prior to transfer to liquid nitrogen for long-term storage.
-
26.
After a week in the −80˚C freezer, transfer cells to a liquid nitrogen freezer at or below −130˚ C for longer storage.
Pause Point: Cells can be stored in the −80˚ C freezer for up to a week and longer term at or below −130˚ C in a liquid nitrogen freezer.
Thawing cells
Cultures must be properly thawed to give the cells the best chance of recovery. Growth may not immediately match that of the culture prior to being frozen but should pick up within a week or two after thawing.
-
27.
Preheat water bath to 37˚ C.
-
28.
Pipet 20 mL of media into a 50 mL conical tube warm to ~24˚ C.
-
29.
Obtain cryovial with cells.
Optional: If thawing more than one or two cryovials at a time, store the other cryovials on dry ice until ready to thaw.
-
30.
Place cryovial in water bath.
-
31.
Incubate for 1 minute then check to see if it has thawed. If not, check in 15–30 s intervals until thawed.
-
32.
Once thawed, pipet contents from cryovial into the media from step 28.
-
33.
Rinse the cryovial with 1,000 μL of the media from step 32 and pipet it back into the conical tube.
-
34.
Screw the conical tube lid tightly and invert to ensure adequate mixing.
Note: This is to ensure the DMSO is diluted adequately.
-
35.
Centrifuge at 340 × g for 5 min.
-
36.
Aspirate off the supernatant.
Optional: Resuspend in 20 mL of media or PBS, centrifuge at 340 × g for 5 min, and aspirate off the supernatant.
-
37.
Resuspend and plate as desired following steps in the Cell Growth Protocol.
Expected outcomes
This protocol describes an effective and minimally invasive method to establish patient-derived cell lines and three-dimensional growth of these cells. Between one to three weeks of culture, single cells and small clusters obtained from FNA grow to create small organoids (Figure 4). Initial organoid culture success rates and long-term propagation rates following cryopreservation can be found in Vilgelm et al., 2020.
Figure 4.

Images of patient-derived cancer organoids
(A) Bright-field images of patient-derived melanoma organoids plated in disc culture 6 days after plating 10×. Scale bars, 100 μm.
(B) Bright-field images of patient-derived thyroid organoids plated in semi-solid culture 21 days after plating 10×. Scale bars, 100 μm.
Limitations
Not all tumors robustly produce organoids, although most tumors we assessed grow with this method including thyroid, colon, pancreas, gastric, renal, and melanoma. We recommend utilizing both culture methods when a sample is collected (semi-solid and disc), as some samples will preferentially grow in the semi-solid culture, while others will preferentially grow as discs. In addition, not all tumor types will generate a robust population of 2D cells. Two-dimensional cultures are typically composed of both fibroblasts and tumor cells, with the fibroblast percentage increasing with each passage. In our hands, thyroid and melanoma grow the most robust 2D cultures, with gastrointestinal and renal tumors having fewer or no 2D cells.
It is vital to obtain the sample directly from surgery or as soon as it reaches surgical pathology. This is to make sure the samples are sterile. If the sample is not obtained during these times, the grossing/dissection in surgical pathology can cause contamination or cell death (i.e., ink, formalin, etc.).
Troubleshooting
Problem 1
Clogged needle during FNA.
Potential solution
-
•
Very carefully unscrew the needle while holding the syringe holder/syringe combination. Pull the stopper back on the syringe holder/syringe combination then screw the needle back on. Put the needle in the rinsing media and press down forcefully on the stopper.
Problem 2
Organoid clumping after plating and during growth.
Potential solution
-
•
To ensure even distribution of cells, pipet the cell suspension very well or invert to mix approximately 10 times.
-
•
Mixing the cells well before adding the Matrigel. While working with Matrigel, make sure to keep it and anything that contains it on ice until it is ready to start solidifying.
-
•
Pipetting vigorously can create heat and cause the Matrigel to start solidifying so mix keep the cell suspension with Matrigel on ice.
-
•
When plating semi-solid culture, pipet the cell suspension in a figure 8 motion while plating.
Problem 3
Media evaporation.
Potential solution
-
•
To grow robust cultures, some lines may need to be incubated for many weeks. Because of this, they can be prone to evaporation of complete media. To decrease evaporation, plate the cultures in the middle of the plate and fill the surrounding empty wells in the plates with sterile PBS.
-
•
Replenish the water pans in the incubator frequently to maintain humidity.
-
•
Place cells in a separate incubator that is not accessed as frequently to decrease exposure to door opening.
-
•
Replace or replenish the media more frequently.
Problem 4
Disc detachment from plate.
Potential solution
-
•
Always use warmed media (37˚ C) when changing the media on disc cultures to prevent disruption of the Matrigel matrix. Cold media causes the Matrigel to dissociate.
-
•
Pipet carefully down the side of each well to prevent disc detachment.
-
•
Pre-warm the plate prior to plating the disc culture ensures better attachment.
-
•
When 2D cells extensively undermine the discs, the disc can detach, so this is a good point to freeze down the organoid disc.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Vivian Weiss, (vivian.l.weiss@vumc.org).
Materials availability
This study did not generate new unique reagents.
Data and code availability
This study did not generate or analyze any datasets or code.
Acknowledgments
The project described was supported by the National Center for Research Resources, grant UL1 RR024975-01, and is now at the National Center for Advancing Translational Sciences, grant 2 UL1 TR000445-06. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. The project described was also supported by American Thyroid Association (2019-0000000090 to V.L.W.), K12 Vanderbilt Clinical Oncology Research Career Development Program (NCI K12CA090625 to V.L.W.), V Foundation Scholar Award (to V.L.W.), Children’s Cancer Research Fund Emerging Scientist Grant (to V.L.W.), ACS (133934-CSDG-19-216-01-TBG to V.L.W.), NIH R37 (CA233770 to A.V.), Training in Pharmacological Sciences NIH T32GM007628-42 (to K.N.B.), NIH K08 (1K08 CA240901-01A1 to V.L.W.), NIH R35GM122516, and R01CA224188 (to E.L.). Figures 1, 2A, and 3 and Video S1 were created with Biorender.com.
Author contributions
C.J.P. wrote the paper; K.N.B. edited the paper and assisted with figures; M.E.B. and E.L. edited the paper; A.V., N.B., O.G.M., and V.L.W. isolated patient tissue, consented patients, designed the methodology, and edited the paper.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.xpro.2020.100220.
References
- Lee M., Bergdorf K., Phifer C., Jones C., Byon S., Sawyer L., Bauer J., Weiss V. Novel three-dimensional cultures provide insights into thyroid cancer behavior. Endocr. Relat. Cancer. 2020;27:111–121. doi: 10.1530/ERC-19-0374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilgelm A., Bergdorf K., Wolf M., Bharti V., Shattuck-Brandt R., Blevins A., Jones C., Phifer C., Lee M., Lowe C. Fine-needle aspiration-based patient-derived cancer organoids. iScience. 2020;23:101408. doi: 10.1016/j.isci.2020.101408. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate or analyze any datasets or code.