

Abstract
Bovine respiratory syncytial virus (BRSV) is an etiologic agent of bovine respiratory disease. The rapid evolutionary rate of BRSV contributes to genetic and antigenic heterogeneity of field strains and causes occasional vaccine failure. We conducted molecular epidemiologic characterization of BRSV circulating in Japan to obtain genetic information for vaccine-based disease control. Phylogenetic analysis of G and F gene sequences revealed that all of the isolated Japanese BRSV strains clustered in the same genetic subgroup, which was distinct from the 9 known groups. We assigned the Japanese group to subgenotype X. The Japanese isolates formed 2 temporal clusters: isolates from 2003 to 2005 clustered in lineage A; isolates from 2017 to 2019 formed lineage B. The alignment of the deduced amino acid sequences of the G gene revealed that the central hydrophobic region responsible for viral antigenicity is conserved in all of the isolates; unique amino acid mutations were found mainly in mucin-like regions. Our results suggest that BRSV has evolved uniquely in Japan to form the new subgenotype X; the antigenic homogeneity of the viruses within this group is inferred.
Keywords: bovine respiratory disease, bovine respiratory syncytial virus, subgenotype X
Bovine respiratory syncytial virus (BRSV; Pneumoviridae, Orthopneumovirus, Bovine orthopneumovirus) is a major cause of bovine respiratory disease, which has significant economic impact on the cattle industry worldwide. Both live-attenuated and inactivated vaccines are used to control the virus in Japan. BRSV has a rapid evolutionary rate and high genetic heterogeneity.11 These divergent viral subpopulations create a broad antigenic spectrum that is, at least partially, responsible for occasional vaccine failures.
BRSV is an enveloped and pleomorphic virus with a single-stranded and negative-sense RNA genome ~15.2 kb in length that encodes 10 proteins. The attachment glycoprotein (G protein) is responsible for viral binding to cell surface receptors and for immunogenicity.13 The G protein has 3 domains: cytoplasmic, located between amino acids (aa) 1–37; transmembrane, between aa 38–65; and the extracellular domain, between aa 66–257.14 This last domain consists of 2 mucin-like regions (MLRs; aa 66–157 and 190–257) and a central hydrophobic region (CHR; aa 158–189). The CHR contains a highly conserved hydrophobic pocket for receptor binding and 4 cysteines that form a so-called “cysteine noose” for formation of 2 disulfide bridges.2 This extracellular domain is a particularly useful target for studying the origins and evolution of BRSV.12 Fusion (F) is the virion surface glycoprotein and is responsible for fusion of the virus with host cell membranes and for syncytia formation.6 The F gene is highly conserved, and its nucleotide sequence variation is lower among BRSV isolates compared with the G gene.13 Several studies have used G sequences, and one study has used both G and F sequences to analyze genetic evolution of BRSV; these studies have shown that BRSV strains can be classified into 9 genetic subgroups, I–IX.1,4,9,12 We used the G gene, in combination with information on the F gene, to analyze Japanese BRSV strains to obtain molecular epidemiologic characterization of the virus to provide genetic information for vaccine-based disease control.
We collected 22 clinical samples (13 nasal swabs and 9 lung tissue samples) from cattle with moderate-to-severe bovine respiratory disease in 21 herds. Samples HK0903/05/JP and HK0904/05/JP (cases 2, 3) were collected from the same farm; the remaining samples were collected from different farms (Table 1). Seven samples (cases 1–7) were obtained between 2004 and 2005, and 15 samples (cases 8–15, 20–26) were collected between 2017 and 2019. Sixteen samples (cases 1–4, 6, 7, 12–15, 20–24, 26) were obtained in autumn and winter (October–March), and 6 samples (cases 5, 8–11, 25) in spring and summer (April–September). The ages of the affected animals were 0–45 mo (mean: 6.8 mo). Two nasal swabs, HK10/18/JP and HK15/19/JP (cases 12 and 14), were inoculated onto Vero cells, which were grown in minimum essential medium (Eagle MEM; Nissui) supplemented with 5% fetal bovine serum. Cells were examined daily, and BRSV isolation was confirmed by the appearance of specific cytopathic effects. Animal treatment and sampling conformed to the institutional guidelines approved by the ethics committee of the National Institute of Animal Health.
Table 1.
Information on the Japanese viral isolates used in our study and their accessions.
| Sampling location/Case | Isolate name | Year/month | Age | Sex | Breed | Sample | Accessions | ||
|---|---|---|---|---|---|---|---|---|---|
| G gene | F gene | Reference | |||||||
| Hokkaido | |||||||||
| 1 | HK0901/05/JP | 2005/01 | 2 mo | F | Hol | Lung | LC549464 | LC549442 | Current study |
| 2 | HK0903/05/JP | 2005/02 | 1 mo | F | Hol | Nasal swab | LC549465 | LC549443 | Current study |
| 3 | HK0904/05/JP | 2005/03 | 4 mo | F | Hol | Nasal swab | LC549466 | LC549444 | Current study |
| 4 | HK1201/04/JP | 2004/10 | 2 mo | M | Hol | Lung | LC549467 | LC549445 | Current study |
| 5 | HK1202/05/JP | 2005/04 | 4 mo | F | Hol | Nasal swab | LC549468 | LC549446 | Current study |
| 6 | HK1204/05/JP | 2005/11 | 3 mo | F | Hol | Nasal swab | LC549469 | LC549447 | Current study |
| 7 | HK1205/05/JP | 2005/12 | 1 mo | M | Cross | Lung | LC549470 | LC549448 | Current study |
| 8 | HK02/18/JP | 2018/05 | 0 mo | F | Hol | Nasal swab | LC549471 | LC549449 | Current study |
| 9 | HK03/18/JP | 2018/06 | 2 mo | F | Cross | Nasal swab | LC549472 | LC549450 | Current study |
| 10 | HK04/18/JP | 2018/06 | 1 mo | M | Hol | Nasal swab | LC549473 | LC549451 | Current study |
| 11 | HK07/18/JP | 2018/08 | 1 mo | M | Hol | Nasal swab | LC549474 | LC549452 | Current study |
| 12 | HK10/18/JP | 2018/10 | 3 mo | M | Cross | Nasal swab | LC549475 | LC549453 | Current study |
| 13 | HK13/18/JP | 2018/12 | 9 mo | F | Hol | Nasal swab | LC549476 | LC549454 | Current study |
| 14 | HK15/19/JP | 2019/02 | 32 mo | F | Hol | Nasal swab | LC549477 | LC549455 | Current study |
| 15 | HK23/19/JP | 2019/02 | 0 mo | F | Hol | Lung | LC549478 | LC549456 | Current study |
| Honsyu | |||||||||
| 16 | IW02 | 2003/02 | 6 mo | NA | JB | Nasal swab | LC499984 | NA | 15 |
| 17 | IW03 | 2003/02 | 2 y | NA | Hol | Nasal swab | LC499985 | NA | 15 |
| 18 | FKI11 | 2003/01 | 4 mo | NA | JB | Nasal swab | LC499994 | NA | 15 |
| 19 | YM01 | 2003/11 | 3 y | NA | Hol | Nasal swab | LC499991 | NA | 15 |
| 20 | HS01/17/JP | 2017/02 | 45 mo | F | Hol | Nasal swab | LC549479 | LC549457 | Current study |
| 21 | TY01/18/JP | 2018/11 | 5 mo | M | Cross | Nasal swab | LC549480 | LC549458 | Current study |
| Kyusyu | |||||||||
| 22 | MY02/19/JP | 2019/02 | 4 mo | F | JB | Lung | LC549481 | LC549459 | Current study |
| 23 | MY01/19/JP | 2019/03 | 9 mo | M | JB | Lung | LC549482 | LC549460 | Current study |
| 24 | MY03/19/JP | 2019/03 | 1 mo | F | JB | Lung | LC549483 | LC549461 | Current study |
| 25 | MY04/19/JP | 2019/04 | 9 mo | M | JB | Lung | LC549484 | LC549462 | Current study |
| 26 | MY05/19/JP | 2019/03 | 11 mo | F | JB | Lung | LC549485 | LC549463 | Current study |
Cross = crossbred of Holstein–Friesian and Japanese Black; F = female; Hol = Holstein–Friesian; JB = Japanese Black; M = male; NA = not available.
The G and F genes were amplified from the clinical samples or from cell culture supernatants by reverse-transcription (RT) nested PCR (RT-nPCR) as described previously, with some minor modifications.12 In brief, a spin-column viral RNA isolation kit (QIAamp viral RNA mini kit; Qiagen) was used for RNA extraction according to the manufacturer’s instructions. The first PCR was performed (PrimeScript one step RT-PCR kit v.2; Takara) with outer primers G2.5 and F2.7 for amplification of the G gene, and F2.1 and F2.2 for amplification of the F gene. The amplification conditions were 50°C for 30 min for RT, denaturation at 94°C for 2 min, followed by 30 cycles at 94°C for 30 s, 55°C for 30 s, and 72°C for 90 s. The second PCR was performed (PrimeSTAR Max DNA polymerase; Takara) with inner primers VG1 and VG4 for amplification of the G gene, and F2.3 and F2.4 for amplification of the F gene; the same amplification conditions were used as in the first PCR except that the extension time was changed to 1 min. The PCR products were separated by electrophoresis on a 1.5% agarose gel, stained with a fluorescent reagent (Midori Green Advance; Nippon Genetics), and detected under blue-green LED light. The expected amplicon sizes were 541 and 833 bp for G and F genes, respectively.
The amplicons were gel purified (QIAquick gel extraction kit; Qiagen) according to the manufacturer’s instructions, and direct sequencing was performed with forward and reverse primers using a sequencing kit (PRISM Big Dye terminator cycle sequencing kit; Applied Biosystems) and a genetic analyzer (ABI 3130; Applied Biosystems). The nucleotide sequences of both strands were obtained for verification and submitted to GenBank (Table 1). The obtained G gene sequences were aligned (Clustal W program; available in MEGA version X, https://www.megasoftware.net/) with 33 reference strains from GenBank including 4 Japanese strains,15 IW02, IW03, FKI11, and YM01, isolated in 2003, and the Japanese vaccine strain rs-52. The analysis of the F gene sequences included 21 representative strains with I–VI and IX subgenotypes. F gene sequences of the strains with the VII and VIII subgenotypes were not available. A phylogenetic tree was constructed using MEGA X with neighbor-joining and Kimura 2-parameter methods using 1,000 bootstrap replicates. Multiple alignments of the deduced amino acid sequences of the G gene were created using MEGA X.
Amplicons of the expected size were obtained from all 22 samples after RT-nPCR. Phylogenetic analysis revealed that all 26 Japanese BRSV strains clustered in the same genetic subgroup and were separate from the known subgenotypes I–IX (Fig. 1). We tentatively assigned the Japanese group to a new subgenotype X. The Japanese BRSVs could be further subdivided into 2 lineages, A and B. Each lineage consisted of 7 analyzed and 4 reference sequences isolated between 2003 and 2005, and 15 analyzed sequences isolated between 2017 and 2019. The comparison of the nucleotide sequences of lineages A and B showed high identities, 94.0–99.7% and 95.1–100% among each group, and 91.2–97.8% between groups (Suppl. Table 1). The level of nucleotide sequence identities between subgenotype X and representative I–IX subgenotypes was 81–95% (Suppl. Table 1). A similar tree topology was obtained from the phylogenetic analysis of the F gene; all of the Japanese isolates grouped in the same branch and were distant from the other representative strains (Suppl. Fig. 1). Divergence of nucleotide sequences of <6.3% was found between the Japanese isolates and the representative BRSVs; the cleavage site and the fusion domain important for their fusion ability were well conserved (Suppl. Table 2).
Figure 1.
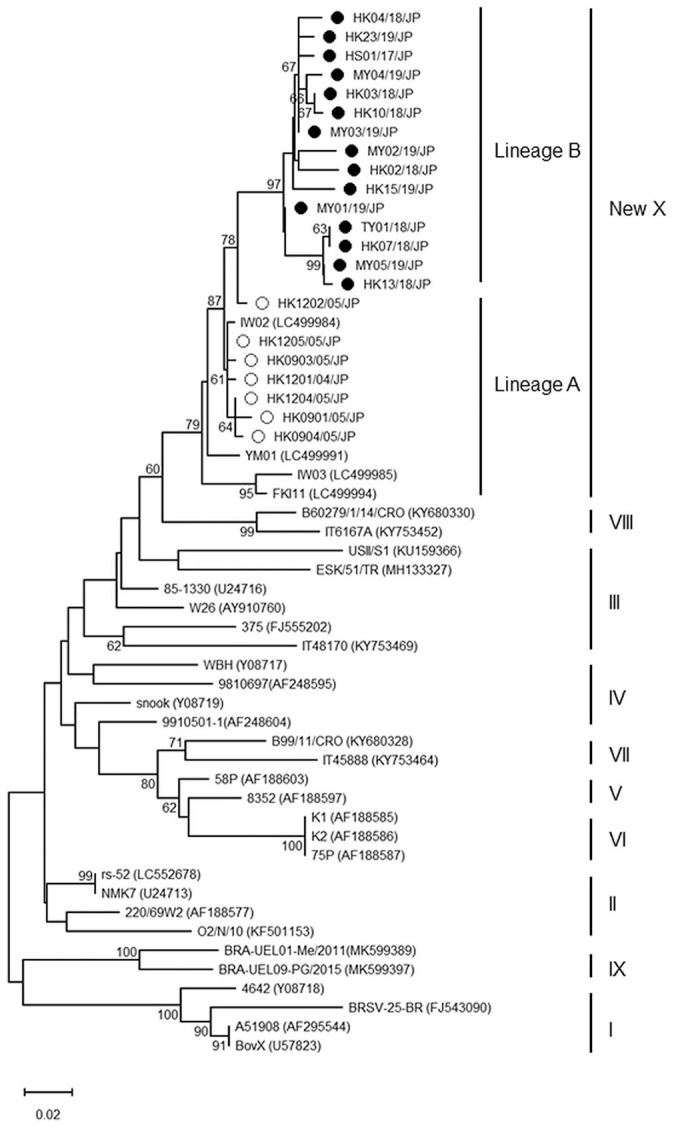
Neighbor-joining phylogenetic tree based on partial nucleotide sequences of bovine respiratory syncytial virus (BRSV) G genes. Numbers are bootstrap values. The scale bar indicates changes per site. Open and closed dots show isolates of lineages A and B, respectively. GenBank accessions of the representative BRSV strains are shown in parentheses.
Alignment of amino acid sequences in the G gene revealed that 4 cysteine residues important for antibody reactivity at positions 173, 176, 182, and 186 were conserved in all 26 Japanese sequences (Suppl. Fig. 2). All of the analyzed strains, except one with single amino acid substitution, had the sequence 173CSTCEGNLACLSLC186 in the CHR. Other unique mutations were found in the extracellular domain of the Japanese BRSVs compared with the prototypic subgenotype II strain NMK7: N113→S113, T120→A120, G123→S123, P125→S125, D127→I127, G128→E128, G144→R144, P150→S150, P155→S155, E157→K157, H162→N162, N163→T163, S165→F165, Y170→H170, P194→S194. Thus, most mutations were observed in the MLRs. Lineage B–specific mutations were also found in the MLRs: G119→R119, I134→T134, N149→I149, P190→S190, P206→S206.
In our study, BRSV infection occurred mostly in calves ≤1-y-old, and during autumn and winter. Epidemiology of the BRSV cases was similar to that described in the literature.6,11 Twenty strains were sequenced directly from clinical samples without prior adaptation to cell culture growth, and 2 strains were sequenced after propagation. No characteristic differences were found in the sequences between the groups. This is consistent with previous reports that cell culture adaptation and several passages in culture do not have a major impact on the nucleotide sequence of the G gene.7
The genetic analysis of the G gene revealed that the Japanese BRSV samples analyzed in our study were homologous (divergence was <8.8%) compared with the American and European isolates that had sequence divergence of 15% and 11%, respectively.3,8 In a previous report, Japanese BRSV strains were found to be genetically related to subgroup III.15 However, the phylogenetic tree in our study showed that Japanese BRSV strains clustered in an apparently different lineage. Although the cutoff values for new genotype definition at both the nucleotide and amino acid levels are not clearly determined, the nucleotide dissimilarity of the Japanese BRSVs against subgenotypes I–IX is extensive,11 suggesting the validity of our newly identified subgenotype. BRSV infection in Japan between 2003 and 2005 involved only lineage A strains and they were replaced by the lineage B strains between 2017 and 2019, resulting in the temporal clustering of Japanese BRSVs. The Japanese BRSVs are likely to be monophyletic with current ongoing evolution. Further phylogenetic studies using samples in the period 2006–2016 may reveal other patterns of subgenotype circulation and shifting of the dominant subpopulations. On the other hand, no geographic clustering was found in the comparison of sequences from different locations in this country. Japanese cattle herds may form a common transmission chain given that the animals are transported from farm to farm without measures to avoid the spread of BRSV. Similar results were obtained from the genetic analysis of the F gene region. Although the F gene sequence was also used for phylogenetic analysis, the resulting tree showed less resolution as a result of low nucleotide variability. Therefore, we did not perform in-depth analysis of this region.
Four antigenic subgroups, A, B, intermediate, and untyped, have been identified from monoclonal antibody and polyclonal sera analyses against G and F proteins.3,5,10 Point substitutions of residues 180, 183, and 184 in the CHR substantially influence the antibody reactivity of BRSV. Combinations of these mutations can form the motifs of LACLS, PACSP, and LACSS at position 180–184, which are typical of antigenic subgroups A, B, and intermediate, respectively. All of the Japanese strains and the vaccine strain rs-52 had the A type motif in this region. The hydrophobic pocket adjacent to this region also plays an important role for receptor binding, and the residues V171, P172, C173, C176, C182, L185, and C186 are essential to this structure.2 These residues, as well as the cysteine noose, were completely conserved among the Japanese and the vaccine strains.
Our results suggest that the present BRSV strains might possess an important structure of immunologic activity with a closer relationship to the vaccine strain, and cattle could be protected against the new BRSV genotype by vaccination based on strain rs-52. However, further in vivo trials will be required to evaluate the efficacy of the present vaccine. On the other hand, 11 amino acid substitutions were found in the extracellular domain mainly in the MLRs. Five substitutions were present only in the isolates obtained in 2017–2019 and not in isolates obtained in 2003–2005 or in the vaccine strain. These mutations may represent a gradual accumulation of amino acid change and continuum of evolution. Our study provides basic information on the epidemiology of BRSV infection in Japan. Our results are relevant to the efficacy of the current vaccine and will be of value to the development or modification of future vaccines.
Supplemental Material
Supplemental material, sj-pdf-1-vdi-10.1177_1040638720975364 for Sequence and unique phylogeny of G genes of bovine respiratory syncytial viruses circulating in Japan by Asuka Kumagai, Kyoko Kawauchi, Kiyohiko Andoh and Shinichi Hatama in Journal of Veterinary Diagnostic Investigation
Acknowledgments
We thank the staff of the Hokkaido local government for providing field samples.
Footnotes
Declaration of conflicting interests: The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The authors received no financial support for the research, authorship, and/or publication of this article.
ORCID iD: Shinichi Hatama  https://orcid.org/0000-0001-7749-0114
https://orcid.org/0000-0001-7749-0114
Supplementary material: Supplementary material for this article is available online.
Contributor Information
Asuka Kumagai, National Institute of Animal Health, National Agriculture and Food Research Organization, Tsukuba, Ibaraki, Japan.
Kyoko Kawauchi, Tokachi Livestock Hygiene Service Center, Obihiro, Hokkaido, Japan.
Kiyohiko Andoh, National Institute of Animal Health, National Agriculture and Food Research Organization, Tsukuba, Ibaraki, Japan.
Shinichi Hatama, National Institute of Animal Health, National Agriculture and Food Research Organization, Tsukuba, Ibaraki, Japan.
References
- 1. Bertolotti L, et al. Genetic characterization of bovine respiratory syncytial virus strains isolated in Italy: evidence for the circulation of new divergent clade. J Vet Diagn Invest 2018;30:300–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Doreleijers JF, et al. Solution structure of the immunodominant region of protein G of bovine respiratory syncytial virus. Biochemistry 1996;35:14684–14688. [DOI] [PubMed] [Google Scholar]
- 3. Furze JM, et al. Antigenically distinct G glycoproteins of BRSV strains share a high degree of genetic homogeneity. Virology 1997;231:48–58. [DOI] [PubMed] [Google Scholar]
- 4. Krešić N, et al. Genetic analysis of bovine respiratory syncytial virus in Croatia. Com Immunol Microbial Infect Dis 2018;58:52–57. [DOI] [PubMed] [Google Scholar]
- 5. Langedijk JP, et al. Antigenic structure of the central conserved region of protein G of bovine respiratory syncytial virus. J Virol 1997;71:4055–4061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Larsen LE. Bovine respiratory syncytial virus (BRSV); a review. Acta Vet Scand 2000;41:4055–4061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Larsen LE, et al. Serological and genetic characterization of bovine respiratory syncytial virus (BRSV) indicates that Danish isolates belong to the intermediate subgroup: no evidence of a selective effect on the variability of G protein nucleotide sequence by prior cell culture adaption and passages in cell culture or calves. Vet Microbiol 1998;62:265–279. [DOI] [PubMed] [Google Scholar]
- 8. Larsen LE, et al. Extensive sequence divergence among bovine respiratory syncytial viruses isolated during recurrent outbreaks in closed herds. J Clin Microbiol 2000;38:4222–4227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Leme RA, et al. Molecular characterization of Brazilian wild-type strains of bovine respiratory syncytial virus reveals genetic diversity and putative new subgroup of virus. Vet Q 2020;40:83–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Prozzi D, et al. Antigenic and molecular analysis of the variability of bovine respiratory syncytial virus G glycoprotein. J Gen Virol 1997;78:359–366. [DOI] [PubMed] [Google Scholar]
- 11. Sarmiento-Silva RE, et al. Epidemiology, molecular epidemiology, and evolution of bovine respiratory syncytial virus. Viruses 2012;4:3452–3467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Valarcher JF, et al. Evolution of bovine respiratory syncytial virus. J Virol 2000;74:10714–10728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Valarcher JF, et al. Bovine respiratory syncytial virus infection. Vet Res 2007;38:153–180. [DOI] [PubMed] [Google Scholar]
- 14. Valentova V. The antigenic and genetic variability of bovine respiratory syncytial virus with emphasis on the G protein. Vet Med 2012;48:254–266. [Google Scholar]
- 15. Yaegashi G, et al. Genetic and antigenic analyses of bovine respiratory syncytial virus detected in Japan. J Vet Med Sci 2005;67:145–150. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material, sj-pdf-1-vdi-10.1177_1040638720975364 for Sequence and unique phylogeny of G genes of bovine respiratory syncytial viruses circulating in Japan by Asuka Kumagai, Kyoko Kawauchi, Kiyohiko Andoh and Shinichi Hatama in Journal of Veterinary Diagnostic Investigation