

Key Points
Question
Can the integration of genomic and clinicopathologic features predict recurrence better than the TNM system after complete resection of early-stage lung adenocarcinoma (LUAD)?
Findings
In this observational study of 426 patients with LUAD, alterations in SMARCA4 and TP53 and fraction of genome altered were independently associated with relapse-free survival. By integrating genomic and clinicopathologic factors, this prediction model outperformed the TNM-based model (concordance probability estimate, 0.73 vs 0.61) for prediction of relapse-free survival and was externally validated using The Cancer Genome Atlas data set.
Meaning
These findings suggest that integration of genomic and clinicopathologic factors are associated with risk of recurrence in surgically resected LUAD, potentially enriching and increasing accrual to adjuvant therapy clinical trials.
This cohort study identifies tumor genomic factors independently associated with recurrence in patients with lung adenocarcinoma and develops a machine-learning prediction model to determine whether genomic and clinicopathologic features are associated with risk of recurrence compared with the TMN system.
Abstract
Importance
Recommendations for adjuvant therapy after surgical resection of lung adenocarcinoma (LUAD) are based solely on TNM classification but are agnostic to genomic and high-risk clinicopathologic factors. Creation of a prediction model that integrates tumor genomic and clinicopathologic factors may better identify patients at risk for recurrence.
Objective
To identify tumor genomic factors independently associated with recurrence, even in the presence of aggressive, high-risk clinicopathologic variables, in patients with completely resected stages I to III LUAD, and to develop a computational machine-learning prediction model (PRecur) to determine whether the integration of genomic and clinicopathologic features could better predict risk of recurrence, compared with the TNM system.
Design, Setting, and Participants
This prospective cohort study included 426 patients treated from January 1, 2008, to December 31, 2017, at a single large cancer center and selected in consecutive samples. Eligibility criteria included complete surgical resection of stages I to III LUAD, broad-panel next-generation sequencing data with matched clinicopathologic data, and no neoadjuvant therapy. External validation of the PRecur prediction model was performed using The Cancer Genome Atlas (TCGA). Data were analyzed from 2014 to 2018.
Main Outcomes and Measures
The study end point consisted of relapse-free survival (RFS), estimated using the Kaplan-Meier approach. Associations among clinicopathologic factors, genomic alterations, and RFS were established using Cox proportional hazards regression. The PRecur prediction model integrated genomic and clinicopathologic factors using gradient-boosting survival regression for risk group generation and prediction of RFS. A concordance probability estimate (CPE) was used to assess the predictive ability of the PRecur model.
Results
Of the 426 patients included in the analysis (286 women [67%]; median age at surgery, 69 [interquartile range, 62-75] years), 318 (75%) had stage I cancer. Association analysis showed that alterations in SMARCA4 (clinicopathologic-adjusted hazard ratio [HR], 2.44; 95% CI, 1.03-5.77; P = .042) and TP53 (clinicopathologic-adjusted HR, 1.73; 95% CI, 1.09-2.73; P = .02) and the fraction of genome altered (clinicopathologic-adjusted HR, 1.03; 95% CI, 1.10-1.04; P = .005) were independently associated with RFS. The PRecur prediction model outperformed the TNM-based model (CPE, 0.73 vs 0.61; difference, 0.12 [95% CI, 0.05-0.19]; P < .001) for prediction of RFS. To validate the prediction model, PRecur was applied to the TCGA LUAD data set (n = 360), and a clear separation of risk groups was noted (log-rank statistic, 7.5; P = .02), confirming external validation.
Conclusions and Relevance
The findings suggest that integration of tumor genomics and clinicopathologic features improves risk stratification and prediction of recurrence after surgical resection of early-stage LUAD. Improved identification of patients at risk for recurrence could enrich and enhance accrual to adjuvant therapy clinical trials.
Introduction
Recurrence after complete resection (R0) of early-stage non–small cell lung cancer (NSCLC) is estimated at 13% to 23% for node-negative disease1,2 and higher for N1 disease. Most recurrences (80%) occur within 2 years of resection, usually at distant sites, and are associated with a dismal 5-year survival of 30%.1,3 Although adjuvant cisplatin-based chemotherapy improves relapse-free survival (RFS) in patients with larger tumors and node-positive disease, the overall survival benefit is minimal, with most patients deriving no benefit.4 Current National Comprehensive Cancer Network (NCCN) recommendations for adjuvant therapy5 are based solely on TNM categories, although a recent study6 suggests RFS is improved in patients with epidermal growth factor receptor–mutant lung adenocarcinoma (LUAD) treated with adjuvant epidermal growth factor receptor–selective tyrosine kinase inhibitors, compared with doublet chemotherapy. Moreover, the emerging role of immunotherapy in induction and adjuvant therapy for early-stage NSCLC argues for more contemporary approaches to identify patients at high risk of recurrence after surgery.
Broad-panel next-generation sequencing (NGS) has increasingly been integrated into clinical care, to identify actionable mutations or genomic determinants of response to systemic therapies in advanced NSCLC.7 However, little is known about the role of NGS in surgically resected early-stage NSCLC, where recurrence, not response to therapy, is the determinant of survival. Moreover, although studies have identified poor prognostic factors in these patients, to our knowledge, no study has developed a prediction model for recurrence using NGS and high-risk pathologic features.
To more accurately stratify patients for risk of recurrence, we performed a prospective observational cohort study in patients with completely resected stages I to III LUAD. We sought to develop a prediction model that integrates genomic and clinicopathologic variables to predict recurrence and investigated the performance of this model compared with the conventional TNM system.
Methods
Study Population and Data Collection
Following institutional review board approval from Memorial Sloan Kettering Cancer Center (MSK), we prospectively observed consecutive patients who underwent complete resection (R0) for stages I to III LUAD and provided informed consent for broad-panel NGS (MSK–IMPACT) on the primary tumor. Patients who underwent induction therapy and incomplete (R1/R2) resection and those with low-quality NGS were excluded (see CONSORT diagram in eFigure 1 in the Supplement). Clinicopathologic and genomic comparisons were made between our cohort and a stage IV LUAD cohort previously reported7; no patients with stage IV cancer were included in subsequent RFS analysis. This study followed the Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) reporting guideline.8
Prospectively collected demographic, imaging, staging (AJCC Cancer Staging Manual, 8th edition), pathologic, genomic, recurrence, and follow-up data were reviewed. Follow-up was performed in accordance with NCCN guidelines.5 Metachronous lesions were distinguished from recurrences using the criteria of Martini and Melamed,9 with confirmation of clonal relatedness using previously published genomic data.10 Predominant histologic subtypes were classified as lepidic, acinar, papillary, micropapillary, or solid.
MSK-IMPACT Sequencing
Tumor genomic profiling was performed in all 426 patients using MSK-IMPACT, with sequencing coverage and genomic alteration classification details listed in the eMethods in the Supplement.11,12 Tumor mutation burden (TMB) was defined as the total number of nonsynonymous coding variants per megabase (Mb) and was normalized by panel size by dividing the total number of mutations by the length of the coding region captured by each panel (0.98 Mb in the 341-gene panel [n = 14], 1.06 Mb in the 410-gene panel [n = 229], and 1.22 Mb in the 468-gene panel [n = 183]). Previous investigators11,12 showed that this method correlates well with TMB derived from whole-exome sequencing. The fraction of genome altered (FGA) was defined as the number of bases in sequenced genomic segments with log2 copy number fold change of greater than 0.2 or no greater than 0.2 for the total number of bases in all sequenced segments.
RFS Association Analysis
Data were analyzed from 2014 to 2018. The primary end point—RFS (defined as the time from surgery to first recurrence or death from any cause)—was estimated using Kaplan-Meier analysis and compared between groups using the log-rank test. Relapse-free survival was assessed using association analysis and prediction models. In the clinicopathologic association analysis, Cox proportional hazards regression was used to quantify associations between clinicopathologic factors and RFS (see the eMethods in the Supplement for additional statistical methods). The association of genomic factors and specific genes with RFS was also assessed using univariable Cox proportional hazards regression, using false-discovery rate–adjusted P values where applicable; 2-sided P < .05 indicated statistical significance. Genes altered at a frequency of at least 1% were considered (n = 47) and adjusted by background genomic factors (TMB, FGA, and whole-genome doubling). Genomic factors significant in univariable analysis were assessed in the presence of the significant factors from the clinicopathologic association model, to generate a clinicopathologic-adjusted multivariable model. Associations between TMB/FGA and aggressive pathologic features were evaluated using the Mann-Whitney or Kruskal-Wallis test.
Prediction Model and External Validation
The prediction model (PRecur) was developed by integrating the same clinicopathologic and genomic variables as the association model using a publicly available ensemble machine-learning framework previously reported (eMethods in the Supplement).13 PRecur builds a collection of gradient boosting survival tree models for RFS. One thousand survival regression trees were generated using repeated training-test splits to assess RFS prediction performance, assigning a predicted risk score to each patient in the test set at each iteration. Patients were divided into 3 risk groups (low, intermediate, and high) using the maximally selected rank statistic approach.14 The concordance probability estimate (CPE) was used to evaluate the prediction accuracy of the model compared with the actual time to recurrence. To avoid the well-known upward biases of concordance indices in estimating concordance probability in highly censored data sets, we report CPE, which uses probability imputation to result in a more unbiased estimator of prediction concordance.15
To externally validate our integrated prediction model, we examined The Cancer Genome Atlas (TCGA) LUAD data set (n = 360).16 We generated a PRecur external validation model for our MSK-IMPACT data set inclusive of overlapping clinical covariates between our data set and the TCGA data set (age, sex, smoking status, and pathologic stage), plus all genes (n = 47) and genomic factors (TMB, FGA, and whole-genome doubling). We then performed PRecur analysis identically to the complete model (trained on the MSK-IMPACT data set then applied to the TCGA data set) (eMethods in the Supplement). An interactive PRecur web application is available at https://axelitomartin.shinyapps.io/OncoCast-NSCLC/.
Results
Demographic and Clinical Characteristics
In total, 426 patients met inclusion criteria. Median follow-up was 2.52 (interquartile range [IQR], 2.08-3.14) years. Median age at surgery was 69 (IQR, 62-75) years. Two hundred eighty-six patients (67%) were women, 140 (33%) were men, and 318 (75%) had pathologic stage I cancer (Table 1). Seventy-five patients (18%) developed a recurrence, of which 57 (76%) were distant (eTable 1 in the Supplement).
Table 1. Clinicopathologic Characteristics of the Stage I-III Cohort, With and Without Recurrence.
| Characteristic | Patient groupa | ||
|---|---|---|---|
| All (n = 426) | Without recurrence (n = 351) | With recurrence (n = 75) | |
| Age at surgery, median (IQR), y | 69 (62-75) | 69 (63-75) | 70 (59-75) |
| Sex | |||
| Female | 286 (67) | 240 (68) | 46 (61) |
| Male | 140 (33) | 111 (32) | 29 (39) |
| Smoking status | |||
| Current | 28 (7) | 26 (7) | 2 (3) |
| Former | 303 (71) | 251 (72) | 52 (69) |
| Never | 95 (22) | 74 (21) | 21 (28) |
| Clinical stage | |||
| IA1 | 36 (8) | 28 (8) | 8 (11) |
| IA2 | 188 (44) | 167 (48) | 21 (28) |
| IA3 | 94 (22) | 77 (22) | 17 (23) |
| IB | 51 (12) | 44 (13) | 7 (9) |
| IIA | 14 (3) | 8 (2) | 6 (8) |
| IIB | 38 (9) | 24 (7) | 14 (19) |
| IIIA | 5 (1) | 3 (1) | 2 (3) |
| Primary tumor SUVmax, median (IQR)b | 3.7 (1.9-7.6) | 3.2 (1.8-6.1) | 7.5 (5.0-10.3) |
| Operative approach | |||
| Open | 71 (17) | 42 (12) | 29 (39) |
| Minimally invasive | 355 (83) | 309 (88) | 46 (61) |
| Operative procedure | |||
| Sublobar | 139 (33) | 118 (34) | 21 (28) |
| Lobectomy | 283 (66) | 229 (65) | 54 (72) |
| Pneumonectomy | 4 (1) | 4 (1) | 0 |
| Pathologic tumor size, median (IQR), cm | 1.8 (1.2-3.0) | 1.7 (1.2-2.6) | 2.9 (1.6-4.0) |
| Predominant histologic subtypec | |||
| Lepidic | 71 (17) | 69 (20) | 2 (3) |
| Acinar | 220 (52) | 181 (52) | 39 (53) |
| Papillary | 29 (7) | 22 (6) | 7 (9) |
| Micropapillary | 23 (5) | 19 (5) | 4 (5) |
| Solid | 52 (12) | 32 (9) | 20 (27) |
| Other | 26 (6) | 24 (7) | 2 (3) |
| pT category | |||
| 1a | 62 (15) | 57 (16) | 5 (7) |
| 1b | 156 (37) | 141 (40) | 15 (20) |
| 1c | 66 (15) | 57 (16) | 9 (12) |
| 2a | 80 (19) | 58 (17) | 22 (29) |
| 2b | 16 (4) | 13 (4) | 3 (4) |
| 3 | 33 (8) | 17 (5) | 16 (21) |
| 4 | 13 (3) | 8 (2) | 5 (7) |
| pN category | |||
| 0 | 344 (81) | 310 (88) | 34 (45) |
| 1 | 33 (8) | 19 (5) | 14 (19) |
| 2 | 34 (8) | 12 (3) | 22 (29) |
| X | 15 (4) | 10 (3) | 5 (7) |
| Pathologic tumor stage | |||
| I | 318 (75) | 290 (83) | 28 (37) |
| II | 61 (14) | 42 (12) | 19 (25) |
| III | 47 (11) | 19 (5) | 28 (37) |
| Normalized TMB, median (IQR) | 4.7 (2.5-8.2) | 4.7 (1.9-8.2) | 5.7 (3.1-8.5) |
| FGA ( × 100), median (IQR)d | 3.8 (0.4-11.1) | 2.9 (0.3-8.9) | 10.8 (3.0-21.9) |
| Whole-genome doublinge | 92 (22) | 68 (19) | 24 (35) |
| Spread through air spacesf | |||
| Present | 213 (61) | 176 (58) | 37 (90) |
| Absent | 134 (39) | 130 (42) | 4 (10) |
| Lymphovascular invasiong | |||
| Present | 151 (36) | 98 (28) | 53 (72) |
| Absent | 271 (64) | 250 (72) | 21 (28) |
| Visceral pleural invasionh | |||
| Present | 63 (15) | 42 (12) | 21 (28) |
| Absent | 362 (85) | 308 (88) | 54 (72) |
| Adjuvant therapyi | |||
| None | 333 (79) | 294 (85) | 39 (52) |
| Systemic therapy | 61 (15) | 42 (12) | 19 (25) |
| PORT with or without systemic therapy | 25 (6) | 8 (2) | 17 (23) |
| Status at last contact, No. of patients | |||
| No evidence of disease | 336 | 335 | 5 |
| Alive with disease | 41 | 0 | 37 |
| Death | |||
| Other cause | 10 | 8 | 2 |
| Disease | 28 | 0 | 28 |
| Unknown cause | 11 | 8 | 3 |
| All | 49 | 16 | 33 |
Abbreviations: FGA, fraction of genome altered; IQR, interquartile range; PORT, postoperative radiation therapy; SUVmax, maximum standardized uptake value; TMB, tumor mutation burden.
Unless otherwise indicated, data are expressed as number (percentage) of patients. Percentages have been rounded and may not total 100.
Includes 366 patients, 296 without and 70 with recurrence.
Includes 421 patients, 347 without and 74 with recurrence.
Includes 415 patients, 345 without and 70 with recurrence.
Includes 418 patients, 349 without and 69 with recurrence.
Includes 347 patients, 306 without and 41 with recurrence.
Includes 422 patients, 348 without and 74 with recurrence.
Includes 425 patients, 350 without and 75 with recurrence.
Includes 419 patients, 344 without and 75 with recurrence.
Genomic Alterations by Stage
Studies have shown that early-stage NSCLC has a different transcriptomic profile than metastatic tumors17; however, little is known about differences in tumor genomics between early- and advanced-stage LUAD. To explore this, we compared the demographic and clinical characteristics of our stages I to III cohort with the previously reported stage IV cohort (eFigure 2A in the Supplement), as well as the alteration frequencies of LUAD-associated genes (eFigure 2B in the Supplement). Although demographic and clinicopathologic factors were similar, KRAS (OMIM 190070) was more frequently altered in the stages I to III cohort, whereas TP53 (OMIM 191170) mutations and fusions in RET (OMIM 164761), ROS1 (OMIM 165020), and ALK (OMIM 105590) were more frequent in the stage IV cohort (eTable 2 in the Supplement). Moreover, among patients with stage IV compared with stages I to III cancer, significantly increased TMB (median, 5.6 vs 4.7; difference, 0.9 [95% CI, 0.11-1.7]; P = .005) and FGA ( × 100) (median, 11.0 vs 3.8; difference, 7.3 [95% CI, 5.0-9.5]; P < .001) were found. Within the stages I to III cohort, higher pathologic stage was associated with solid or micropapillary histologic subtypes (stage I, 39 of 318 [12%]; stage II, 18 of 61 [30%]; stage III, 18 of 47 [38%]), lymphovascular invasion (stage I, 79 of 315 [25%]; stage II, 35 of 61 [57%]; stage III, 37 of 47 [79%]), and spread through air spaces (stage I, 150 of 270 [56%]; stage II, 34 of 45 [76%]; stage III, 29 of 32 [91%]) (Figure 1). As expected, the fraction of mutations associated with a smoking signature was increased in those with smoking history (median, 22.6% [IQR, 3.4%-66.8%] in current smokers, 4.2% [IQR, 1.0%-14.8%] in former smokers, and 0.8% [IQR, 0.5%-1.2%] in never smokers; P < .001) (eFigure 3 in the Supplement). Results of NGS revealed that a greater proportion of patients with stage II or III cancer had alterations in SMARCA4 (OMIM 603254) (stage I, 4 of 318 [1%]; stage II, 2 of 61 [3%]; stage III, 6 of 47 [13%]) than those with stage I disease. In contrast, stage I tumors harbored more truncating mutations in RBM10 (OMIM 300080), an RNA-binding protein and splicing regulator (stage I, 49 of 318 [15%]; stage II, 3 of 61 [5%]; stage III, 0 of 47).
Figure 1. Oncoprint of the Study Cohort by Pathologic Stage With Annotated Clinicopathologic Variables.
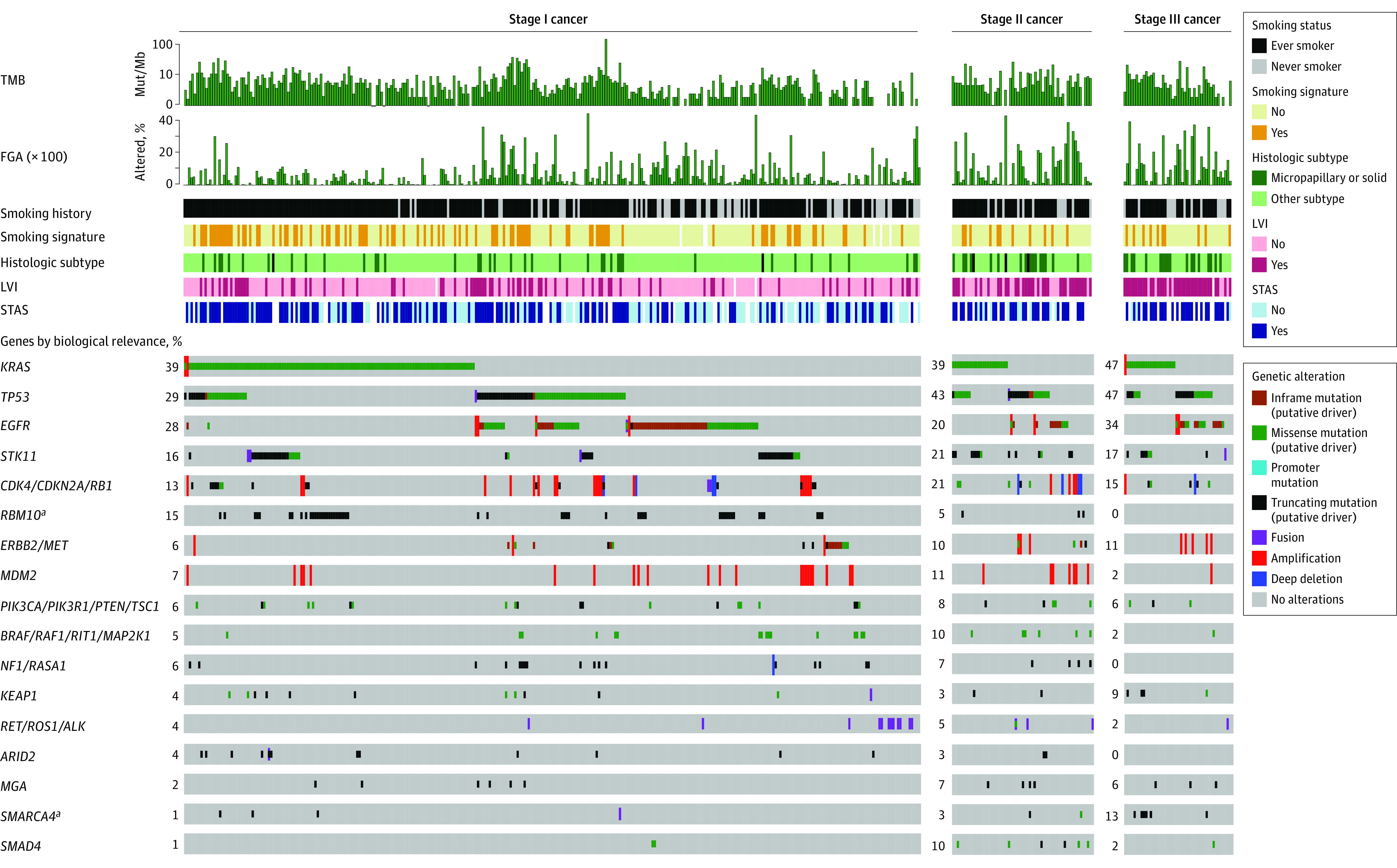
Genes are grouped by biological relevance in lung adenocarcinoma. FGA indicates fraction of genome altered; LVI, lymphovascular invasion; Mut/Mb, mutations per megabase; STAS, spread through air spaces; and TMB, tumor mutation burden.
aP < .05, false-discovery rate, for difference in alteration frequency between stages using Fisher exact test.
RFS Association Analysis
Multivariable analysis of clinicopathologic variables revealed that solid histologic subtype (hazard ratio [HR], 1.74; 95% CI, 1.05-2.89; P = .03), lymphovascular invasion (HR, 2.44; 95% CI, 1.48-4.05; P = .001), pathologic stages II (HR, 2.39; 95% CI, 1.28-4.45; P = .006) and III (HR, 3.64; 95% CI, 1.92-6.90; P < .001), open (thoracotomy) approach (HR, 1.83; 95% CI, 1.14-2.94; P = .01), and sublobar resection (HR, 1.96; 95% CI, 1.16-3.31; P = .01) were associated with worse RFS (eTable 3 in the Supplement). The association of genomic factors with RFS was similarly examined. On univariable analysis, alterations in ERBB2 (OMIM 164870) (HR, 2.94; 95% CI, 1.47-5.88; P = .002), SMARCA4 (3.57; 95% CI, 1.55-8.19; P = .003), and TP53 (HR, 2.32; 95% CI, 1.53-3.51; P < .001) as well as increasing TMB (HR, 1.02; 95% CI, 1.01-1.03; P < .001) and FGA (HR, 1.04; 95% CI, 1.02-1.05; P < .001) were associated with worse RFS (eTable 4 in the Supplement). We then determined which factors were independently associated with RFS in the presence of significant factors observed in our clinicopathologic association model. Only SMARCA4 (HR, 2.44; 95% CI, 1.03-5.77; P = .04), TP53 (HR, 1.73;95% CI, 1.09-2.73; P = . 02), and FGA (HR, 1.03; 95% CI, 1.01-1.04; P = .005) retained significance (Table 2). The specific alterations in SMARCA4 and TP53 are listed in eTable 5 in the Supplement.
Table 2. Univariable and Clinicopathologic-Adjusted Multivariable Cox Proportional Hazards Model for RFSa.
| Genomic factor/gene | Data | Univariable analysis | Clinicopathologic-adjusted multivariable analysis | |||
|---|---|---|---|---|---|---|
| HR (95% CI) | P value | FDR-corrected P value | HR (95% CI) | P value | ||
| TMB, median (IQR) | 4.7 (2.5-8.2) | 1.02 (1.01-1.03) | <.001 | NA | 1.01 (1.00-1.03) | .07 |
| FGA ( × 100), median (IQR) | 3.8 (0.4-11.1) | 1.04 (1.02-1.05) | <.001 | NA | 1.03 (1.01-1.04) | .005 |
| Gene, No. (%) | ||||||
| ERBB2 | 20 (5) | 2.94 (1.47-5.88) | .002 | .03 | 1.99 (0.96-4.16) | .07 |
| SMARCA4 | 12 (3) | 3.57 (1.55-8.19) | .003 | .03 | 2.44 (1.03-5.77) | .04 |
| TP53 | 138 (32) | 2.32 (1.53-3.51) | <.001 | .003 | 1.73 (1.09-2.73) | .02 |
Abbreviations: FDR, false discovery rate; FGA, fraction of genome altered; HR, hazard ratio; IQR, interquartile range; NA, not applicable; RFS, relapse-free survival; TMB, normalized tumor mutation burden.
Full univariable analysis is presented in eTable 4 in the Supplement.
Next, we explored whether genes independently associated with RFS exhibited co-occurrence with known targetable drivers or KRAS (eFigure 4 in the Supplement). Oncogenic alterations in SMARCA4 and TP53 were enriched in tumors that recurred but were not found to demonstrate co-occurrence with any of the 4 genes with level 1 evidence of clinical actionability (EGFR [OMIM 131550], ALK, ROS1, and BRAF [OMIM 164757])18 or KRAS. eTable 6 in the Supplement shows the number of actionable alterations for EGFR, ALK, ROS1, and BRAF in our cohort. In addition, although a significant increase was found for TMB (median, 5.7 vs 4.7; difference, 0.9 [95% CI, −0.3 to 2.2]; P = .02) and FGA ( × 100) (median, 10.8 vs 2.8; difference, 8.2 [95% CI, 5.8-10.5]; P < .001) in tumors that recurred, only FGA was independently associated with RFS (HR, 1.03; 95% CI, 1.01-1.04; P = .005) (Table 2).
Association of Aggressive Pathologic Features With TMB and FGA
Although TMB was not independently associated with RFS, we a priori hypothesized that TMB may be associated with aggressive clinicopathologic features (eFigure 5 in the Supplement). Higher TMB was significantly associated with smoking (P < .001), solid or micropapillary histologic subtype (P = .003), lymphovascular invasion (P = .002), spread through air spaces (P = .002), node-positive tumors (P = .03), and increased tumor maximum standardized uptake value (SUVmax) (P < .001). Interestingly, TMB was not associated with tumor size or pathologic stage. The association of FGA with aggressive pathologic features was also explored (eFigure 6 in the Supplement). Increasing FGA was associated with micropapillary-predominant subtype (P < .001), lymphovascular invasion (P < .001), node-positive tumors (P = .005), and increased tumor SUVmax (P < .001). Furthermore, increasing FGA correlated with increasing TMB (ρ = 0.454; P < .001; eFigure 7 in the Supplement). We found that higher TMB and FGA were associated with aggressive clinicopathologic features in surgically resected LUAD, but only FGA is independently associated with RFS.
PRecur Prediction Model for RFS
Given the heterogeneity of LUAD and the variability in clinical outcomes with pTNM staging to guide adjuvant therapy recommendations, we used PRecur for risk stratification and prediction of RFS. Based on statistically optimized risk score cutoffs, patients were divided into low-, intermediate-, and high-risk groups (eFigure 8 in the Supplement). The low- and intermediate-risk groups had significantly better 3-year RFS of 93% (95% CI, 89%-98%) and 72% (95% CI, 64%-81%), respectively, compared with 33% (95% CI, 22%-50%) for the high-risk group (log-rank statistic, 88.4; P < .001) (Figure 2A). In our PRecur prediction model, the 5 clinicopathologic variables in our clinicopathologic association model, plus tumor SUVmax and visceral pleural invasion, were associated with higher risk scores and worse RFS. We also found that alterations in SMARCA4 and TP53 were strongly predictive of worse RFS, whereas RBM10 mutation was associated with better RFS (eFigure 9A in the Supplement). Importantly, our prediction model for RFS outperformed TNM-based models (CPE, 0.73 vs 0.61; difference, 0.12 [95% CI, 0.05-0.19]; P < .001) (eFigure 9B in the Supplement) in discriminating which patients are most likely to experience recurrence. In addition, we found that 19 of 23 patients (83%) with stage I cancer who had recurrences were classified as intermediate or high risk by PRecur analysis. In contrast, using TNM- and NCCN-based criteria for high-risk features, only 18 of 28 patients (64%) with stage I cancer and recurrence would be classified as intermediate or high risk (eTable 7 in the Supplement), confirming the improved capability of PRecur to predict recurrence, compared with TNM, in stage I tumors.
Figure 2. Computational Machine-Learning Prediction Model (PRecur) for Relapse-Free Survival (RFS) Using Integrated Clinicopathologic and Genomic Variables for Risk Stratification.
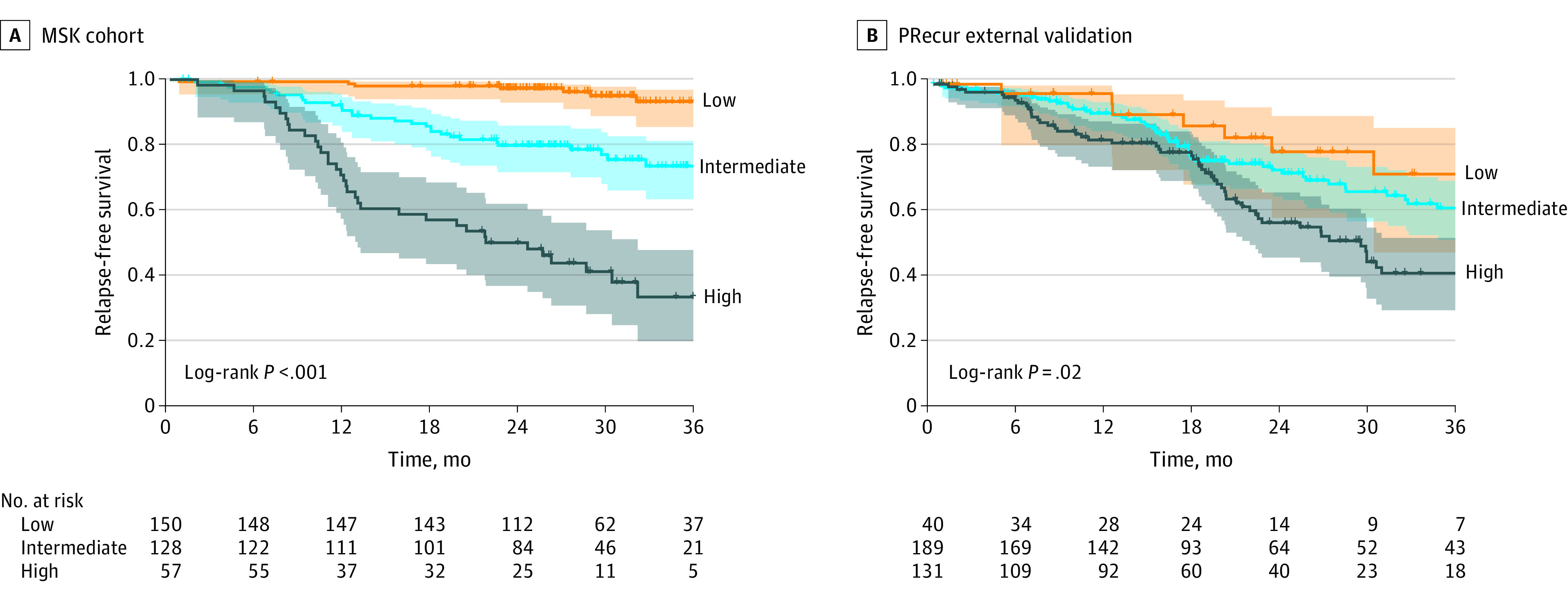
A, Kaplan-Meier plot of 3-year RFS by risk group for the Memorial Sloan Kettering (MSK) cohort (n = 426). B, Kaplan-Meier plot of 3-year RFS by risk group using the PRecur external validation prediction model for the Cancer Genome Atlas lung adenocarcinoma cohort (n = 360). Shaded areas indicate 95% CIs.
Most patients who developed a recurrence (55 of 75 [73%]) had a primary tumor without a level 1 actionable mutation, were not candidates for an adjuvant targeted therapeutic approach, and may have had a worse prognosis. Therefore, as an exploratory analysis, we repeated our PRecur prediction model, excluding patients with a level 1 actionable mutation (n = 117). The RFS prediction performance of this new PRecur model (n = 309) was equal to the complete model (CPE, 0.73) (eFigure 10A in the Supplement), and again a clear RFS separation was noted between risk groups (log-rank statistic, 54.1; P < .001) (eFigure 10B in the Supplement).
External Validation of the Prediction Model
We next externally validated our prediction model using the TCGA LUAD database.16 Because the TCGA data set lacks selected variables found in our full prediction model, we generated a PRecur external validation model that was trained on our MSK data set and includes clinicopathologic factors available in both data sets (age, sex, smoking status, pathologic stage), in addition to all genomic variables from our full prediction model. The PRecur external validation model performed similarly to our full PRecur model for RFS prediction (CPE, 0.70 vs 0.73) and risk group separation (log-rank statistic, 68.7; P < .001) (eFigure 11 in the Supplement). The PRecur external validation model was then applied to the TCGA data set (145 recurrence events), and the 3 risk groups (distribution shown in eFigure 12 in the Supplement) again demonstrated a clear RFS separation (log-rank statistic, 7.5; P = .02) (Figure 2B), thus validating our prediction model. The predicted RFS for each risk group in the MSK and TCGA cohorts is shown in eTable 8 in the Supplement.
Clinical Applications of the PRecur Prediction Model
Two hypothetical clinical scenarios highlight the difference in predicted RFS between the TNM and PRecur prediction models at an individual patient level. The first scenario depicts a patient with a 1.8-cm tumor (Figure 3A) that is low risk by TNM classification (pT1bN0M0, stage IA2). However, after PRecur analysis (Figure 3C), the patient’s 3-year predicted RFS was 51%, substantially worse than the 3-year RFS of 87% predicted by TNM only (Figure 3E). This patient may be a candidate for adjuvant therapy outside of current NCCN guidelines.
Figure 3. Computational Machine-Learning Prediction Model (PRecur) for Relapse-Free Survival (RFS) Applied to 2 Patient Scenarios.

A, Patient with a small, 1.8-cm tumor (pT1bN0M0, stage IA). Three-year RFS curves were predicted by PRecur vs the TNM model for all patients with pT1bN0M0 in our cohort (n = 136). B, Patient with a large, 5.1-cm tumor (pT3N0M0, stage IIB). Three-year RFS curves were predicted by PRecur vs the TNM model for all patients with pT3N0M0 in our cohort (n = 53). FGA indicates fraction of genome altered; L, left; P, posterior; R, right; and TMB, tumor mutation burden.
In contrast, the second scenario depicts a patient with a 5.1-cm tumor (Figure 3B) that is high-risk for recurrence using TNM classification (pT3N0M0, stage IIB). After PRecur analysis (Figure 3D), this patient’s 3-year predicted RFS is 94%, in contrast to the 61% predicted by TNM (Figure 3F), and thus adjuvant therapy may be deferred.
Discussion
Currently, identification of patients at increased risk of recurrence after complete resection of LUAD relies solely on tumor size and nodal involvement and is agnostic to tumor genomics and clinicopathologic variables. In this study, we found FGA, as well as alterations in SMARCA4 and TP53, to be independently associated with worse RFS, even after adjustment for clinicopathologic factors associated with RFS. Using a statistical machine-learning tool to integrate genomic and clinicopathologic data, we created PRecur, a prediction model that predicts RFS substantially better than TNM classification. Moreover, we externally validated our prediction model for RFS using the TCGA LUAD data set.
Although similar genomic alterations exist between primary and metastatic NSCLC, heterogeneous driver mutations have been observed during tumor evolution. This emphasizes intratumoral heterogeneity, clonality, and chromosome instability between the primary tumor and metastases.19 We identified notable genomic differences, including a greater proportion of KRAS-mutated tumors in early-stage LUAD and more TP53 mutations and RET/ROS1/ALK fusions in advanced-stage LUAD. In addition, TMB and FGA were significantly higher in late-stage LUAD, confirming that more mutations and copy number aberrations exist in advanced-stage disease.
Although TP53 mutations occur in 40% to 50% of cases of LUAD, they had no prognostic value and did not predict response to adjuvant chemotherapy in the LACE (Lung Adjuvant Cisplatin Evaluation)-Bio cohort.20 Similarly, deficiency of SMARCA4 protein, a catalytic subunit of the SWI/SNF chromatin remodeling complex, is associated with poor prognosis in NSCLC.21 We observed that TP53 and SMARCA4 mutations were independently associated with worse RFS and were predictive of recurrence in our PRecur risk-stratification model.
Studies examining the predictive and prognostic value of TMB in surgically resected NSCLC have yielded conflicting results.22,23 We show that FGA (a measure of total copy number aberrations and chromosomal instability)—not TMB—is independently associated with worse RFS and is a principal component of our PRecur prediction model. In support of our observation, a recent TCGA analysis suggests that tumor copy number aberrations are a pancancer prognostic factor associated with recurrence and death in several solid tumor malignant neoplasms.24
The well-known genomic and pathologic heterogeneity of LUAD is related to challenges in determining risk of recurrence and treating recurrence after surgical resection. Thirty-nine of the 75 patients (52%) in our study who experienced recurrence received no adjuvant therapy. Moreover, 28 recurrences (37%) occurred in patients with pathologic stage I tumors, and PRecur analysis identified 83% of these patients as having an intermediate- or high-risk of recurrence.
Broad-panel NGS for tumor profiling and the development of increasingly sophisticated machine-learning algorithms have resulted in genomically annotated risk-stratification models for renal and breast cancers.25,26 We found our PRecur model predicted RFS better than the anatomically based TNM criteria (CPE, 0.73 vs 0.55-0.61). The CPE of 0.73 for PRecur can be interpreted as 73% concordance between the predicted and actual outcomes, compared with 55% to 61% using the TNM system. This double-digit increase in CPE represents a substantial improvement in model prediction performance. Patient benefits of contemporary risk-stratification models include increased appropriate use of adjuvant therapies, or conversely that no other therapy is indicated. In addition, circulating tumor DNA analyses to detect minimal residual disease have shown promise,27,28 although challenges in early-stage NSCLC are well described.29 It is plausible that tumor genomic and pathologic risk models, such as PRecur, combined with circulating tumor DNA analysis may offer the best risk assessment for recurrence in the postoperative setting.
Strengths and Limitations
Strengths of our study include the focus on LUAD histologic subtypes, the prospectively collected, well-annotated clinicopathologic and genomic data, complete follow-up information, and external validation of our findings. Limitations of our study include a limited number of recurrence events, with a median follow-up of 2.5 years; however, 80% of recurrences in patients with completely resected early-stage LUAD occur within 2 years.1,3 In addition, we were able to identify patients with stages II and III cancer with low recurrence risk as well as those with high-risk stage I cancer who otherwise would be incorrectly risk stratified by TNM staging alone. Furthermore, no prespecified power calculation was conducted, which could lead to model overfitting. Finally, although the predictive ability of the PRecur external validation model remains high, despite the inclusion of only 4 clinicopathologic variables, our findings need further validation.
Conclusions
This prospective observational cohort study uncovers the association between tumor genomics and recurrence in surgically resected early-stage LUAD. We show that integration of tumor genomics and high-risk clinicopathologic factors performs better than traditional TNM classifications to risk stratify patients for recurrence. Our work moves beyond prognostication to an enhanced ability to predict recurrence, which could then be used to enrich accrual to clinical trials of adjuvant therapy in patients with surgically resected LUAD.
eMethods. Sequencing and Analysis
eFigure 1. CONSORT Diagram for the Stage I-III Cohort
eFigure 2. Distribution of Demographic, Clinicopathologic, and Genomic Characteristics in Early- and Late-Stage Lung Adenocarcinoma (LUAD)
eFigure 3. Fraction and Distribution of Mutations Associated With a Smoking Signature
eFigure 4. Genomic Patterns of Recurrence
eFigure 5. Association Between Tumor Mutation Burden (TMB) and Aggressive Clinicopathologic Features
eFigure 6. Association Between Fraction of Genome Altered (FGA) and Aggressive Clinicopathologic Features
eFigure 7. Association Between Fraction of Genome Altered and Tumor Mutation Burden
eFigure 8. Histogram of the Relapse-Free Survival (RFS) Risk Score Computed Using PRecur, Integrating Clinical and Next-Generation Sequencing Data
eFigure 9. PRecur Prediction Model for Relapse-Free Survival (RFS) Using Integrated Clinicopathologic and Genomic Variables for Risk Stratification
eFigure 10. PRecur Prediction Model Including Only Patients in the MSK Cohort Whose Primary Tumor Did Not Harbor a Level 1 Actionable Mutation (n = 309)
eFigure 11. PRecur-ExVal Prediction Model for the MSK Cohort (n = 426)
eFigure 12. Histogram of the Relapse-Free Survival (RFS) Risk Score Computed Using PRecur-ExVal for the TCGA Data Set (n = 360)
eTable 1. Summary of Patterns of Recurrence (n = 75)
eTable 2. Genes in the 468-Gene MSK-IMPACT Panel
eTable 3. Univariable and Multivariable Cox Proportional Hazards Models for Relapse-Free Survival, Using Clinicopathologic Variables
eTable 4. Univariable and Clinicopathologic (CP)-Adjusted Multivariable Cox Model for Relapse-Free Survival
eTable 5. Types of Alteration and Recurrence Rates for Patients With Alterations in Genes Associated With Relapse-Free Survival
eTable 6. Number of Level 1 Actionable Alterations
eTable 7. Comparison of Proportion of Patients With Pathologic Stage I Cancer Who Recurred by TNM Risk Group
eTable 8. Predicted Relapse-Free Survival at 1, 2, and 3 Years by PRecur Risk Group for the MSK and TCGA Data Sets
eReferences.
References
- 1.Brandt WS, Bouabdallah I, Tan KS, et al. . Factors associated with distant recurrence following R0 lobectomy for pN0 lung adenocarcinoma. J Thorac Cardiovasc Surg. 2018;155(3):1212-1224.e3. doi: 10.1016/j.jtcvs.2017.09.151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thornblade LW, Mulligan MS, Odem-Davis K, et al. . Challenges in predicting recurrence after resection of node-negative non–small cell lung cancer. Ann Thorac Surg. 2018;106(5):1460-1467. doi: 10.1016/j.athoracsur.2018.06.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lou F, Huang J, Sima CS, Dycoco J, Rusch V, Bach PB. Patterns of recurrence and second primary lung cancer in early-stage lung cancer survivors followed with routine computed tomography surveillance. J Thorac Cardiovasc Surg. 2013;145(1):75-81. doi: 10.1016/j.jtcvs.2012.09.030 [DOI] [PubMed] [Google Scholar]
- 4.Pignon JP, Tribodet H, Scagliotti GV, et al. ; LACE Collaborative Group . Lung adjuvant cisplatin evaluation: a pooled analysis by the LACE Collaborative Group. J Clin Oncol. 2008;26(21):3552-3559. doi: 10.1200/JCO.2007.13.9030 [DOI] [PubMed] [Google Scholar]
- 5.Campbell JD, Alexandrov A, Kim J, et al. ; Cancer Genome Atlas Research Network . Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas. Nat Genet. 2016;48(6):607-616. doi: 10.1038/ng.3564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhong WZ, Wang Q, Mao WM, et al. ; ADJUVANT investigators . Gefitinib versus vinorelbine plus cisplatin as adjuvant treatment for stage II-IIIA (N1-N2) EGFR-mutant NSCLC (ADJUVANT/CTONG1104): a randomised, open-label, phase 3 study. Lancet Oncol. 2018;19(1):139-148. doi: 10.1016/S1470-2045(17)30729-5 [DOI] [PubMed] [Google Scholar]
- 7.Jordan EJ, Kim HR, Arcila ME, et al. . Prospective comprehensive molecular characterization of lung adenocarcinomas for efficient patient matching to approved and emerging therapies. Cancer Discov. 2017;7(6):596-609. doi: 10.1158/2159-8290.CD-16-1337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.von Elm E, Altman DG, Egger M, Pocock SJ, Gøtzsche PC, Vandenbroucke JP; STROBE Initiative . The Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) statement: guidelines for reporting observational studies. J Clin Epidemiol. 2008;61(4):344-349. doi: 10.1016/j.jclinepi.2007.11.008 [DOI] [PubMed] [Google Scholar]
- 9.Martini N, Melamed MR. Multiple primary lung cancers. J Thorac Cardiovasc Surg. 1975;70(4):606-612. doi: 10.1016/S0022-5223(19)40289-4 [DOI] [PubMed] [Google Scholar]
- 10.Chang JC, Alex D, Bott M, et al. . Comprehensive next-generation sequencing unambiguously distinguishes separate primary lung carcinomas from intrapulmonary metastases: comparison with standard histopathologic approach. Clin Cancer Res. 2019;25(23):7113-7125. doi: 10.1158/1078-0432.CCR-19-1700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rizvi H, Sanchez-Vega F, La K, et al. . Molecular determinants of response to anti-programmed cell death (PD)-1 and anti-programmed death-ligand 1 (PD-L1) blockade in patients with non–small-cell lung cancer profiled with targeted next-generation sequencing. J Clin Oncol. 2018;36(7):633-641. doi: 10.1200/JCO.2017.75.3384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sanchez-Vega F, Mina M, Armenia J, et al. ; Cancer Genome Atlas Research Network . Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell. 2018;173(2):321-337.e10. doi: 10.1016/j.cell.2018.03.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shen R, Martin A, Ni A, et al. . Harnessing clinical sequencing data for survival stratification of patients with metastatic lung adenocarcinomas. JCO Precis Oncol. 2019;3:3. doi: 10.1200/PO.18.00307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lausen B, Schumacher M. Maximally selected rank statistics. Biometrics. 1992;48(1):73-85. doi: 10.2307/2532740 [DOI] [Google Scholar]
- 15.Gonen M, Heller G. Concordance probability and discriminatory power in proportional hazards regression. Biometrika. 2005;92(4):965-970. doi: 10.1093/biomet/92.4.965 [DOI] [Google Scholar]
- 16.Eguchi T, Kameda K, Lu S, et al. . Lobectomy is associated with better outcomes than sublobar resection in spread through air spaces (STAS)–positive T1 lung adenocarcinoma: a propensity score-matched analysis. J Thorac Oncol. 2019;14(1):87-98. doi: 10.1016/j.jtho.2018.09.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Beer DG, Kardia SL, Huang CC, et al. . Gene-expression profiles predict survival of patients with lung adenocarcinoma. Nat Med. 2002;8(8):816-824. doi: 10.1038/nm733 [DOI] [PubMed] [Google Scholar]
- 18.Dworakowska D, Jassem E, Jassem J, et al. . MDM2 gene amplification: a new independent factor of adverse prognosis in non-small cell lung cancer (NSCLC). Lung Cancer. 2004;43(3):285-295. doi: 10.1016/j.lungcan.2003.09.010 [DOI] [PubMed] [Google Scholar]
- 19.Jamal-Hanjani M, Wilson GA, McGranahan N, et al. ; TRACERx Consortium . Tracking the evolution of non–small-cell lung cancer. N Engl J Med. 2017;376(22):2109-2121. doi: 10.1056/NEJMoa1616288 [DOI] [PubMed] [Google Scholar]
- 20.Ma X, Le Teuff G, Lacas B, et al. ; LACE-Bio Collaborative Group . Prognostic and predictive effect of TP53 mutations in patients with non–small cell lung cancer from adjuvant cisplatin-based therapy randomized trials: a LACE-Bio pooled analysis. J Thorac Oncol. 2016;11(6):850-861. doi: 10.1016/j.jtho.2016.02.002 [DOI] [PubMed] [Google Scholar]
- 21.Rekhtman N, Montecalvo J, Chang JC, et al. . SMARCA4-deficient thoracic sarcomatoid tumors represent primarily smoking-related undifferentiated carcinomas rather than primary thoracic sarcomas. J Thorac Oncol. 2020;15(2):231-247. doi: 10.1016/j.jtho.2019.10.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Devarakonda S, Rotolo F, Tsao MS, et al. . Tumor mutation burden as a biomarker in resected non–small-cell lung cancer. J Clin Oncol. 2018;36(30):2995-3006. doi: 10.1200/JCO.2018.78.1963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Owada-Ozaki Y, Muto S, Takagi H, et al. . Prognostic impact of tumor mutation burden in patients with completely resected non–small cell lung cancer: brief report. J Thorac Oncol. 2018;13(8):1217-1221. doi: 10.1016/j.jtho.2018.04.003 [DOI] [PubMed] [Google Scholar]
- 24.Hieronymus H, Murali R, Tin A, et al. . Tumor copy number alteration burden is a pan-cancer prognostic factor associated with recurrence and death. Elife. 2018;7:e37294. doi: 10.7554/eLife.37294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Voss MH, Reising A, Cheng Y, et al. . Genomically annotated risk model for advanced renal-cell carcinoma: a retrospective cohort study. Lancet Oncol. 2018;19(12):1688-1698. doi: 10.1016/S1470-2045(18)30648-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sparano JA, Gray RJ, Ravdin PM, et al. . Clinical and genomic risk to guide the use of adjuvant therapy for breast cancer. N Engl J Med. 2019;380(25):2395-2405. doi: 10.1056/NEJMoa1904819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tie J, Wang Y, Tomasetti C, et al. . Circulating tumor DNA analysis detects minimal residual disease and predicts recurrence in patients with stage II colon cancer. Sci Transl Med. 2016;8(346):346ra92. doi: 10.1126/scitranslmed.aaf6219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Garcia-Murillas I, Schiavon G, Weigelt B, et al. . Mutation tracking in circulating tumor DNA predicts relapse in early breast cancer. Sci Transl Med. 2015;7(302):302ra133. doi: 10.1126/scitranslmed.aab0021 [DOI] [PubMed] [Google Scholar]
- 29.Abbosh C, Birkbak NJ, Swanton C. Early stage NSCLC - challenges to implementing ctDNA-based screening and MRD detection. Nat Rev Clin Oncol. 2018;15(9):577-586. doi: 10.1038/s41571-018-0058-3 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
eMethods. Sequencing and Analysis
eFigure 1. CONSORT Diagram for the Stage I-III Cohort
eFigure 2. Distribution of Demographic, Clinicopathologic, and Genomic Characteristics in Early- and Late-Stage Lung Adenocarcinoma (LUAD)
eFigure 3. Fraction and Distribution of Mutations Associated With a Smoking Signature
eFigure 4. Genomic Patterns of Recurrence
eFigure 5. Association Between Tumor Mutation Burden (TMB) and Aggressive Clinicopathologic Features
eFigure 6. Association Between Fraction of Genome Altered (FGA) and Aggressive Clinicopathologic Features
eFigure 7. Association Between Fraction of Genome Altered and Tumor Mutation Burden
eFigure 8. Histogram of the Relapse-Free Survival (RFS) Risk Score Computed Using PRecur, Integrating Clinical and Next-Generation Sequencing Data
eFigure 9. PRecur Prediction Model for Relapse-Free Survival (RFS) Using Integrated Clinicopathologic and Genomic Variables for Risk Stratification
eFigure 10. PRecur Prediction Model Including Only Patients in the MSK Cohort Whose Primary Tumor Did Not Harbor a Level 1 Actionable Mutation (n = 309)
eFigure 11. PRecur-ExVal Prediction Model for the MSK Cohort (n = 426)
eFigure 12. Histogram of the Relapse-Free Survival (RFS) Risk Score Computed Using PRecur-ExVal for the TCGA Data Set (n = 360)
eTable 1. Summary of Patterns of Recurrence (n = 75)
eTable 2. Genes in the 468-Gene MSK-IMPACT Panel
eTable 3. Univariable and Multivariable Cox Proportional Hazards Models for Relapse-Free Survival, Using Clinicopathologic Variables
eTable 4. Univariable and Clinicopathologic (CP)-Adjusted Multivariable Cox Model for Relapse-Free Survival
eTable 5. Types of Alteration and Recurrence Rates for Patients With Alterations in Genes Associated With Relapse-Free Survival
eTable 6. Number of Level 1 Actionable Alterations
eTable 7. Comparison of Proportion of Patients With Pathologic Stage I Cancer Who Recurred by TNM Risk Group
eTable 8. Predicted Relapse-Free Survival at 1, 2, and 3 Years by PRecur Risk Group for the MSK and TCGA Data Sets
eReferences.