

Abstract
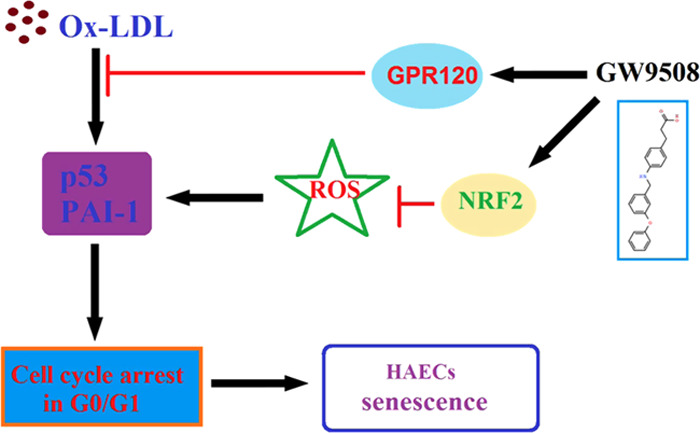
Introduction Oxidized low-density lipoprotein (ox-LDL)-induced endothelial senescence is involved in the pathogenesis of atherosclerosis and many cardiovascular diseases. G-protein-coupled receptor 120 (GPR120), a type of orphan G-protein-coupled receptors (GPRs), plays a vital role in mediating anti-inflammatory and insulin-sensitizing effects. The biological function of GPR120 in vascular endothelial cells is largely unknown. Methods The human aortic endothelial cells (HAECs) were treated with ox-LDL (100 μg/mL) in the presence or absence of GW9508 (50 μM) or AH9614 (1 μM) for 24 h. The LDH assay was used to determine cell death. The dihydroethidium (DHE) staining assay was used to measure intracellular levels of reactive oxidative species (ROS), and a senescence β-galactosidase assay kit was used to determine endothelial senescence. Gene and protein expressions were measured using real-time polymerase chain reaction (PCR) and western blot analysis, respectively. Results Ox-LDL treatment decreased the expression of GPR120 by more than half in HAECs. Typically, 100 μg/mL of ox-LDL- induced 35.2% LDH release, which was reduced to 16.9% by 50 μM GW9508, the agonist of GPR120. Importantly, GW9508 relieved cytotoxicity and suppressed the ox-LDL-induced increase in the activity of senescence-associated β-galactosidase (SA-β-Gal) (from 3.3-fold to 1.6-fold of the control group) and the generation of cellular reactive oxidative species (ROS) (from 3.8-fold to 1.6-fold of the control group). Furthermore, we found that GW9508 ameliorated ox-LDL-induced endothelial cell cycle arrest at the G0/G1 phase and the expression of key senescence proteins, including p53 and plasminogen activator inhibitor-1(PAI-1). Mechanistically, we showed that GW9508 promoted ox-LDL-induced transcriptional factor NF-E2-related factor 2 (NRF2) (increase by 47.3%) translocation into the nucleus. The effect of GW9508 is dependent on its receptor GPR120, the blockage of which by its specific antagonist, AH7614, abolished the antisenescence effect of GW9508. Conclusion Collectively, this study revealed the protective effect of GPR120 activation in vascular endothelial cells, implying that GPR120 is a promising therapeutic target for the treatment of cardiovascular diseases.
1. Introduction
Aging has been considered as one of the most significant risk factors in the development of cardiovascular diseases.1,2 Endothelial cells (ECs) are the main component of the inner lining of blood vessels. Accumulated pieces of evidence have shown that senescence of ECs plays a critical role in the development of cardiovascular diseases, including atherosclerosis,3,4 and that the senescent phenotype of vascular ECs is featured in atherosclerotic lesions.5,6 ECs are the direct response to hemodynamic stress and biochemical stimuli.7 Low-density lipoprotein (LDL), especially the oxidized form (ox-LDL), plays an important role in the initiation and progression of atherosclerosis by inducing EC dysfunction.8 Notably, ox-LDL normally provokes quiescent endothelium and is easily deposited on the vascular wall by immune cells.9 Ox-LDL-induced endothelial senescence directly contributes to the accumulation of vascular ROS.10 Senescent endothelial cells stay at an irreversible growth arrest, exhibiting a flattened and enlarged morphology, and express cell cycle regulators such as p53, p16, and PAI-1. Senescent endothelial cells acquire prothrombotic, antifibrinolytic, and proinflammatory phenotypes, which are risk factors for atherosclerosis.6 Endothelial senescence-caused endothelial dysfunction and vascular abnormality are hallmarks of atherosclerosis.7 Exploration of the underlying mechanism and potential interventions aimed at modulating ECs are essential for preventing or delaying atherosclerosis.
The complexity of atherosclerosis suggests that its pathogenesis involves many biological processes, including the cell surface receptors, which sense extracellular signals. G-protein-coupled receptors (GPCRs) are large groups of proteins used for many aspects of cellular function. A recent study showed that several orphan GPCRs are activated by free fatty acids (FFAs) and act as the receptors for these lipid molecules. GPR40 and GPR120 are the two types of receptors activated by long-chain fatty acids (LCFAs).11,12 Specifically, GPR120 is the putative receptor of omega-3 fatty acids, which are essential for cardiovascular health, and found in many natural supplements and foods. GPR120 plays an essential role in anti-inflammation, insulin signaling pathways, glucose metabolism, and the secretion of intestinal hormones.13 GPR120 has been a potential novel therapeutic target for the treatment of type 2 diabetes mellitus.14 In vascular endothelial cells, GPR120 is involved in cell migration and tumor angiogenesis.15,16 Based on this knowledge, we hypothesized that GPR120-mediated signals could play an essential role in the regulation of vascular cells. In this study, we sought to investigate its role in ox-LDL-stimulated vascular endothelial senescence.
2. Results
2.1. Ox-LDL Represses Endothelial GPR120 Expression
To determine the involvement of GPR120 in endothelial senescence, we treated human aortic endothelial cells (HAECs) with ox-LDL and evaluated GPR120 expression. In our time-course experiment, HAECs were treated with 100 μg/mL of ox-LDL for 12, 24, and 48 h. Compared to the nontreated cells, we found that ox-LDL repressed GPR120 mRNA (Figure 1A) and protein expression (Figure 1B) in a time-dependent manner. These data suggest that GPR120 could be a responsive factor to the ox-LDL treatment in endothelial cells.
Figure 1.

Ox-LDL reduced the expression of GPR120 in human aortic endothelial cells (HAECs). Cells were treated with 100 μg/mL ox-LDL for 12, 24, and 48 h. (A) mRNA of GPR120 as determined by real-time polymerase chain reaction (PCR). (B) Protein of GPR120 as determined by western blot analysis (*, P < 0.01 vs the vehicle group; # and ##, P < 0.01 and 0.001 vs the 12 h ox-LDL group).
2.2. GPR120 Agonist GW9508 Relieves Ox-LDL-Induced Cytotoxicity
Subsequently, we investigated the role of GPR120 in ox-LDL-induced cytotoxicity. We added a compound agonist GW9508 in our ox-LDL treatment experiment and assessed cellular LDH release levels. The molecular structure of GW9508 is shown in Figure 2A. Results in Figure 2B demonstrate that ox-LDL stimulation induced LDH release in a dose-dependent manner from 25 to 100 μg/mL. Compared to nontreated cells, 24 h ox-LDL stimulation (100 μg/mL) induced a 6-fold increase in LDH release, but the presence of 50 μM GW9508 reduced more than half of the LDH release (Figure 2C).
Figure 2.

Agonism of GPR120 with GW9508 prevented the ox-LDL-induced release of lactate dehydrogenase (LDH) in human aortic endothelial cells (HAECs). Cells were treated with ox-LDL (100 μg/mL) in the presence or absence of GW9508 (50 μM) for 24 h. (A) Molecular structure of GW9508. (B) Cells were treated with ox-LDL (25, 50, and 100 μg/mL) for 24 h. The release of LDH was assayed (*, P < 0.01 vs the vehicle group; # and ##, P < 0.01 and 0.001 vs the 25 μg/mL ox-LDL group); (C) Cells were treated with ox-LDL (100 μg/mL) in the presence or absence of GW9508 (50 μM) for 24 h. The release of LDH was measured. The release of LDH (%) = 100× (experimental LDH release-medium background)/(maximum LDH release control-medium background) (***, P < 0.0001 vs the vehicle group; ##, P < 0.001 vs the ox-LDL group).
2.3. GW9508 Inhibits ox-LDL-Induced Endothelial Senescence
The protective effect of GW9508 on ox-LDL-induced endothelial toxicity indicates that the activation of the GPR120 receptor could have a beneficial role to counter ox-LDL. We then determined its influence on endothelial senescence by measuring SA-β-gal activity. Compared to nontreated cells, ox-LDL treatment for 7 days induced more than a 3-fold increase in SA-β-gal activity, but the presence of the same dose of GW9508 inhibited more than half of SA-β-gal activity (Figure 3).
Figure 3.
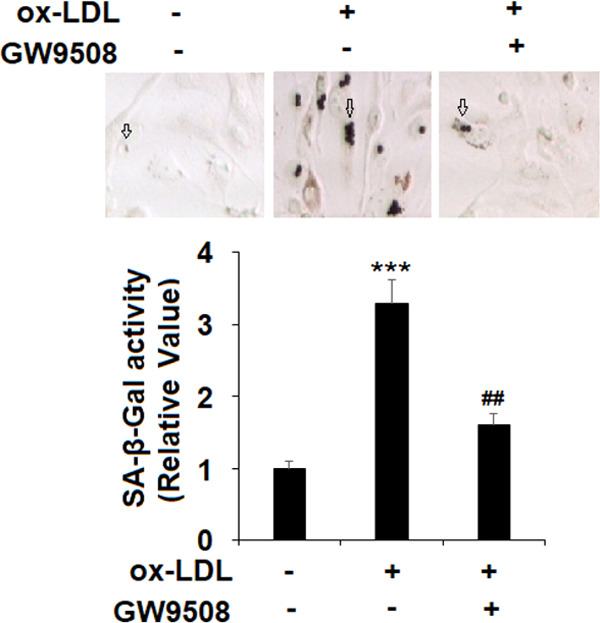
GW9508 prevented ox-LDL-induced cellular senescence in human aortic endothelial cells (HAECs). Cells were treated with ox-LDL (100 μg/mL) in the presence or absence of GW9508 (50 μM) for 7 days. The SA-β-Gal activity of HAECs was measured to determine cellular senescence (***, P < 0.0001 vs the vehicle group; ##, P < 0.001 vs the ox-LDL group).
2.4. GW9508 Protects ox-LDL-Induced Cell Cycle G0/G1 Phase Growth Arrest
Subsequently, we analyzed the cell cycle profile with the ox-LDL treatment. The normal growing HAEC population had about 51% G0/G1 phase cells at arrest, and while ox-LDL induced a 79% G1/G0 phase cell population, the presence of GW9508 significantly attenuated ox-LDL-induced cell cycle arrest, and roughly 62% population of the cells was at the G1/G0 phase arrest (Figure 4).
Figure 4.

GW9508 prevented ox-LDL-induced cell cycle arrest in the G0/G1 phase in human aortic endothelial cells (HAECs). Cells were treated with ox-LDL (100 μg/mL) in the presence or absence of GW9508 (50 μM) for 7 days. (A) Representative histograms showing control and treated cell populations in G0/G1, S, and G2/M phases. (B) Cell cycle distribution in HAECs treated with ox-LDL or GW9508 (*, P < 0.01 vs the vehicle group; #, P < 0.01 vs the ox-LDL group).
2.5. GW9508 Inhibits ox-LDL-Induced p53 and PAI-1 Expressions
Mechanistically, we tested several key cell cycle and senescence regulators, including transcription factor p53 and coagulation factor PAI-1. In the same 24 h ox-LDL experiment, we found that GW9508 inhibited both p53 and PAI-1 ox-LDL-induced expressions at mRNA (Figure 5A) and protein levels (Figure 5B), respectively.
Figure 5.

GW9508 reduced ox-LDL-induced expression of p53 and PAI-1 in human aortic endothelial cells (HAECs). Cells were treated with ox-LDL (100 μg/mL) in the presence or absence of GW9508 (50 μM) for 24 h. (A) mRNA of p53 and PAI-1 and (B) protein of wild-type p53 and PAI-1 (***, P < 0.0001 vs vehicle control; ##, P < 0.001 vs the ox-LDL group).
2.6. GW9508 Suppresses ox-LDL-Induced ROS Generation
Ox-LDL-induced ROS production is one of the fundamental mechanisms to endothelial senescence. We predicted that GW9508 would have a suppressive role in ROS generation. Indeed, the dihydroethidium (DHE) staining experiment showed that cotreatment with GW9508 suppressed more than half of ROS production by ox-LDL (Figure 6).
Figure 6.
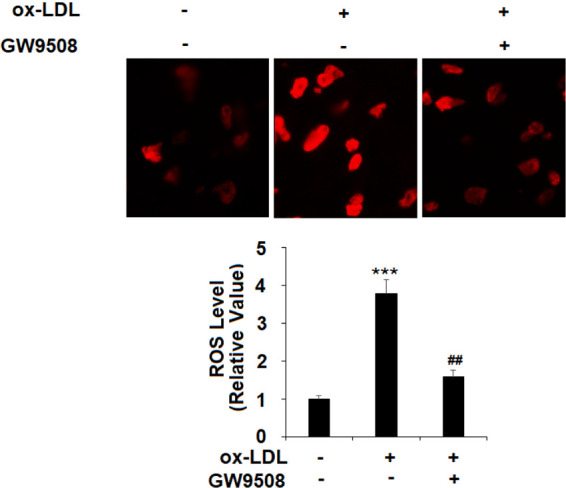
GW9508 decreased ox-LDL-induced production of reactive oxygen species (ROS) in human aortic endothelial cells (HAECs). Cells were treated with ox-LDL (100 μg/mL) in the presence or absence of GW9508 (50 μM) for 24 h. Production of ROS was determined using dihydroethidium (DHE) staining (***, P < 0.0001 vs vehicle control; ##, P < 0.001 vs the ox-LDL group).
2.7. GW9508 Promotes Nuclear Translocation of NRF2
Furthermore, we found that GW9508 exhibited robust promotion of ox-LDL-induced nuclear translocation of NRF2 in the 24 h treatment experiment. Compared to the nontreated cells, ox-LDL induced increased NRF2 translocation into nuclei by 1.9-fold and GW9508 cotreated cells induced about a 2.8-fold increase of NRF2 translocated to nuclei (Figure 7).
Figure 7.

GW9508 increased nuclear translocation of nuclear factor erythroid-2-related factor 2 (NRF2). Cells were treated with ox-LDL (100 μg/mL) in the presence or absence of GW9508 (50 μM) for 24 h. Nuclear translocation of NRF2 was measured using western blot (***, P < 0.0001 vs vehicle control; ##, P < 0.001 vs the ox-LDL group).
2.8. Protective Role of GW9502 is Dependent on GPR120
Finally, we examined the effect of GW9502 when GPR120 was blocked with its selective antagonist, AH9614. In the 24 h ox-LDL treatment experiment, blockage of GPR120 not only abolished the suppressive effect of GW9502 (Figure 8A) on ox-LDL-induced cellular senescence but also demolished its inhibitory effect on p53 and PAI-1 at both mRNA (Figure 8B) and protein levels (Figure 8C).
Figure 8.

Effects of GW9508 in inhibiting cellular senescence in human aortic endothelial cells (HAECs). Cells were treated with ox-LDL (100 μg/mL) with or without GW9508 (50 μM) or the GPR120 antagonist AH7614 (1 μM) for 24 h. (A) SA-β-Gal activity; (B) mRNA of p53 and PAI-1; and (C) protein of p53 and PAI-1 (***, P < 0.0001 vs vehicle control; ##, P < 0.001 vs the ox-LDL group; and $$$, P < 0.0001 vs the ox-LDL + AH7614 group).
3. Discussion
Endothelial dysfunction has been recognized as an important factor in the progression of atherosclerosis.7 Endothelial cells express LOX-1 as the receptor for ox-LDLs to mediate their effects. Ox-LDL activates the endothelial apoptosis pathway and increases the production of ROS, which are the features of endothelial senescence.17 Ox-LDL is the biomarker of oxidative stress and vascular aging. The ox-LDL-caused endothelial senescence is detrimental to vascular function and has been proposed as an essential indicator of atherosclerosis.18
G-protein-coupled receptors (GPCRs) are the largest and most diverse group of membrane receptors in eukaryotes and play a role in an incredible array of functions in the human body.19
GPCRs have become the molecular targets of a wide range of medicines employed clinically to treat metabolic and inflammatory diseases. Among them, both GPR40 and GPR120 are GPCRs that can be activated by saturated and unsaturated long-chain fatty acids (LCFAs). GPR40 is expressed at high levels in islets and mediates the ability of LCFAs to potentiate glucose-stimulated insulin secretion (GSIS). GPR120 is expressed at high levels in the colon, adipose, and pituitary and at more modest levels in pancreatic islets.20 Although activated by the same group of free fatty acids, GPR40 and GPR120 are not closely related.21 In our initial investigation, we found that the ox-LDL treatment repressed the expression of endothelial GPR120 in a time-dependent manner, suggesting that endothelial GPR120 could mediate ox-LDL-responsive signals and possibly play a role in the pathogenesis of atherosclerosis. We then assessed the involvement of GPR120 in ox-LDL-induced cellular stress and senescence through agonizing its activation. We employed the compound agonist GW9508, which activates GPR120, and showed that GW9508 attenuated ox-LDL-induced cytotoxicity as revealed by its suppression of cellular LDH release. Senescence-associated β-galactosidase (SA-β-gal) is the cellular marker of senescence. After the measurement of endothelial SA-β-Gal activity, we found that GW9508 robustly suppressed ox-LDL-induced cellular senescence.
Further, in this study, we found that GW9508 protected endothelial cells from ox-LDL-induced production of cellular ROS and G0/G1 phase growth arrest. While ox-LDL treatment elongated the quiescent/gap phase (G0/G1), shortened the DNA synthesis phase (S phase), and induced senescent phenotype, the activation of GPR120 by GW9508 partially reversed the effect of ox-LDL on cell cycle arrest and the senescence marker SA-β-Gal. These findings indicate that activation of GPR120 by GW9508 exhibits a protective effect to counter ox-LDL-induced vascular senescence.
Mechanistically, we found that GW9508 suppressed ox-LDL-induced cell cycle regulators p53 and PAI-1 but induced NRF2. Predictably, the blockage of GPR120 by its selective antagonist AF7614 completely abolished all of the protective effects of GW9508. GW9508 is the agonist of both GPR40 and GPR120 but only increases the transcription of GPR40.22 However, AF7614 is a selective GPR120-specific antagonist.23 These facts indicate the role of GW9508 in endothelial cells, which is solely dependent on GPR120 signals. Stress-induced premature senescence of endothelial cells is characterized by cell cycle arrest and linked to the pathogenesis of atherosclerosis.24,25
Several cellular regulators are known to contribute to the senescence of endothelial cells, including p53 and PAI-1. P53 serves as a survival factor as a response to cell cycle arrest in endothelial cells.26 PAI-1 acts as a mediator and marker of vascular aging,27 and senescent endothelial cells increase the expression of plasminogen activator inhibitor-1 (PAI-1).28 NRF2 is the molecular link between oxidative stress and vascular aging.29 Oxidative stress-induced NRF2 translocation into the nucleus promotes the transcription of antioxidant proteins, which establish detoxification to protect the endothelial cells in the setting of aging and atherosclerosis.30 Consistently, the most recent study shows omega-3 fatty acid (putative GPR120 ligand) via the upregulation of NRF2-mediated antioxidant response.31
In in vitro-cultured vascular endothelial cells, GPR120 signals have been linked to extracellular regulated protein kinases (ERKs) and endothelial NO synthase (eNOS) signaling pathways, which are critical to endothelial function.15 The most recent study indicates that the GPR120-linked AMP-activated protein kinase (AMPK) pathway is involved in the protective role of the fatty acid metabolite in endothelial cells.32 Therefore, endothelial GPR120 could play a significant role in the regulation of vascular function. This study demonstrated that the activation of GPR120 by GW9504 had a protective effect on oxidative stress-induced endothelial senescence by regulating several cellular pathways, including p53, PAI-1, and NRF2. This is consistent with a recent study showing that the activation of GPR120 with its agonists GW9508 and TUG-891 prevents ox-LDL-induced attachment of monocytes to endothelial cells by reducing the expression of VCAM-1 and E-selectin but elevating the expression level of KLF2.26 These data indicate that GPR120 and its agonists could have potential implications in the treatment of atherosclerosis.
Several limitations exist in this study. First, we only assessed the beneficial effects of GPR120 and its agonist GW9508 against ox-LDL-induced cellular senescence in an in vitro primary HAEC culture model. Although HAECs have been widely used in previous studies to investigate the mechanism of the endothelial toxicity of ox-LDL, it should be noted that a complex heterogeneity exists between different vascular beds.33,34 Therefore, it is necessary to study the effects of GPR120 in endothelial cells from other vascular beds, such as the coronary artery, carotid artery, and brachiocephalic artery. Second, the pathophysiological mechanism of atherosclerosis is complicated. A variety of risk factors have been reported to be involved in atherosclerosis, including genetics, diabetes, aging, and obesity.35 In addition to ox-LDL, there are various chemokines and cytokines involved in the pathophysiology of atherosclerosis.36 Third, a diversity in types of cells, including smooth muscle cells, fibroblasts, monocytes, and the interactions between these cells participate in the development of atherosclerosis.37 The physiological function of GPR120 in these cells is still unknown. Animal models are important tools for exploring the pathogenesis and treatment of atherosclerosis. Further in vivo studies with animal experiments will be helpful to verify the pharmacological function of GPR120 in atherosclerosis.
4. Materials and Methods
4.1. Endothelial Cell Culture
The primary human aortic endothelial cells (HAECs) were purchased from a commercial source (Lonza). The cells were maintained in low serum (2% fetal bovine serum (FBS)) growth media (EGM2) with multiple growth factors. All of the cultures were housed in 5% (v/v) CO2 and humidified conditions at 37 °C. The experiments were performed within 3–7 passages of culture time.
4.2. Ox-LDL, GPR120 Agonist, and Antagonist Treatment
To conduct the cell treatment experiment, the subconfluent HAECs were incubated with 100 μg/mL38,39 growth media incorporated with freshly prepared ox-LDL (Thermo Fisher Scientific) at various times. To activate GPR120, the cells were treated with compound agonist GW9508 (50 μM, Sigma-Aldrich).40 To inactivate GPR120, the cells were treated with the specific antagonist AH7614 (1 μM).41
4.3. LDH Assay
The LDH assay kit was from Thermo Fisher Scientific (c20300). The assay was to capture LDH levels from cultured media, which are released from damaged cells. Briefly, the cell media were collected and cleared to remove cell debris. To set up a Maximum LDH Release Control, 2 μL of 10% Triton X-100 was added to the vehicle-only wells, mixed, and incubated for at least 15 min before sample removal. The collected samples were reacted with the substrate in a 96-well plate to have an enzymatic reaction. The reacted plate was measured using a microplate reader at 450 nM. Blank wells and a series of LDH standard diluent wells were performed in parallel to calculate sample LDH activity. The release of LDH (%) =100 × (experimental LDH release-medium background)/ (maximum LDH release control-medium background).
4.4. ROS Assay
The cellular reactive oxygen (ROS) production was measured by staining the living cells with dihydroethidium (DHE) dye in serum-free media for 30 min. The unbound dyes were washed away with phosphate-buffered saline (PBS), and cells were also counter-stained with DAPI for 10 min. The digital images from 20 different fields were immediately taken on the Leica DVM6 platform (Leica microsystem) and quantified using ImageJ software (NIH). The final data were presented as fold change.
4.5. SA-β-Gal Assay
The cellular senescence was monitored by measuring the activity of SA-β-gal using the senescence β-galactosidase assay kit (Cell signal Technology), following the product’s instruction sheet. Briefly, cells were fixed with 4% paraformaldehyde for 15 min and then incubated with an SA-β-gal reaction solution overnight at 37 °C. The SA-β-gal activity was determined using the percentage of positive staining cells in 20 random fields under a microscope.
4.6. Cell Cycle Analysis
The cell cycle profile was assessed using flow cytometry (Beckman Coulter). In brief, the cells were fixed in cold 95% ethanol for 30 min. To conduct staining, the cell pellet was resuspended in 50 μg/mL propidium iodide (PI) and 50 μL of RNase A at 37 °C for 3 h. The proportion of each phase of the cell cycle was analyzed.
4.7. Real-Time PCR Analysis
The total RNAs from HAECs were isolated using an RNeasy mini kit (Qiagen, German). The extraction steps were followed according to the instructions of the manufacturer. The purities and concentrations of RNAs were monitored using a Nanodrop 2000 spectrophotometer (Cole Parmer). cDNA was synthesized with an iScript one-step reverse transcription kit (Bio-Rad Laboratories) according to the product’s manual. The synthesized cDNA mix was further diluted by 1:10. The real-time PCR reaction was performed with SYBR Supermix and 1 μL of cDNA mix for 40 PCR cycles on an ABI Prism 7500 system (Applied Biosystems Inc.). The relative expressions of these genes were calculated by the double delta Ct method and normalized to control GAPDH.
4.8. Western Blot
To obtain total protein extract, the cultured cells were lysed using radio immunoprecipitation assay (RIPA) buffer with protease inhibitor (Sigma-Aldrich). The lysates were then centrifuged for soluble proteins. In the NRF2 translocation experiment, the cytosol and nuclear portion of proteins were separated by a commercial NE-PER nuclear and cytoplasmic isolation kit (Thermo Fisher Scientific). A total of 10–20 μg of proteins was loaded to 4–12% precasted polyacrylamide gel electrophoresis (PAGE) gel to separate the protein mix. After the optimal separation, the gel was transferred to a PVDF membrane, and the blot was blocked using 5% nonfat milk-PBST for 1 h at room temperature. This was followed by incubation with the specific primary antibodies for 2 h and the final reaction with associated chemiluminescence-coupled second antibodies for 1 h, respectively. The antibodies used in this study include GPR120 (1:1000, Santa Cruz #sc-390752), p53 (1:1000, Santa Cruz #sc-126), PAI-1 (1:2000, Santa Cruz #sc-5297), NRF2 (1:500, Santa Cruz #sc-518033), and β-actin (1:5000, Merck Millipore, #MAB1501R). The reacted blots were processed with the iBright western blot image system (Thermo Fisher Scientific), and the digital images were taken for the densitometric analysis.
4.9. Statistics
All experiments in this study were repeated at least three times. The data were expressed as mean ± standard error of the mean (S.E.M). The comparisons between different groups were performed by analysis of variance (ANOVA). A P value < 0.05 was considered statistically significant.
Acknowledgments
This study is funded by the “Dongguan Science and Technology Support Project (No. DGSTSP-20180325)”.
The authors declare no competing financial interest.
References
- Sabbatinelli J.; Prattichizzo F.; Olivieri F.; Procopio A. D.; Rippo M. R.; Giuliani A. Where Metabolism Meets Senescence: Focus on Endothelial Cells. Front. Physiol. 2019, 10, 1523 10.3389/fphys.2019.01523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Head T.; Daunert S.; Goldschmidt-Clermont P. J. The Aging Risk and Atherosclerosis: A Fresh Look at Arterial Homeostasis. Front. Genet. 2017, 8, 216 10.3389/fgene.2017.00216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minamino T.; Komuro I. Vascular cell senescence: contribution to atherosclerosis. Circ. Res. 2007, 100, 15–26. 10.1161/01.RES.0000256837.40544.4a. [DOI] [PubMed] [Google Scholar]
- Minamino T.; Miyauchi H.; Yoshida T.; Ishida Y.; Yoshida H.; Komuro I. Endothelial cell senescence in human atherosclerosis: role of telomere in endothelial dysfunction. Circulation 2002, 105, 1541–1544. 10.1161/01.CIR.0000013836.85741.17. [DOI] [PubMed] [Google Scholar]
- Gimbrone M. A. Jr.; García-Cardeña G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ. Res. 2016, 118, 620–636. 10.1161/CIRCRESAHA.115.306301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bochenek M. L.; Schütz E.; Schäfer K. Endothelial cell senescence and thrombosis: Ageing clots. Thromb. Res. 2016, 147, 36–45. 10.1016/j.thromres.2016.09.019. [DOI] [PubMed] [Google Scholar]
- Cahill P. A.; Redmond E. M. Vascular endothelium - Gatekeeper of vessel health. Atherosclerosis 2016, 248, 97–109. 10.1016/j.atherosclerosis.2016.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Y.; Song D.; Wu J.; Wang J. Long Non-Coding RNAs Link Oxidized Low-Density Lipoprotein with the Inflammatory Response of Macrophages in Atherogenesis. Front. Immunol. 2020, 11, 24 10.3389/fimmu.2020.00024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trpkovic A.; Resanovic I.; Stanimirovic J.; Radak D.; Mousa S. A.; Cenic-Milosevic D.; Jevremovic D.; Isenovic E. R. Oxidized low-density lipoprotein as a biomarker of cardiovascular diseases. Crit. Rev. Clin. Lab. Sci. 2015, 52, 70–85. 10.3109/10408363.2014.992063. [DOI] [PubMed] [Google Scholar]
- Wang J.; Bai Y.; Zhao X.; Ru J.; Kang N.; Tian T.; Tang L.; An Y.; Li P. oxLDL-mediated cellular senescence is associated with increased NADPH oxidase p47phox recruitment to caveolae. Biosci Rep. 2018, 38, BSR20180283 10.1042/BSR20180283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh Y.; Kawamata Y.; Harada M.; Kobayashi M.; Fujii R.; Fukusumi S.; Ogi K.; Hosoya M.; Tanaka Y.; Uejima H.; Tanaka H.; Maruyama M.; Satoh R.; Okubo S.; Kizawa H.; Komatsu H.; Matsumura F.; Noguchi Y.; Shinohara T.; Hinuma S.; Fujisawa Y.; Fujino M. Free fatty acids regulate insulin secretion from pancreatic beta cells through GPR40. Nature 2003, 422, 173–176. 10.1038/nature01478. [DOI] [PubMed] [Google Scholar]
- Hirasawa A.; Tsumaya K.; Awaji T.; Katsuma S.; Adachi T.; Yamada M.; Sugimoto Y.; Miyazaki S.; Tsujimoto G. Free fatty acids regulate gut incretin glucagon-like peptide-1 secretion through GPR120. Nat. Med. 2005, 11, 90–94. 10.1038/nm1168. [DOI] [PubMed] [Google Scholar]
- Karakuła-Juchnowicz H.; Róg J.; Juchnowicz D.; Morylowska-Topolska J. GPR120: Mechanism of action, role and potential for medical applications. Postepy Hig. Med. Dosw. 2017, 71, 942–953. 10.5604/01.3001.0010.5809. [DOI] [PubMed] [Google Scholar]
- Milligan G.; Alvarez-Curto E.; Hudson B. D.; Prihandoko R.; Tobin A. B. FFA4/GPR120: Pharmacology and Therapeutic Opportunities. Trends Pharmacol. Sci. 2017, 38, 809–821. 10.1016/j.tips.2017.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao C.-Y.; Lii C. K.; Ye S. Y.; Li C. C.; Lu C. Y.; Lin A. H.; Liu K. L.; Chen H. W. Docosahexaenoic acid inhibits vascular endothelial growth factor (VEGF)-induced cell migration via the GPR120/PP2A/ERK1/2/eNOS signaling pathway in human umbilical vein endothelial cells. J. Agric. Food Chem. 2014, 62, 4152–4158. 10.1021/jf5007165. [DOI] [PubMed] [Google Scholar]
- Wu Q.; Wang H.; Zhao X.; Shi Y.; Jin M.; Wan B.; Xu H.; Cheng Y.; Ge H.; Zhang Y. Identification of G-protein-coupled receptor 120 as a tumor-promoting receptor that induces angiogenesis and migration in human colorectal carcinoma. Oncogene 2013, 32, 5541–5550. 10.1038/onc.2013.264. [DOI] [PubMed] [Google Scholar]
- Gradinaru D.; Borsa C.; Ionescu C.; Prada G. I. Oxidized LDL and NO synthesis--Biomarkers of endothelial dysfunction and ageing. Mech. Ageing Dev. 2015, 151, 101–113. 10.1016/j.mad.2015.03.003. [DOI] [PubMed] [Google Scholar]
- Tian X.-L.; Li Y. Endothelial cell senescence and age-related vascular diseases. J. Genet. Genomics 2014, 41, 485–495. 10.1016/j.jgg.2014.08.001. [DOI] [PubMed] [Google Scholar]
- Duan J.; Shen D.; Zhou X. E.; Bi P.; Liu Q.; Tan Y.; Zhuang Y.; Zhang H.; Xu P.; Huang S.; Ma S.; He X.; Melcher K.; Zhang Y.; Xu H. E.; Jiang Y. Cryo-EM structure of an activated VIP1 receptor-G protein complex revealed by a NanoBiT tethering strategy. Nat. Commun. 2020, 11, 4121 10.1038/s41467-020-17933-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suckow A. T.; Polidori D.; Yan W.; Chon S.; Ma J. Y.; Leonard J.; Briscoe C. P. Alteration of the Glucagon Axis in GPR120 (FFAR4) Knockout Mice. J. Biol. Chem. 2014, 289, 15751–15763. 10.1074/jbc.M114.568683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan G.; Alvarez-Curto E.; Watterson K. R.; Ulven T.; Hudson B. D. Characterizing pharmacological ligands to study the long-chain fatty acid receptors GPR40/FFA1 and GPR120/FFA4. Br. J. Pharmacol. 2015, 172, 3254–3265. 10.1111/bph.12879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briscoe C. P.; Peat A. J.; McKeown S. C.; Corbett D. F.; Goetz A. S.; Littleton T. R.; McCoy D. C.; Kenakin T. P.; Andrews J. L.; Ammala C.; Fornwald J. A.; Ignar D. M.; Jenkinson S. Pharmacological regulation of insulin secretion in MIN6 cells through the fatty acid receptor GPR40: identification of agonist and antagonist small molecules. Br. J. Pharmacol. 2006, 148, 619–628. 10.1038/sj.bjp.0706770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moonwiriyakit A.; Wattanaphichet P.; Chatsudthipong V.; Muanprasat C. GPR40 receptor activation promotes tight junction assembly in airway epithelial cells via AMPK-dependent mechanisms. Tissue Barriers 2018, 6, 1–12. 10.1080/21688370.2018.1480741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J.; Patschan S.; Goligorsky M. S. Stress-induced premature senescence of endothelial cells. J. Nephrol. 2008, 21, 337–344. [PubMed] [Google Scholar]
- Goligorsky M. S.; Chen J.; Patschan S. Stress-induced premature senescence of endothelial cells: a perilous state between recovery and point of no return. Curr. Opin. Hematol. 2009, 16, 215–219. 10.1097/MOH.0b013e32832a07bd. [DOI] [PubMed] [Google Scholar]
- Gudkov A. V.; Komarova E. A. Pathologies associated with the p53 response. Cold Spring Harbor Perspect. Biol. 2010, 2, a001180 10.1101/cshperspect.a001180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comi P.; Chiaramonte R.; Maier J. A. Senescence-dependent regulation of type 1 plasminogen activator inhibitor in human vascular endothelial cells. Exp. Cell Res. 1995, 219, 304–308. 10.1006/excr.1995.1232. [DOI] [PubMed] [Google Scholar]
- Vaughan D. E.; Rai R.; Khan S. S.; Eren M.; Ghosh A. K. Plasminogen Activator Inhibitor-1 Is a Marker and a Mediator of Senescence. Arterioscler., Thromb., Vasc. Biol. 2017, 37, 1446–1452. 10.1161/ATVBAHA.117.309451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair N.; Gongora E. Oxidative Stress and Cardiovascular Aging: Interaction Between NRF-2 and ADMA. Curr. Cardiol. Rev. 2017, 13, 183–188. 10.2174/1573403X13666170216150955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B.; Lu Y.; Chen Y.; Cheng J. The role of Nrf2 in oxidative stress-induced endothelial injuries. J. Endocrinol. 2015, 225, R83–R99. 10.1530/JOE-14-0662. [DOI] [PubMed] [Google Scholar]
- Sakai C.; Ishida M.; Ohba H.; Yamashita H.; Uchida H.; Yoshizumi M.; Ishida T. Fish oil omega-3 polyunsaturated fatty acids attenuate oxidative stress-induced DNA damage in vascular endothelial cells. PLoS One 2017, 12, e0187934 10.1371/journal.pone.0187934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang H. J.; Jung T. W.; Kim J. W.; Kim J. A.; Lee Y. B.; Hong S. H.; Roh E.; Choi K. M.; Baik S. H.; Yoo H. J. Protectin DX prevents H(2)O(2)-mediated oxidative stress in vascular endothelial cells via an AMPK-dependent mechanism. Cell. Signalling 2019, 53, 14–21. 10.1016/j.cellsig.2018.09.011. [DOI] [PubMed] [Google Scholar]
- Jambusaria A.; Hong Z.; Zhang L.; Srivastava S.; Jana A.; Toth P. T.; Dai Y.; Malik A. B.; Rehman J. Endothelial heterogeneity across distinct vascular beds during homeostasis and inflammation. eLife 2020, 9, e51413 10.7554/eLife.51413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tse D.; Stan R. V. Morphological heterogeneity of endothelium. Semin. Thromb. Hemostasis 2010, 36, 236–245. 10.1055/s-0030-1253447. [DOI] [PubMed] [Google Scholar]
- Herrero-Fernandez B.; Gomez-Bris R.; Somovilla-Crespo B.; Gonzalez-Granado J. M. Immunobiology of Atherosclerosis: A Complex Net of Interactions. Int. J. Mol. Sci. 2019, 20, E5293 10.3390/ijms20215293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramji D. P.; Davies T. S. Cytokines in atherosclerosis: Key players in all stages of disease and promising therapeutic targets. Cytokine Growth Factor Rev. 2015, 26, 673–685. 10.1016/j.cytogfr.2015.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabas I.; García-Cardeña G.; Owens G. K. Recent insights into the cellular biology of atherosclerosis. J. Cell Biol. 2015, 209, 13–22. 10.1083/jcb.201412052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanuti M.; Talamonti E.; Maccarrone M.; Chiurchiù V. Activation of GPR55 Receptors Exacerbates oxLDL-Induced Lipid Accumulation and Inflammatory Responses, while Reducing Cholesterol Efflux from Human Macrophages. PLoS One 2015, 10, e0126839 10.1371/journal.pone.0126839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A.; Srinivasan A. K.; Chakrapani L. N.; Kalaiselvi P. LOX-1, the Common Therapeutic Target in Hypercholesterolemia: A New Perspective of Antiatherosclerotic Action of Aegeline. Oxid. Med. Cell. Longevity 2019, 2019, 8285730 10.1155/2019/8285730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anbazhagan A. N.; Priyamvada S.; Gujral T.; Bhattacharyya S.; Alrefai W. A.; Dudeja P. K.; Borthakur A. A novel anti-inflammatory role of GPR120 in intestinal epithelial cells. Am. J. Physiol. Cell Physiol. 2016, 310, C612–21. 10.1152/ajpcell.00123.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheshmehkani A.; Senatorov I. S.; Dhuguru J.; Ghoneim O.; Moniri N. H. Free-fatty acid receptor-4 (FFA4) modulates ROS generation and COX-2 expression via the C-terminal β-arrestin phosphosensor in Raw 264.7 macrophages. Biochem. Pharmacol. 2017, 146, 139–150. 10.1016/j.bcp.2017.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]