

Abstract

Diclofenac (DCF) is widely used as a nonsteroidal anti-inflammatory drug; however, it is associated with severe liver injury. This adverse reaction is thought to be related to the reactive quinone imine (QI) and acyl glucuronide (AG) metabolites of DCF, but it remains controversial which reactive metabolites mainly contribute to DCF-induced toxicity. In this study, we synthesized five types of DCF analogs that were designed to mitigate the formation of reactive QI and/or AG metabolites and evaluated their metabolic stability, cyclooxygenase (COX) inhibitory activity, and toxicity to cryopreserved human hepatocytes. Compounds with fluorine at the 5- and 4′-positions of aromatic rings exhibited modest and high metabolic stability to oxidation by cytochrome P450, respectively, but induced cytotoxicity comparable to DCF. Replacing the carboxylic group of DCF with its bioisosteres was effective in terms of stability to oxidative metabolism and glucuronidation; however, sulfonic acid and sulfonamide groups were not preferable for COX inhibition, and tetrazole-containing analogs induced strong cytotoxicity. On the other hand, compounds that have fluorine at the benzylic position were resistant to glucuronidation and showed little toxicity to hepatocytes. In addition, among these compounds, those with hydrogen at the 4′-position (2a and 2c) selectively inhibited the COX-2 enzyme. Throughout these data, it was suggested that compounds 2a and 2c might be novel safer and more efficacious drug candidates instead of DCF.
Introduction
Diclofenac (DCF) is a nonsteroidal anti-inflammatory drug widely used for the treatment of rheumatoid disorders worldwide. Despite its efficacy, DCF is associated with several adverse reactions, such as gastrointestinal toxicity and liver injury. The incidence of DCF-induced liver injury is rare (approximately 6–18 cases per 100,000 person-years1 or 23 per 100,000 patients2), and it is thought to be idiosyncratic drug-induced liver injury3 (IDILI). Although the concrete mechanism of DCF-induced liver injury is still unclear, similar to other IDILI,4 reactive metabolites of DCF are believed to contribute to the emergence of hepatotoxicity. Indeed, DCF protein adducts were detected in mouse,5,6 rat,7−9 and human samples10,11 both in vitro and in vivo. Recently, candidate proteins targeted by reactive metabolites of DCF have also been characterized using human hepatocytes.12
Previous studies revealed two important metabolic pathways of DCF in humans: oxidative metabolism by cytochrome P450 (CYP) and glucuronidation. The major oxidative metabolite, 4′-hydroxydiclofenac (4′-OH-DCF), is predominantly formed by CYP2C9.13,14 5-Hydroxydiclofenac (5-OH-DCF) is formed mainly by CYP3A4, and other minor mono- or dihydroxylated metabolites are also detected.3,15 The other metabolic pathway of DCF is glucuronidation to yield diclofenac 1-O-β-acyl glucuronide (DCF-AG) mainly by UGT2B7.16
4′-OH and 5-OH-DCF are thought to be key metabolites for the toxicity of DCF. They are further oxidized by CYP2C9 to yield reactive DCF-1′,4′-quinone imine (DCF-1′,4′-QI) and DCF-2,5-quinone imine (DCF-2,5-QI) metabolites17 (Scheme 1). Both of these reactive metabolites are shown to form glutathione adducts in human liver microsomes18,19 (HLM) and human hepatocytes,20 and these findings suggest that both DCF-1′,4′-QI and DCF-2,5-QI may contribute to the hepatotoxicity of DCF. However, Shen et al. have demonstrated that DCF forms protein adducts in HLMs by CYP3A421 and Poon et al. have shown that only the N-acetylcysteine adduct of DCF-2,5-QI was detected in human urine samples,9 implying that DCF-2,5-QI plays an important role in DCF-induced toxicity. In addition, Naisbitt et al. reported that 5-OH-DCF and DCF-2,5-QI induced immune cell activation in mice.22 Some studies have also shown that other reactive metabolites generated by CYP might contribute to the toxicity of DCF. It was demonstrated that arene oxides, which turn into 4′- or 5-OH-DCF, might lead to liver toxicity.23,24
Scheme 1. Proposed Bioactivation Mechanisms of DCF.

AGs are unstable and react with nucleophiles such as amino acids to give acylation and glycation adducts; thus, AGs are believed to be a risk factor for idiosyncratic toxicity.25 DCF-AG has been shown to form both types of adducts (Scheme 1) with rat hepatic microsomes fortified with UDP-glucuronic acid (UDPGA).26 It was also reported that protein adducts derived from DCF-AG were identified from DCF-treated patients.11 Oda et al. demonstrated that DCF-AG is at least partly involved in DCF-induced liver injury in mice.27 However, there are also some reports indicating that DCF-AG might not be related to cytotoxicity.28,29
To our knowledge, only one study has investigated the structure–metabolism relationships of DCF analogs by Pang et al.(30) They prepared 2′-halogen and/or 5-alkyl DCF analogs and assessed their phase I metabolic stability and electrophilic reactivity. It provides some useful insights into future drug design, but considering the metabolic profile of DCF, the contribution of phase II metabolism should be taken into account. In addition, the pharmacological activity of almost all of these compounds is weaker than DCF,31 which gives limited motivations to apply these structural changes in drug discovery.
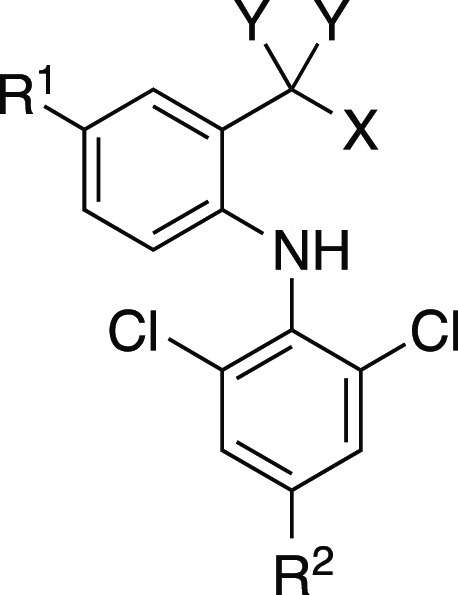
In the present study, from the viewpoints of both medicinal chemistry and drug metabolism, we report the synthesis of a series of DCF analogs that were designed to mitigate the formation of reactive QI and AG metabolites without losing the pharmacological activity of DCF (Chart 1). Compounds 1b–1d, which have fluorines at the 5-(R1) and/or 4′- (R2) position(s) of aromatic rings, were designed to avoid the formation of 4′- and/or 5-OH-DCF and the subsequent generation of reactive QI intermediates. The common method for reducing AG metabolites is thought to be introducing methyl group(s) into the benzylic position, as exemplified by the examples of ibufenac and ibuprofen. However, our preliminary data showed that such mono- and di-methylated DCF derivatives lost COX inhibitory activity. This made us synthesize compound 2a, which has fluorine at the benzylic position instead of methyl groups. It was designed to investigate the electronic effect of fluorine on glucuronidation of carboxylic acid and expected to retain the COX inhibitory activity. The carboxy group of DCF was replaced with its bioisosteres in compounds 3a, 4a, and 5a, and these analogs were expected to reduce the generation of AG metabolites without losing the COX inhibitory activity. Throughout evaluations of their metabolic stability, hepatocyte toxicity, and COX inhibitory activity, it has been demonstrated that the introduction of two fluorines at the benzylic position of DCF is effective in terms of both hepatocyte toxicity and COX inhibition.
Chart 1. Structures of DCF and Its Analogs Synthesized in the Present Study.

Results
Chemistry
Fluorinated DCF derivatives 1b–1d were synthesized using the reported procedure32 with slight modifications (Scheme 2). After protection of the carboxy group of 6a or 6b, the protected amides 7a or 7b were coupled with corresponding amines followed by deprotection to give target compounds 1b–1d. To synthesize 2a–2d, DCF (1a) or synthesized 1b–1d were converted to ethyl ester 9a–9d, fluorinated using N-fluorobenzenesulfonimide (NFSI), and subsequent hydrolysis gave compounds 2a–2d (Scheme 3).
Scheme 2. Synthesis of DCF Derivatives 1b–1d.

Reagents and conditions: (a) SOCl2, CH2Cl2, rt–50 °C, 1–3 h, then 40% NH(CH3)2-H2O, 0 °C, 30 min, 71–83%; (b) 2,6-dichloroaniline (for 8c) or 2,6-dichloro-4-fluoroaniline (for 8b and 8d), K2CO3, Cu powder, CuI, toluene, reflux, 62–64 h, 66–90%; (c) NaOH, C2H5OH, H2O, reflux, 2.5–4 h, 90–99%.
Scheme 3. Synthesis of DCF Analogs 2a–2d.

Reagents and conditions: (a) H2SO4, C2H5OH, reflux, 0.5–2 h, 70–92%; (b) LiHMDS, NFSI, THF, −78 °C, 1–2 h, 49%–quant.; (c) NaOH, CH3OH, H2O, rt, 2 h, 73–96%.
Bertinaria’s synthetic strategy33 was adopted to synthesize tetrazole analogs 3a–3d (Scheme 4). Briefly, the carboxy group of the starting materials was protected with dimethylamine, and coupling with the 2,6-dichloroaniline moiety gave compounds 12a–12d. After deprotection, the carboxy group was reduced using LiAlH4. The alcohols 14a–14d were transferred to the nitrile via chloride intermediates without isolation, and the construction of a tetrazole ring with sodium azide gave tetrazole 3a–3d. Sulfonic analog 4a was synthesized from alcohol 14a in two steps.
Scheme 4. Synthesis of DCF Analogs 3a–3d and 4a.

Reagents and conditions: (a) SOCl2, CH2Cl2, 50 °C, 2–2.5 h, then 40% NH(CH3)2-H2O, 0 °C, 30 min, 94–95%; (b) 2,6-dichloroaniline (for 12a and 12c) or 2,6-dichloro-4-fluoroaniline (for 12b and 12d), K2CO3, Cu powder, CuI, toluene, reflux, 84–96 h, 64–88%; (c) NaOH, C2H5OH, H2O, reflux, 12–18 h, 84%–quant.; (d) LiAlH4, THF, 45 °C–reflux, 0.5–9 h, 26–97%; (e) SOCl2, pyridine, THF, rt, 30 min, then NaCN, DMSO, 40 °C, 1–1.5 h, 54–69%; (f) NaN3, NH4Cl, DMF, 120 °C, 8.5–24 h, 58–81%; (g) (1) PBr3, CH2Cl2, 0 °C–rt, 15 h; (2) Na2SO3, acetone, H2O, reflux, 24 h, 26% in two steps.
The synthetic route of sulfonamide analog 5a is shown in Scheme 5. 2-Nitrobenzylsulfonyl chloride was prepared from 2-nitrobenzyl chloride using the reported method34 and converted to sulfonamide 16. After the reduction of the nitro group, compound 17 was coupled with 2,6-dichloroboronic acid to yield 5a.
Scheme 5. Synthesis of Sulfonamide Analog 5a.

Reagents and conditions: (a) thiourea, C2H5OH, reflux, 4.5 h; (b) NCS, 2 M HCl, CH3CN, rt, 16 h; (c) NH4OH aq, THF, rt, 1 h, 43% in three steps; (d) Fe powder, NH4Cl, C2H5OH, H2O, reflux, 1.5 h, 83%; (e) 2,6-dichloroboronic acid, DBU, Cu(OAc)2, 1,4-dioxane, rt, 3 h, 22%.
Detailed synthetic and purification procedures of each compound can be found in the Supporting Information.
Metabolic Stability of DCF and Its Analogs
DCF and synthesized analogs were incubated with pooled human liver microsomes fortified with an NADPH-generating system (for CYP-mediated oxidation) or UDPGA (for glucuronidation). The metabolic stability of the test compounds was defined as the percentage of parent compounds remaining during the incubation and is shown in Table 1. For CYP-mediated oxidation, compounds that have fluorine at the 4′-position (R2) were more metabolically stable than compounds without fluorine (1b, 1d vs 1a, 1c; 2b, 2d vs 2a, 2c; 3b, 3d vs 3a, 3c). In addition, the stabilities of tetrazole-, sulfonic acid-, and sulfonamide-type DCF analogs (3a, 4a, and 5a), in which the carboxy group of DCF was replaced by its bioisosteres, were higher than that of DCF. Compounds 2a–2d, 3a–3d, 4a, and 5a, which were designed to mitigate bioactivation via glucuronidation, were more resistant to glucuronidation than DCF. Compounds 1c and 1d, which have fluorine at the 5-position (R1) of DCF, tended to be less susceptible to glucuronidation than compounds 1a and 1b.
Table 1. COX Inhibitory Activity and Metabolic Stability of DCF and Its Analogs.
| compounds |
IC50 (μM) |
metabolic
stability (% remaining)a |
||||||
|---|---|---|---|---|---|---|---|---|
| no. | X | Y | R1 | R2 | COX-1 | COX-2 | oxidation | glucuronidation |
| DCF (1a) | –COOH | H | H | H | 0.44 | 7.5 | 33 ± 4.9 | 38 ± 3.2 |
| 1b | –COOH | H | H | F | 0.67 | 27 | 99 ± 1.2 | 43 ± 0.8 |
| 1c | –COOH | H | F | H | 0.48 | 7.8 | 44 ± 1.0 | 58 ± 3.3 |
| 1d | –COOH | H | F | F | 0.73 | 26 | 107 ± 2.2 | 57 ± 3.2 |
| 2a | –COOH | F | H | H | >100 | 23 | 28 ± 5.0 | 108 ± 2.8 |
| 2b | –COOH | F | H | F | >100 | >100 | 95 ± 5.1 | 105 ± 1.5 |
| 2c | –COOH | F | F | H | >100 | 21 | 25 ± 0.4 | 110 ± 6.4 |
| 2d | –COOH | F | F | F | >100 | >100 | 107 ± 3.7 | 96 ± 16 |
| 3a | –tetrazole | H | H | H | >100 | 12 | 61 ± 2.8 | 100 ± 8.4 |
| 3b | –tetrazole | H | H | F | >100 | >100 | 90 ± 8.3 | 107 ± 2.2 |
| 3c | –tetrazole | H | F | H | >100 | 48 | 74 ± 6.4 | 91 ± 12 |
| 3d | –tetrazole | H | F | F | >100 | >100 | 103 ± 1.3 | 99 ± 3.0 |
| 4a | –SO3H | H | H | H | >100 | >100 | 53 ± 4.3 | 111 ± 7.6 |
| 5a | –SO2NH2 | H | H | H | >100 | >100 | 82 ± 1.5 | 108 ± 1.9 |
For testing metabolic stability, the test compounds were incubated with HLM for 30 min. The remaining amount of test compounds is expressed as percentages relative to the incubation (−) sample. Values represent the mean ± S.D. (n = 3).
COX Inhibitory Activity
The COX-1 (ovine) and COX-2 (human recombinant) inhibitory activities of DCF and its analogs were assessed using a COX Fluorescent Inhibitor Screening Assay Kit (Table 1). Interestingly, only compounds 1a–1d inhibited the COX-1 enzyme (Table 1). The IC50 values among 1a–1d were almost identical, which is consistent with the results of a previous study.32 The COX-2 inhibitory activity was different for each compound. The inhibitory activities of compounds 1b and 1d, which have fluorine at the 4′-position, were weaker than that of compound 1a (DCF), although compound 1c inhibited COX-2 as strongly as 1a did. This tendency was also observed in compounds 2a–2d and 3a–3d. Compounds 2a and 3a inhibited COX-2, but their IC50 values were approximately two to four times higher than that of DCF. Compounds 4a and 5a, which have sulfo or sulfonamide groups instead of the carboxy group of DCF, did not inhibit COX-2 up to a concentration of 100 μM.
Hepatocyte Toxicity of DCF and Its Analogs
The cytotoxicity of DCF and its analogs (1b–1d, 2a–2d, and 3a–3d) was assessed using cryopreserved human hepatocytes (Figure 1). DCF derivatives containing fluorinated aromatic rings 1b–1d induced cytotoxicity comparable to DCF, and similarly, the toxicities of tetrazole-type analogs 3a–3d were comparable to or slightly stronger than that of DCF. On the other hand, compounds 2a–2d induced little toxicity at a concentration of 200 μM, and there was no significant difference from the control group.
Figure 1.

Cytotoxicity of DCF and its analogs in cryopreserved human hepatocytes. Each bar shows the cell viability after incubating with test compounds (200 μM) for 2 h with shaking. Cell viability was assessed by WST-8 assay, and the data are expressed relative to the control group. The control group was treated with 0.25 v/v% DMSO. Each value represents the mean ± S.D. of three samples. **p < 0.01; significantly different from control, ††p < 0.01, †p < 0.05; significantly different from DCF, Dunnett’s post hoc test.
Discussion
The purpose of the present study is to develop novel DCF analogs that avoid the metabolic activation of DCF and therefore reduce hepatotoxicity. As shown in Scheme 1, several bioactivation pathways have been proposed to explain the toxicity of DCF-induced liver injury. Although many studies have shown evidence of metabolic activation of DCF both in vitro and in vivo, it has been incompletely understood which reactive metabolite mainly contributes to the toxicity of DCF, and this made us design DCF analogs that avoid the generation of both reactive QI and AG metabolites. To apply the structural modification found in this study to future drug discovery research, especially for drug candidates containing carboxy groups, this work also aimed to maintain the pharmacological activity of DCF from the viewpoint of medicinal chemistry.
Because 4′-OH-DCF and 5-OH-DCF, which are believed to be converted into reactive DCF-1′,4′-QI and DCF-2,5-QI, are the key metabolites, we first synthesized compounds 1b–1d by introducing fluorine at the 4′- and/or 5-position(s) of DCF. It is well known that the C–F bond (98–115 kcal/mol) is stronger than the C–H bond (89–110 kcal/mol) and thus enhances metabolic stability.35 Although introducing fluorine into the 5-position of DCF exhibited a modest increase in stability against CYP-mediated oxidation (see 1c in Table 1), introducing fluorine into the 4′-position made the compound highly metabolically stable (see 1b and 1d in Table 1), and this is consistent with previous reports where 4′-OH DCF is the major phase I metabolite of DCF. These data indicate that compounds 1b–1d might avoid the formation of 1′,4′- and/or 2,5-QI intermediates. All of these compounds inhibited both COX-1 and COX-2 enzymes, but compounds 1b and 1d showed higher IC50 values than DCF and 1c. This indicates that fluorine at the 4′-position of the aromatic ring is not preferable for COX inhibition.
Next, we synthesized compounds 2a, 3a, 4a, and 5a to mitigate the generation of DCF-AG. As we expected, all of these compounds were highly resistant to glucuronidation (Table 1). These data indicate that reactive AG metabolites may not be generated from these compounds and might contribute to their lower hepatotoxicity. Moreover, compounds 3a, 4a, and 5a were also more metabolically stable against CYP-mediated oxidation than DCF. This may be because the bioisosteres of carboxylic acid change the lipophilicity and electron density of aromatic rings of these compounds. On the other hand, although compounds 2a and 3a inhibited the COX-2 enzyme, compounds 4a and 5a did not inhibit it. Interestingly, all of these compounds lost their COX-1 inhibitory activity. The reason why these compounds did not inhibit COX-1 remains unclear, but this point is favorable rather than a problem, as they selectively inhibit COX-2, an enzyme responsible for inflammation and pain, leading to a lower risk of peptic ulceration caused by DCF. It should be noted that Lee et al. have reported that fluorinated rutaecarpine inhibits COX-2 enzymes more selectively compared to the parent compound,36 although this bioactive alkaloid is structurally different from DCF. Since compounds 4a and 5a lost their inhibitory activity against both COX enzymes, further structural modification and toxicity assays were not performed.
We then synthesized compounds 2b–2d and 3b–3d to avoid CYP-mediated metabolic activation in addition to the prevention of glucuronidation. Similar to compounds 2a and 3a, all of these compounds had great stability in glucuronidation metabolism. Stability against oxidative metabolism was correlated with the results of compounds 1a–1d. This suggests that the 4′-hydroxylated metabolite might also be the major oxidative metabolite of these types of DCF analogs, and at least, compounds that have fluorine at the 4′-position (2b, 2d, 3b, and 3d) might avoid the generation of 1′,4′-QI-type reactive intermediates. The COX-2 inhibitory activity of such compounds was strongly decreased, but compounds 2c and 3c inhibited COX-2 selectively. This tendency was also consistent with the results of 1a–1d.
The hepatocyte toxicity of DCF and its analogs was also assessed using cryopreserved human hepatocytes (Figure 1). Interestingly, compounds 1b–1d showed toxicity comparable to DCF. In contrast, compounds 2a–2d, which mitigated the formation of reactive AG metabolites, displayed little cytotoxicity and showed little difference in toxicity among these compounds. In addition, compounds 2a–2c showed significantly lower toxicity than DCF. Considering the susceptibility of 1b–1d to glucuronidation, it could be thought that their reactive AG metabolites induced toxicity. On the other hand, although compounds 2a–2c have the probability of being converted into 2,5- and/or 1′,4′-QI metabolites, they did not induce hepatocyte toxicity and showed little difference from 2d. These results indicate that reactive AG metabolites might play an important role in DCF-induced hepatocyte toxicity. The toxicities of tetrazole-type analogs 3a–3d were comparable to or slightly stronger than that of DCF despite resistance to glucuronidation, suggesting that the tetrazole analogs might induce cytotoxicity through a different mechanism from QI or AG formation and that replacing the carboxy group of DCF by tetrazole is not favorable in terms of cytotoxicity. In this study, we used hepatocyte suspension as a screening system and for ease of use. However, it is widely accepted that DILI has very complex mechanisms and is difficult to evaluate by a simple in vitro model. To further discuss the key reactive metabolites that contribute to the hepatotoxicity of DCF and the possibility of clinical DILI induced by synthesized DCF analogs, it will be necessary to use a more complicated in vitro assay system such as 2D-sandwich culture or 3D culture or to conduct an in vivo experiment.
Conclusions
In conclusion, we synthesized five types of DCF analogs and evaluated their metabolic stability, COX inhibitory activity, and hepatocyte toxicity. Compounds 2a and 2c were highly resistant to glucuronidation, and their cytotoxicity in cryopreserved hepatocytes was much weaker than that of DCF. In addition, they underwent phase I metabolism by CYP to the same extent as DCF and inhibited COX-2 selectively with IC50 values three times higher than that of DCF. Although the slightly weaker inhibition against COX-2 is an unfavorable profile, we propose that compounds 2a and 2c might serve as drug candidates that could be developed for use as efficacious anti-inflammatory drugs with much safer profiles than DCF, with the aim of enabling dose escalation in vivo and overcoming the attenuation of potency in vitro.
Experimental Procedures
Materials
NADP+, glucose-6-phosphate (G6P), and G6P dehydrogenase (G6PDH) were purchased from Roche Diagnostics (Basel, Switzerland). DCF was obtained from Tokyo Chemical Industry (Tokyo, Japan). All other reagents used for synthetic, microsomal, and cytotoxicity experiments were of analytical grade.
Metabolic Stability to CYP-Mediated Oxidation in Liver Microsomes
Stability to CYP-mediated oxidation was evaluated using pooled human (200 donors) liver microsomes (HLM) purchased from XenoTech (Lenexa, KS) fortified with the NADPH-generating system according to our previously reported procedure37 with slight modifications. All incubations (0.5 mL total volume) were conducted at 37 °C in a water bath and stopped by the addition of 1 mL of an ice-cold mixed acetonitrile/methanol (2/1) solution containing 1 μM aprepitant as an internal standard. After centrifugation, the supernatant was injected and analyzed by liquid chromatography with mass spectrometry (LC-MS, 6120; Agilent Technologies, Palo Alto, CA). HPLC mobile phase A was water with 0.1% formic acid, and mobile phase B was acetonitrile with 0.1% formic acid. The chromatographic separation conditions were the same as those in our previous report as follows: 30% solvent B for 1 min, 30–100% B in 5 min, 100% B for 4 min, and then 100–30% B in 0.1 min followed by 30% B for another 3.9 min (15 min in total) at a flow rate of 0.5 mL/min. Test compounds were ionized via electrospray ionization using the negative ion mode and detected using selected ion monitoring mode.
Metabolic Stability to Glucuronidation in Liver Microsomes
The metabolic stability to glucuronidation was calculated by the % remaining test compounds after incubating with HLM fortified with UDPGA based on the reported procedure.38 The reaction mixtures containing HLM (1.0 mg protein/mL), test compounds (100 μM), MgCl2 (10 mM), d-saccharic acid 1,4-lactone (5.0 mM), and alamethicin (25 μg/mg microsomal protein) in 0.1 M K-Pi buffer (pH 7.4) were preincubated for 15 min on ice. The reaction was initiated by adding UDPGA (2.0 mM), incubated for 30 min at 37 °C and then quenched by the addition of 1 mL of ice-cold acetonitrile containing 1 μM aprepitant and 1% v/v formic acid. The mixture was centrifuged at 10,000g for 10 min. The supernatant (10 μL) was injected and analyzed by LC-MS, and chromatographic separations were performed at a flow rate of 0.5 mL/min under the following gradient elution profile: 30% solvent B for 1 min, 30–100% B in 7 min, 100% B for 8 min, and then 100–30% B in 0.1 min followed by 30% B for another 3.9 min (20 min in total).
COX Inhibition Assay
The COX inhibitory activities of DCF analogs were assessed with a COX Fluorescent Inhibitor Screening Assay Kit (Cayman Chemical, Ann Arbor, MI) according to the manufacturer’s instructions. In brief, the mixtures of COX-1 (ovine) or COX-2 (human recombinant), hemin, test compounds, and Tris–HCl buffer (100 mM, pH 8.0) in a 96-well black plate were incubated at room temperature for 5 min. Reactions of COX-1 or COX-2 were initiated by the addition of arachidonic acid and 10-acetyl-3,7-dihydroxyphenoxazine and incubated at room temperature for 2 min. The plate was read using the VICTOR Nivo Multimode Plate Reader (PerkinElmer, Waltham, MA) with an excitation wavelength of 530–540 nm and an emission wavelength of 585–595 nm.
Cell Cultures
Cryopreserved human hepatocytes (H1000. H15B, HC5-9), OptiThaw Hepatocyte Media and OptiIncubate Hepatocyte Media, were purchased from XenoTech. These hepatocytes were thawed according to the manufacturer’s instructions and incubated in a 5% CO2 incubator under a humidified atmosphere at 37 °C.
Cytotoxicity Assay
Hepatocyte toxicity was assessed by WST-8 assay using a cell counting kit-8 (CCK-8, Dojindo Laboratories, Kumamoto, Japan). The procedure of this assay was adopted from a previous study.39 Cells were seeded (5.0 × 104 cells/200 μL/well) on a 96-well plate (Iwaki/Asahi Techno Glass Corporation, Tokyo, Japan), and the test compounds dissolved in DMSO were added. DMSO only was added as a control (final DMSO concentration was 0.25% v/v). The cells were incubated with shaking for 2 h, and then the CCK-8 solution was added. After another 4 h of incubation, the absorbance at 450 nm was measured (reference 600 nm) using Infinite M200 PRO Microplate Readers (TECAN, Ma€nnedorf, Switzerland), and cell viability was determined.
Statistical Analysis
Statistical analysis was performed using one-way ANOVA with Dunnett’s post hoc test by JMP 15 (SAS Institute Inc., Cary, NC) at significance levels of p < 0.05.
Acknowledgments
This research was supported by the Platform Project for Supporting Drug Discovery and Life Science Research (Basis for Supporting Innovative Drug Discovery and Life Science Research) from AMED under grant number JP20am0101089. This work was also supported by JSPS KAKENHI (grant number 16K08379), the Sasakawa Scientific Research Grant from The Japan Science Society, and a special grant generously provided by the Hoansha Foundation. We thank Akiko Yamada, Miki Kohei, and Yu Kamimura for helping with the synthesis of DCF analogs. Y.T. is grateful to Dr. Takumi Yasuno for his technical advice regarding the COX inhibition assay and cell-based assay.
Glossary
Abbreviations
- AG
acyl glucuronide
- COX
cyclooxygenase
- CYP
cytochrome P450
- DCF
diclofenac
- DMSO
dimethyl sulfoxide
- IDILI
idiosyncratic drug-induced liver injury
- G6P
glucose-6-phosphate
- G6PDH
glucose-6-phosphate dehydrogenase
- HLM
human liver microsome
- LC-MS
liquid chromatography–mass spectrometry
- NADP+
nicotinamide adenine dinucleotide phosphate
- UDP
uridine diphosphate
- UDPGA
UDP-glucuronic acid
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.0c04942.
Synthetic procedures of DCF analogs, analytical data, and NMR spectra (PDF)
Author Contributions
Y.T. carried out almost all of the experimental work and wrote the paper. M.O. performed the experiment. T.O. and T.M. participated in the conception of the experimental work as well as in interpreting the data. K.T. and S.N. participated in discussing the results. All the authors contributed to manuscript revision and approved the final revision of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Walker A. M. Quantitative studies of the risk of serious hepatic injury in persons using nonsteroidal antiinflammatory drugs. Arthritis Rheum. 1997, 40, 201–208. 10.1002/art.1780400204. [DOI] [PubMed] [Google Scholar]
- Laine L.; Goldkind L.; Curtis S. P.; Connors L. G.; Yanqiong Z.; Cannon C. P. How common is diclofenac-associated liver injury? Analysis of 17,289 arthritis patients in a long-term prospective clinical trial. Am. J. Gastroenterol. 2009, 104, 356–362. 10.1038/ajg.2008.149. [DOI] [PubMed] [Google Scholar]
- Boelsterli U. A. Diclofenac-induced liver injury: A paradigm of idiosyncratic drug toxicity. Toxicol. Appl. Pharmacol. 2003, 192, 307–322. 10.1016/S0041-008X(03)00368-5. [DOI] [PubMed] [Google Scholar]
- Uetrecht J.; Naisbitt D. J. Idiosyncratic Adverse Drug Reactions: Current Concepts. Pharmacol. Rev. 2013, 65, 779–808. 10.1124/pr.113.007450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wade L. T.; Kenna J. G.; Caldwell J. Immunochemical identification of mouse hepatic protein adducts derived from the nonsteroidal anti-inflammatory drugs diclofenac, sulindac, and ibuprofen. Chem. Res. Toxicol. 1997, 10, 546–555. 10.1021/tx960153t. [DOI] [PubMed] [Google Scholar]
- Pumford N. R.; Myers T. G.; Davila J. C.; Highet R. J.; Pohl L. R. Immunochemical Detection of Liver Protein Adducts of the Nonsteroidal Antiinflammatory Drug Diclofenac. Chem. Res. Toxicol. 1993, 6, 147–150. 10.1021/tx00032a002. [DOI] [PubMed] [Google Scholar]
- Hargus S. J.; Martin B. M.; George J. W.; Pohl L. R. Covalent Modification of Rat Liver Dipeptidyl Peptidase IV (CD26) by the Nonsteroidal Anti-Inflammatory Drug Diclofenac. Chem. Res. Toxicol. 1995, 8, 993–996. 10.1021/tx00050a001. [DOI] [PubMed] [Google Scholar]
- Hargus S. J.; Amouzedeh H. R.; Pumford N. R.; Myers T. G.; McCoy S. C.; Pohl L. R. Metabolic Activation and Immunochemical Localization of Liver Protein Adducts of the Nonsteroidal Anti-inflammatory Drug Diclofenac. Chem. Res. Toxicol. 1994, 7, 575–582. 10.1021/tx00040a014. [DOI] [PubMed] [Google Scholar]
- Poon G. K.; Chen Q.; Teffera Y.; Ngui J. S.; Griffin P. R.; Braun M. P.; Doss G. A.; Freeden C.; Stearns R. A.; Evans D. C.; Baillie T. A.; Tang W. Bioactivation of diclofenac via benzoquinone imine intermediates - Identification of urinary mercapturic acid derivatives in rats and humans. Drug Metab. Dispos. 2001, 29, 1608–1613. [PubMed] [Google Scholar]
- Gil M. L.; Ramirez M. C.; Terencio M. C.; Castell J. Immunochemical detection of protein adducts in cultured human hepatocytes exposed to diclofenac. Biochim. Biophys. Acta, Mol. Basis Dis. 1995, 1272, 140–146. 10.1016/0925-4439(95)00078-X. [DOI] [PubMed] [Google Scholar]
- Hammond T. G.; Meng X.; Jenkins R. E.; Maggs J. L.; Castelazo A. S.; Regan S. L.; Bennett S. N. L.; Earnshaw C. J.; Aithal G. P.; Pande I.; Kenna J. G.; Stachulski A. V.; Park B. K.; Williams D. P. Mass spectrometric characterization of circulating covalent protein adducts derived from a drug Acyl glucuronide metabolite: Multiple albumin adductions in diclofenac patients. J. Pharmacol. Exp. Ther. 2014, 350, 387–402. 10.1124/jpet.114.215079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue K.; Mizuo H.; Ishida T.; Komori T.; Kusano K. Bioactivation of diclofenac in human hepatocytes and the proposed human hepatic proteins modified by reactive metabolites. Xenobiotica 2020, 50, 919–928. 10.1080/00498254.2020.1728592. [DOI] [PubMed] [Google Scholar]
- Stierlin H.; Faigle J. W.; Sallmann A.; Kung W.; Richter W. J.; Kriemler H. P.; Alt K. O.; Winkler T. Biotransformation of diclofenac sodium voltaren® in animals and in man: I. Isolation and identification of principal metabolites. Xenobiotica 1979, 9, 601–610. 10.3109/00498257909042327. [DOI] [PubMed] [Google Scholar]
- Leemann T.; Transon C.; Dayer P. Cytochrome P450TB (CYP2C): A major monooxygenase catalyzing diclofenac 4′-hydroxylation in human liver. Life Sci. 1993, 52, 29–34. 10.1016/0024-3205(93)90285-B. [DOI] [PubMed] [Google Scholar]
- Bort R.; MacÉ K.; Boobis A.; Gómez-Lechón M. J.; Pfeifer A.; Castell J. Hepatic metabolism of diclofenac: Role of human CYP in the minor oxidative pathways. Biochem. Pharmacol. 1999, 58, 787–796. 10.1016/S0006-2952(99)00167-7. [DOI] [PubMed] [Google Scholar]
- King C.; Tang W.; Ngui J.; Tephly T.; Braun M. Characterization of rat and human UDP-glucuronosyltransferases responsible for the in Vitro glucuronidation of diclofenac. Toxicol. Sci. 2001, 61, 49–53. 10.1093/toxsci/61.1.49. [DOI] [PubMed] [Google Scholar]
- Den Braver M. W.; den Braver-Sewradj S. P.; Vermeulen N. P. E.; Commandeur J. N. M. Characterization of cytochrome P450 isoforms involved in sequential two-step bioactivation of diclofenac to reactive p-benzoquinone imines. Toxicol. Lett. 2016, 253, 46–54. 10.1016/j.toxlet.2016.04.022. [DOI] [PubMed] [Google Scholar]
- Tang W.; Stearns R. A.; Wang R. W.; Chiu S. H. L.; Baillie T. A. Roles of human hepatic cytochrome P450s 2C9 and 3A4 in the metabolic activation of diclofenac. Chem. Res. Toxicol. 1999, 12, 192–199. 10.1021/tx9802217. [DOI] [PubMed] [Google Scholar]
- Yu L. J.; Chen Y.; Deninno M. P.; O’Connell T. N.; Hop C. E. C. A. Identification of a novel glutathione adduct of diclofenac, 4′-hydroxy-2′-glutathion-deschloro-diclofenac, upon incubation with human liver microsomes. Drug Metab. Dispos. 2005, 33, 484–488. 10.1124/dmd.104.002840. [DOI] [PubMed] [Google Scholar]
- Tang W.; Stearns R. A.; Bandiera S. M.; Zhang Y.; Raab C.; Braun M. P.; Dean D. C.; Pang J.; Leung K. H.; Doss G. A.; Strauss J. R.; Kwei G. Y.; Rushmore T. H.; Chiu S. H.; Baillie T. A. Studies on cytochrome P-450-mediated bioactivation of diclofenac in rats and in human hepatocytes: Identification of glutathione conjugated metabolites. Drug Metab. Dispos. 1999, 27, 365–372. [PubMed] [Google Scholar]
- Shen S.; Marchick M. R.; Davis M. R.; Doss G. A.; Pohl L. R. Metabolic activation of diclofenac by human cytochrome P450 3A4: Role of 5-hydroxydiclofenac. Chem. Res. Toxicol. 1999, 12, 214–222. 10.1021/tx9802365. [DOI] [PubMed] [Google Scholar]
- Naisbitt D.; Sanderson L.; Meng X.; Stachulski A.; Clarke S.; Park B. Investigation of the immunogenicity of diclofenac and diclofenac metabolites. Toxicol. Lett. 2007, 168, 45–50. 10.1016/j.toxlet.2006.10.014. [DOI] [PubMed] [Google Scholar]
- Masubuchi Y.; Ose A.; Horie T. Diclofenac-induced inactivation of CYP3A4 and its stimulation by quinidine. Drug Metab. Dispos. 2002, 30, 1143–1148. 10.1124/dmd.30.10.1143. [DOI] [PubMed] [Google Scholar]
- Yan Z.; Li J.; Huebert N.; Caldwell G. W.; Du Y.; Zhong H. Detection of a novel reactive metabolite of diclofenac: Evidence for CYP2C9-mediated bioactivation via arene oxides. Drug Metab. Dispos. 2005, 33, 706–713. 10.1124/dmd.104.003095. [DOI] [PubMed] [Google Scholar]
- Iwamura A.; Nakajima M.; Oda S.; Yokoi T. Toxicological potential of acyl glucuronides and its assessment. Drug Metab. Pharmacokinet. 2017, 32, 2–11. 10.1016/j.dmpk.2016.11.002. [DOI] [PubMed] [Google Scholar]
- Kretz-Rommel A.; Boelsterli U. A. Mechanism of covalent adduct formation of diclofenac to rat hepatic microsomal proteins: Retention of the glucuronic acid moiety in the adduct. Drug Metab. Dispos. 1994, 22, 956–961. [PubMed] [Google Scholar]
- Oda S.; Shirai Y.; Akai S.; Nakajima A.; Tsuneyama K.; Yokoi T. Toxicological role of an acyl glucuronide metabolite in diclofenac-induced acute liver injury in mice. J. Appl. Toxicol. 2017, 37, 545–553. 10.1002/jat.3388. [DOI] [PubMed] [Google Scholar]
- Kretzrommel A.; Boelsterli U. A. Diclofenac covalent protein binding is dependent on acyl glucuronide formation and is inversely related to P450-mediated acute cell injury in cultured rat hepatocytes. Toxicol. Appl. Pharmacol. 1993, 120, 155–161. 10.1006/taap.1993.1097. [DOI] [PubMed] [Google Scholar]
- Koga T.; Fujiwara R.; Nakajima M.; Yokoi T. Toxicological evaluation of acyl glucuronides of nonsteroidal anti-inflammatory drugs using human embryonic kidney 293 cells stably expressing human UDP-glucuronosyltransferase and human hepatocytes. Drug Metab. Dispos. 2011, 39, 54–60. 10.1124/dmd.110.035600. [DOI] [PubMed] [Google Scholar]
- Pang Y. Y.; Tan Y. M.; Chan E. C. Y.; Ho H. K. Phase I Metabolic Stability and Electrophilic Reactivity of 2-Phenylaminophenylacetic Acid Derived Compounds. Chem. Res. Toxicol. 2016, 29, 1118–1131. 10.1021/acs.chemrestox.6b00042. [DOI] [PubMed] [Google Scholar]
- Pang Y. Y.; Yeo W. K.; Loh K. Y.; Go M. L.; Ho H. K. Structure-toxicity relationship and structure-activity relationship study of 2-phenylaminophenylacetic acid derived compounds. Food Chem. Toxicol. 2014, 71, 207–216. 10.1016/j.fct.2014.06.013. [DOI] [PubMed] [Google Scholar]
- Moser P.; Sallmann A.; Wiesenberg I. Synthesis and Quantitative Structure-Activity Relationships of Diclofenac Analogues. J. Med. Chem. 1990, 33, 2358–2368. 10.1021/jm00171a008. [DOI] [PubMed] [Google Scholar]
- Bertinaria M.; Shaikh M. A. A. G.; Buccellati C.; Cena C.; Rolando B.; Lazzarato L.; Fruttero R.; Gasco A.; Hoxha M.; Capra V.; Sala A.; Rovati G. E. Designing Multitarget Anti-inflammatory Agents: Chemical Modulation of the Lumiracoxib Structure toward Dual Thromboxane Antagonists-COX-2 Inhibitors. ChemMedChem 2012, 7, 1647–1660. 10.1002/cmdc.201200272. [DOI] [PubMed] [Google Scholar]
- Yang Z.; Xu J. Convenient and environment-friendly synthesis of sulfonyl chlorides from S-alkylisothiourea salts via N-chlorosuccinimide chlorosulfonation. Synthesis 2013, 45, 1675–1682. 10.1055/s-0033-1338743. [DOI] [Google Scholar]
- Johnson B. M.; Shu Y. Z.; Zhuo X.; Meanwell N. A. Metabolic and Pharmaceutical Aspects of Fluorinated Compounds. J. Med. Chem. 2020, 63, 6315–6386. 10.1021/acs.jmedchem.9b01877. [DOI] [PubMed] [Google Scholar]
- Lee C.; Liao J.; Chen S.; Yen C.; Lee Y.; Huang S.; Huang S.; Lin C.; Chang V. H. Fluorine-Modified Rutaecarpine Exerts Cyclooxygenase-2 Inhibition and Anti-inflammatory Effects in Lungs. Front. Pharmacol. 2019, 10, 1–12. 10.3389/fphar.2019.00091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tateishi Y.; Ohe T.; Yasuda D.; Takahashi K.; Nakamura S.; Kazuki Y.; Mashino T. Synthesis and evaluation of nevirapine analogs to study the metabolic activation of nevirapine. Drug Metab. Pharmacokinet. 2020, 35, 238–243. 10.1016/j.dmpk.2020.01.006. [DOI] [PubMed] [Google Scholar]
- Kumar S.; Samuel K.; Subramanian R.; Braun M. P.; Stearns R. A.; Chiu S.-H. L.; Evans D. C.; Baillie T. A. Extrapolation of Diclofenac Clearance from in Vitro Microsomal Metabolism Data: Role of Acyl Glucuronidation and Sequential Oxidative Metabolism of the Acyl Glucuronide. J. Pharmacol. Exp. Ther. 2002, 303, 969–978. 10.1124/jpet.102.038992. [DOI] [PubMed] [Google Scholar]
- Richert L.; Baze A.; Parmentier C.; Gerets H. H. J.; Sison-Young R.; Dorau M.; Lovatt C.; Czich A.; Goldring C.; Park B. K.; Juhila S.; Foster A. J.; Williams D. P. Cytotoxicity evaluation using cryopreserved primary human hepatocytes in various culture formats. Toxicol. Lett. 2016, 258, 207–215. 10.1016/j.toxlet.2016.06.1127. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.